Abstract
Eleven peptides of the general formula H-d-Ser-Ala-Arg-NH-X, where X = (CH2)n-NH2, n = 2–9, (CH2)m-OH, m = 2–4, were obtained and tested for their effect on the amidolytic activities of urokinase, thrombin, trypsin, plasmin, t-PA, and kallikrein. H-d-Ser-Ala-Arg-NH-(CH2)5-NH2 inhibited urokinase with a Ki value of 6.3 μM.
Introduction
Plasminogen activators are highly specific serine proteases capable of activating plasminogen to plasmin. Two kinds have been identified in mammals, urokinase-type (u-PA) and tissue-type (t-PA) plasminogen activators. Urokinase is important in tissue remodeling and t-PA in vascular fibrinolysis. They share the same primary physiological substrate (plasminogen) and inhibitors (plasminogen activator inhibitor types I and II). Studies have indicated that urokinase has an ability to degrade the extracellular matrix, and is a key mediator in cellular invasion, growth, and the metastasis of tumorsCitation1–4. Elevated levels of u-PA in cancer cells usually indicate a poor prognosis for patient survivalCitation5,Citation6. Consequently, a selective inhibitor for u-PA may be therapeutically useful in cancer treatment. Because t-PA and u-PA possess an extremely high degree of structural similarity, it is difficult to find inhibitors with specificity exclusively toward urokinaseCitation7–9. Structural studies of small molecule inhibitors of urokinase (residues of inhibitor abbreviated as P3-P2-P1) are based on the structure of the enzyme. It is desirable that a synthetic u-PA inhibitor has adequate potency and selectivity for u-PA relative to t-PA, plasmin, thrombin, and trypsin, to avoid the possibility of antifibrinolytic side effects.
Initial studies of enzyme–inhibitor complexes by Spraggon et al., Nienaber et al., and Katz et al. provided information about the binding of a covalent peptidic inhibitor Glu-Gly-Arg-chloromethyl ketoneCitation10–12. These studies showed that the guanidine moiety of arginine (P1) forms an ionic interaction with Asp-189 carboxylate at the S1 site of urokinase. The other sub-site S2 is limited by His-57. The residues of inhibitors larger than glycine or alanine (P2) cannot be accommodated in this site without rearrangement of the histidine moietyCitation13–15. Tamura et al. reported an inhibitor of urokinase containing a d-serine as P3 residue: d-Ser-Ala-Arg-alCitation16. The P3 side-chain of peptidic inhibitors is normally oriented into the solvent and does not interact with trypsin-like proteinases. When position P3 is occupied by unnatural d-amino acid, the side-chain of the amino acid projects into the S4 pocket. The sub-site S4 contains His-99, and the residue of d-Ser could form a favorable interaction with this.
Recently, we described the first analogs of a series of tripeptides as inhibitors of urokinase containing an N-terminal d-Ser and Ac-d-Ser moieties, Gly and Ala as P2 residues, and a C-terminal Arg-OH and Arg-NH2 in the P1 positionCitation17. Compounds with a free amino group: H-d-Ser-Ala-Arg-OH and H-d-Ser-Gly-Arg-OH, inhibited the amidolytic activity of urokinase, thrombin, plasmin, and trypsin. The acetylated acids of tripeptides showed some selectivity: Ac-d-Ser-Ala-Arg-OH inhibited the amidolytic activities of thrombin and trypsin, but Ac-d-Ser-Gly-Arg-OH inhibited the amidolytic activities of urokinase and plasmin. Among all the compounds with an amide as a C-terminal group, only H-d-Ser-Gly-Arg-NH2 showed inhibitory activity on plasmin.
We present the synthesis of peptides of the general formula H-d-Ser-Ala-Arg-NH-X, where X = (CH2)n-NH2, n = 2–9, (CH2)m-OH, m = 2–4. Six trypsin-like serine proteases were used to determine the amidolytic activity of potential inhibitors. Trypsin was used as a standard enzyme for this protease class, whereas thrombin, plasmin, and kallikrein were selected to predict a possible influence on blood coagulation and fibrinolysis. We expected that these kinds of peptide derivatives would show a high selectivity toward urokinase versus t-PA.
Materials and methods
Materials
Fmoc-Arg(Pbf)-OH (Fmoc, 9-fluorenylmethyloxycarbonyl; Pbf, pentamethyldihydrobenzofuran), Fmoc-Ala-OH, chloranil, TNBS (2,4,6-trinitrobenzenesulfonic acid; 1% solution in dimethylformamide (DMF)), acetaldehyde, HOBt (1- hydroxybenzotriazole), 3-amino-1-propanol 2-chloritrityl resin, and 4-amino-1-butanol 2-chlorotrityl resin were purchased from Fluka. Fmoc-d-Ser(t-Bu)-OH (t-Bu, t-butyl), 1,2-diaminoethane-, 1,5-diaminopentane-, 1,6-diaminohexane-, 1,7-diaminoheptane-, 1,9-diaminononanetrityl resins, and glycinol 2-chlorotrityl resin were purchased from Merck (Novabiochem). TFA (trifluoroacetic acid), DIPEA (diisopropylethylamine), piperidine, TBTU (tetrafluoroborate salt of the O-(7-azabenzotriazolyl)-tetramethyl uronium cation), DIC (diisopropylcarbodiimide), NMP (1-methyl-2-pirrolidon), and 2-chlorotrityl chloride were obtained from Iris Biotech GmbH. DCM (dichloromethane) and DMF were products of Chempur. DCM was used without further purification; DMF was distilled over ninhydrin and stored under molecular sieves type 4A. High performance liquid chromatography (HPLC) solvent, acetonitrile, was purchased from Merck. 1,8- Diaminooctane was obtained from Aldrich.
Urokinase, trypsin, kallikrein, and Bzl-l-Arg-pNA·HCl (Bzl, benzyl) were purchased from Sigma. Plasmin, S-2444 (pyro-Glu-Gly-Arg-pNA·HCl), S-2238 (H-d-Phe-Pip-Arg-pNA), S-2251 (H-d-Val-Leu-Lys-pNA), S-2266 (H-d-Val-Leu-Arg-pNA·2HCl, and S-2288 (H-d-Ile-Pro-Arg-pNA) were obtained from Chromogenix. Thrombin was purchased from Lubelska Wytwórnia Szczepionek, and t-PA was obtained from Boehringer Ingelheim.
Synthesis of inhibitors
The peptides shown in were synthesized manually using a standard Fmoc-based strategy. Fmoc deprotection steps were carried out with 20% (v/v) piperidine in DMF/NMP (1:1) for 15 min. Coupling reactions of Fmoc amino acids were performed in DMF/NMP/DCM (1:1:1) using a molar ratio of amino acid/DIC/HOBt/resin of 3:3:3:1 in the case of coupling of Fmoc-Arg(Pbf)-OH, Fmoc-Ala-OH, and Fmoc-Gly-OH. In the case of coupling of Fmoc-d-Ser(t-Bu)-OH, the molar ratio of amino acid/TBTU/HOBt/DIPEA/resin was 2:2:2:4:1. Reactions were monitored with the Stewart chloranil test for amino alcohol resin and with the TNBS test for diamine resins.
Table 1. Structures of obtained peptides: H-d-Ser-Ala-Arg-NH-X.
Cleavage from the resin was carried out with TFA (thiohydroxamic acid)/water (95:5). After 2.5 h stirring, the resin was filtered and washed with TFA. The combined filtrates were concentrated under reduced pressure. The crude peptide was washed with cold diethyl ether, filtered, dissolved in glacial acetic acid, and lyophilized.
The Shimadzu LC-10A system was used for analytical and semipreparatory HPLC (Phenomenex C18, Jupiter 90A, 4 μm, 250 × 10 mm; Phenomenex C18, Jupiter 300A, 5 μm, 250 × 4 mm; solvents: A, 0.1% aqueous TFA; B, 0.1% TFA in acetonitrile, gradient 0% B to 100% B in A in 30 min, flow rate 1 mL/min, monitored at 220 nm). The major peak fraction was pooled and lyophilized. Molecular weight determination was performed by mass spectrometry using a Bruker Daltonics Esquire 6000 with electrospray ionization (ESI). Mass spectrometry analysis confirmed purity and identity ().
Table 2. Analytical data of the synthesized compounds.
Enzymatic investigations
Determination of amidolytic activity was performed as described previouslyCitation18. Detailed description of the method is given below. To 0.2 cm3 of examined compound (1–11; 0.15 M NaCl as control), buffer, and 0.1 cm3 of enzyme solution was added:
a. tris buffer—0.6 cm3 (pH = 8.8); enzyme: urokinase (50 units/cm3); synthetic substrate: S-2444 (0.1 cm3, 3 mM/dm3);
b. tris buffer—0.5 cm3 (pH = 8.4); enzyme: thrombin (1 unit/cm3); synthetic substrate: S-2238 (0.2 cm3, 0.75 mM/dm3);
c. tris buffer—0.5 cm3 (pH = 7.4); enzyme: plasmin (0.4 unit/cm3); synthetic substrate: S-2251 (0.2 cm3, 3 mM/dm3);
d. borane buffer—0.5 cm3 (pH = 7.5); enzyme: trypsin (0.4 unit/cm3), synthetic substrate: Bzl-l-Arg-pNA·HCl (0.2 cm3, 8 mM/dm3);
e. tris buffer—0.6 cm3 (pH = 9.0); enzyme: kallikrein (3 units/cm3); synthetic substrate: S-2266 (0.1 cm3, 7.5 mM/dm3);
f. tris buffer—0.6 cm3 (pH = 8.4); enzyme: t-PA (1.67 mg/cm3); synthetic substrate: S-2288 (0.1 cm3, 10 mM/dm3).
The mixture was incubated at 37°C for 3 min then synthetic substrate solution in the same buffer was added. After 20 min of incubation, adding 0.1 cm3 of 50% acetic acid stopped the reaction and absorbance of the released p-nitroaniline was measured at 405 nm. Every value represents the average of triplicate determination. The IC50 value was considered as the concentration of inhibitor that decreased the absorbance at 405 nm by 50%, compared with the absorbance measured under the same conditions without inhibitor. Inhibition constant Ki was calculated from IC50 based on the Cheng–Prusoff equationCitation19. Results are given in .
Table 3. Inhibition of H-d-Ser-Ala-Arg-NH-X on the amidolytic activity of enzymes.
Our results were compared with the data obtained by Tamura et al.Citation16 for 2-phenethyl-SO2-d-Ser-Ala-Arg-al, the irreversible urokinase inhibitor with the same tripeptide sequence. The determination methods were identical.
Results and discussion
The examined compound did not influence the enzymatic activity of kallikrein and t-PA.
According to the obtained results, the compounds containing amide residues substituted with a short aliphatic amine chain (CH2)n-NH2 (n = 2, 3, 4) as a C-terminal group did not have an influence on the amidolytic activity of the investigated enzymes. Compound 4 containing an amide group substituted with (CH2)5-NH2 residue inhibited urokinase and plasmin. Compound 5 with the (CH2)6-NH2 group showed weak activity toward trypsin and plasmin. Compounds 6 and 8 with (CH2)7-NH2 and (CH2)9-NH2 inhibited urokinase, trypsin, and plasmin. Compound 7 containing (CH2)8-NH2 residue inhibited urokinase, thrombin, and trypsin, but not plasmin; 7 (H-d-Ser-Ala-Arg-NH-(CH2)8-NH2) with Ki value 0.24 μM was the most active compound toward trypsin. Compounds 9, 10, and 11 with (CH2)2-OH, (CH2)3-OH, and (CH2)4-OH residues were poor inhibitors of plasmin, but compounds 10 and 11 were better inhibitors of trypsin. All compounds with a hydroxyl residue of an amide group did not inhibit urokinase and thrombin.
We expected that the use of a specific tripeptide sequence would cause much higher selectivity with regard to urokinase. Compound 4 contained a fragment of pentamethylenediamine (cadaverine) as an amide residue in its structure, which seems to have a significant influence on selectivity. The moiety of cadaverine is a form of decarboxylated derivative of lysine. In the literature, there are peptides described with a C-terminus lysine amideCitation20 and also an amide residue of lysine substituted with cadaverine, as inhibitors of plasminCitation21. These kinds of compounds were designed to be similar to a natural substrate sequence hydrolyzed by plasminCitation22. However, H-d-Ser-Ala-Arg-NH2, tested previously by Markowska et al.Citation17, showed no activity toward plasmin and urokinase, but H-d-Ser-Gly-Arg-NH2 showed weak antiplasmin activity (IC50 = 1 mM, Ki = 90 nM; unpublished data).
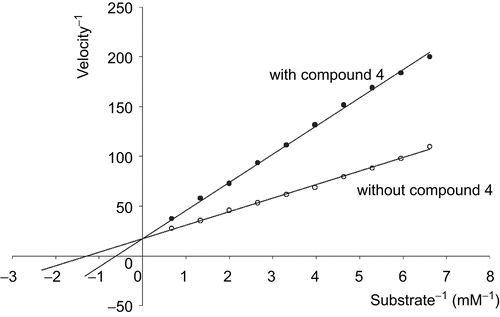
The most active inhibitor of urokinase was 4, H-d-Ser-Ala-Arg-NH-(CH2)5-NH2, with Ki value 6.3 μM. The obtained values of Ki were higher than those of the earlier described inhibitorsCitation15,Citation16. However, 2-phenethyl-SO2-d-Ser-Ala-Arg-al and i-Boc-d-Ser-Ala-Arg-al are alkylating agents, and irreversibly inhibit urokinase by forming a covalent adduct with an active site of the enzyme. presents the Lineweaver–Burk analysis for the effect of 4 on the activity of urokinase. The results show that compound 4 competitively inhibited urokinase.
Figure 1. Lineweaver–Burk analysis of compound 4 inhibition of urokinase was performed in amidolytic assays with S-2444 as described in “Enzymatic investigations.” S-2444 substrate concentration was 3 mM. Compound 4 concentration was 2 mM. Data represent the means of triplicate determinations.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest.
Related Research Data
References
- Duffy MJ. The urokinase plasminogen system: role in malignancy. Curr Pharm Design 2004;10:39–49.
- Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Met Rev 2003;22:205–22.
- Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signalling for cell adhesion, migration and growth. Curr Opin Cell Biol 2000;12:613–20.
- Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogen 1999;3:15–23.
- Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Envolving role of uPA/uPAR system in human cancers. Cancer Treat Res 2008;34:122–36.
- Binder BR, Mihaly J. The plasminogen activator inhibitor “paradox” in cancer. Immun Lett 2008;118:116–124.
- Ke SH, Coombs GS, Tachias K, Corey DR, Madison EL. Optimal subsite occupancy and design of a selective inhibitor of urokinase. J Biol Chem 1997;272:20456–62.
- Rockway TW, Nienaber V, Giranda VL. Inhibitors of protease domain of urokinase-type plasminogen activator. Curr Pharm Design 2002;8:2541–58.
- Rockway TW, Giranda VL. Inhibitors of the proteolytic activity of urokinase type plasminogen activator. Curr Pharm Design 2003;9:1483–98.
- Spraggon G, Philips C, Nowak UK, Ponting CP, Saunders D. The crystal structure of the catalytic domain of human urokinase-type plasminogen activator. Structure 2000;8:681–91.
- Nienaber VL, Davidson D, Edalji R, Giranda VL, Klinghofer V, Henkin J. Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Struct Fold Des 2000;8:553–63.
- Katz BA, Mackman R, Luong C, Radika K, Martelli A, Sprengeler PA, et al. Structural basis for selectivity of small molecule, S1-binding, submicromolar inhibitor of urokinase-type plasminogen activator. Chem Biol 2000;7:299–312.
- Schweinitz A, Steimetzer T, Banke IJ, Arlt MJE, Stürzebecher A, Schuster O, et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J Biol Chem 2004;279:33613–22.
- Zeslawska E, Schweinitz A, Karcher A, Sondermann P, Sperl S, Stürzebecher J, et al. Crystals of urokinase type plasminogen activator variant βc-uPA in complex with small molecule inhibitors open the way towards structure-based drug design. Mol Biol 2000;301:465–75.
- Zeslawska E, Jacob U, Schweinitz A, Coombs G, Bode W, Madison EJ. Crystals of urokinase type plasminogen activator complexes reveal the binding mode of peptidomimetic inhibitors. Mol Biol 2003;328:109–18.
- Tamura S, Weinhouse MI, Roberts CA, Goldman EA, Masukawa K, Anderson SM, et al. Synthesis and biological activity of peptidyl aldehyde urokinase inhibitors. Biorg Med Chem Lett 2000;10:983–7.
- Markowska A, Bruzgo I, Midura-Nowaczek K. Effects of tripeptides on the amidolytic activities of urokinase, thrombin, plasmin and trypsin. Int J Pept Res Ther 2008;14:215–18.
- Okada Y, Tsuda Y, Teno N, Wanaka K, Bohgaki M, Hijikata-Okunomiya A, et al. Synthesis of active center-directed peptide inhibitors of plasmin. Chem Pharm Bull 1988;36:1289–97.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973; 22:3099–108.
- Markowska A, Bruzgo I, Midura-Nowaczek K. Low molecular peptides as potential inhibitors of plasmin. Acta Pol Pharm 2007;64:355–8.
- Midura-Nowaczek K, Lepietuszko I, Bruzgo I. Synthesis of alkylamides of dipeptides as potential plasmin inhibitors. Acta Pol Pharm 2006;63:33–7.
- Hervio LS, Coombs GS, Bergstrom RC, Trivedi K, Corey DR, Madison EL. Negative selectivity and the evolution of protease cascades: the specificity of plasmin for peptide and protein substrates. Chem Biol 2000;7:443–53.