Abstract
Studies on carbonic anhydrase (CA, EC 4.2.1.1) inhibitors have increased due to several therapeutic applications while there are few investigations on activators. Here we investigated CA inhibitory and activatory capacities of a series of dopaminergic compounds on human carbonic anhydrase (hCA) isozymes I, II, and VI. 2-Amino-1,2,3,4-tetrahydronaphthalene-6,7-diol hydrobromide and 2-amino-1,2,3,4-tetrahydronaphthalene-5,6-diol hydrobromide were found to show effective inhibitory action on hCA I and II whereas 2-amino-5,6-dibromoindan hydrobromide and 2-amino-5-bromoindan hydrobromide exhibited only moderate inhibition against both isoforms, being more effective inhibitors of hCA VI. Ki values of the molecules 3–6 were in the range of 41.12–363 μM against hCA I, of 0.381–470 μM against hCA II and of 0.578–1.152 μM against hCA VI, respectively. Compound 7 behaved as a CA activator with KA values of 27.3 μM against hCA I, of 18.4 μM against hCA II and of 8.73 μM against hCA VI, respectively.
Introduction
Carbonic anhydrase (EC 4.2.1.1., CA) is a pH regulatory/metabolic enzyme in all life kingdoms, found in organisms all over the phylogenetic treeCitation1–3 catalyzing the hydration of carbon dioxide to bicarbonate and the corresponding dehydration of bicarbonate in acidic medium with regeneration of CO2Citation4. Sixteen isozymes have been described up to now in mammals, the most active ones as catalysts for carbon dioxide hydration being CA II and CA IXCitation4–7. The sixteen isozymes differ in their subcellular localization, catalytic activity and susceptibility to different classes of inhibitors. Some of them are cytosolic (CA I, CA II, CA III, CA VII and CA XIII), others are membrane bound (CA IV, CA IX, CA XII and CA XIV), two are mitochondrial (CA VA and CA VB), and one is secreted in saliva (CA VI). It has been reported that CA XV isoform is not expressed in humans or in other primates, but it is abundant in rodents and other vertebratesCitation6–15. CAs are produced in a variety of tissues where they participate in several important biological processes such as acid-base balance, respiration, carbon dioxide and ion transport, bone resorption, ureagenesis, gluconeogenesis, lipogenesis and body fluid generationCitation1–10. CA isozymes involved in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders such as edema, glaucoma, obesity, cancer, epilepsy and osteoporosisCitation16–18.
Our groups have recently investigated the interaction of all 16 mammalian CA isozymes with several types of phenols, such as the simple phenol and several of its substituted derivatives, e.g., clioquinol, diphenols, paracetamol, salicyclates and some of their derivatives as well as halogen included phenolic compoundsCitation11–15.
The neurotransmitter dopamine (1) plays a central role in central nervous system-related disorders such as schizophrenia and Parkinson’s diseaseCitation19. In recent years many chemical compounds have been found to possess dopamine-like actions. It has been suggested that 6,7-ADTN (3), a dopamine-like compound, interacts with the dopamine receptor with slightly greater affinity than dopamine itself. Cannon et al. have synthesized a series of 2-amino-4,5-dihydroxyindans, including compounds 2, 4, and reported that certain N-alkylated 4,5-dihydroxyindanes were violent emetics in the dog, and were potent in blockade of the effect of stimulation of the cardioaccelarator nerve in the catCitation20. Aminoindan 2 has been reported to have adrenergic effectsCitation21 and covalent bindingCitation22 to Src family SH2 domains. The hydrochloride salt of 4 has been reported to be useful as an analgesicCitation23. 5,6-Dimethoxy-2-(N-dipropyl)-aminoindan, PNU-99194A, has been reported to be a selective dopamine D3 receptor antagonist with potential antipsychotic properties in animal modelsCitation24.
In the present study, we aimed to purify human CA I, II and VI (hCA I, hCA II and hCA VI) from human blood and examine the in vitro inhibition and activation effects of some dopaminergic compounds on these enzymes, using the esterase activity of hCA I, II and VI, with 4-nitrophenyl acetate as substrate.
Materials and methods
All chemicals were of analytical grade and obtained from either Sigma or Merck
CA purification assay
Purification of hCA I and hCA II were previously describedCitation6. Serum were obtained from fresh human blood obtained from the Blood Center of the Research Hospital at Atatürk University. The blood samples were centrifuged at 5000 rpm for 15 min and precipitant were removed. The serum was isolated. The pH was adjusted to 8.7 with solid Tris. Sepharose-4B-aniline-sulfanylamide affinity column equilibrated with 25 mM Tris-HCl/0.1M Na2SO4 (pH 8.7). The affinity gel was washed with 25 mM Tris-HCl/22 mM Na2SO4 (pH 8.7). The human carbonic anhydrase (hCA VI) isozyme was eluted with 0,25 M H2NSO3H/25 mM Na2HPO4 (pH = 6,7). All procedures were performed at 4°CCitation25.
CA activity assay and kinetic studies
CA activity was assayed by following the change in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer (CHEBIOS UV-VIS) according to the method described by Verpoorte et alCitation26.. The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4 mL 0.05 M Tris-SO4 buffer (pH 7.4), 1 mL 3 mM 4-NPA, 0.5 mL H2O and 0.1 mL enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution. The inhibitory and activatory effects of the dopaminergic compounds were examined. All compounds were tested in triplicate at each concentration used. Different concentrations of the compounds were used. Control cuvette activity in the absence of inhibitor was taken as 100%. For the compounds, an Activity (%)-[Inhibitor] or Activity (%)-[Activator] graphs were drawn. To determine Ki and Ka values, three different concentrations of the compounds were tested. In these experiments, 4-NPA was used as substrate at five different concentrations (0.15–0.75 mM). The Lineweaver–Burk curves were drawnCitation27.
Protein determination
Protein quantity was determined spectrophotometrically at 595 nm according to the Bradford method during the purification steps, using bovine serum albumin as the standardCitation28.
SDS polyacrylamide gel electrophoresis
SDS polyacrylamide gel electrophoresis was performed after purification of the enzymes. It was carried out in 10% and 3% acrylamide for the running and the stacking gel, respectively, containing 0.1% SDS according to Laemmli procedure. A 20 μg sample was applied to the electrophoresis medium. Gels were stained for 1.5 h in 0.1% Coommassie Brilliant Blue R-250 in 50% methanol and 10% acetic acid, then destained with several changes of the same solvent without the dyeCitation29.
Results and discussion
CA isoenzymes, particularly hCA II, have been observed to be inhibited by phenolic compounds in many studiesCitation10–15. The reason for investigating the inhibitory effects of phenols on CA isoenzymes is that phenol is the unique competitive inhibitor with CO2 as substrate of CA IICitation10. Christianson’s group demonstrated by X-ray crystallography that phenol anchors its OH moiety to the zinc-bound water/hydroxide ion of the enzyme active site through a hydrogen bond and through a second hydrogen bond to the NH amide of Thr199, which is a conserved amino acid in all α-CAs and crucial for its catalytic activity and binding of inhibitorsCitation10. The phenyl moiety of phenol was found to lay in the hydrophobic part of the hCA II active site, where CO2, the physiologic substrate of the CAs, binds in the precatalytic complex, explaining the behaviour of phenol as a unique CO2 competitive inhibitor. Only recently, our group investigated the interactions of phenol and some of its substituted derivatives with all mammalian isozymes, CA I–XVCitation11–15, demonstrating inhibition of the enzymes in very low concentrations and the possibility to design isozyme selective CA inhibitors. Indeed, the inhibition profile of various isozymes with this class of agents is very variable, with inhibition constants ranging from the millimolar to the submicromolar range for many simple phenolsCitation11–15.
The purification of the three CA isozymes was performed using a simple one step method with a Sepharose-4B-aniline-sulfanilamide affinity column chromatoghrapy. Human erythrocyte CA I isoenzyme was purified, 121-fold with a specific activity of 874 EUmg−1 and overall yield of 43%; CA II isoenzyme was purified, 810-fold with a specific activity of 6342 EUmg−1 and overall yield of 53%; CA VI isoenzyme was purifed,63-fold with a specific activity of 242 EUmg−1 and overall yield of 28%.
We report here the first study on the inhibitory or activatory effects of dopaminergic compounds of type 3–7 on the esterase activity of hCA I, II and VI. The sulfonamide acetazolamide (AZA) was used as a negative control in our experiments, and for comparison reasons. Previous reports of our groups investigated other phenol-based derivatives (including salicylic acid derivatives and paracetamol) by using a stopped flow, CO2 hydration assay for monitoring CA inhibition. Data of show the following inhibition of hCA I, II and VI with phenols 3–6, by an esterase assay, with 4-NPA as substrate:
Table 1. hCA I, II and VI inhibition data with compounds 3–6, 10–11 and acetazolamide (AZA), by an esterase assay with 4-nitrophenylacetate as substrate
(i) Against the slow cytosolic isozyme hCA I, compounds 5, 6 behaved as weak inhibitors (), with Ki values of 161 and 363 μM. A second group of derivatives, including 3 and 4 showed better inhibitory activity compared to the previously mentioned methoxy aromatic compounds, with Ki values of 41.12 and 45.08 μM (). Acetazolamide is also a medium CAI with this assay and substrate against hCA I (KI of 36.37 μM). Kinetic investigations (Lineweaver–Burk plots, data not shown) indicate that similar to sulfonamides and inorganic anionsCitation1–3, all the investigated phenols act as noncompetitive inhibitors with 4-NPA as substrate, i.e., they bind in different regions of the active site cavity as compared to the substrate.
Scheme 1. Dopaminergic compounds investigated in the present study.
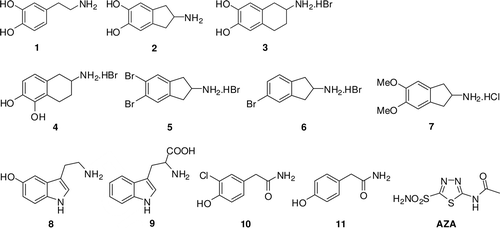
(ii) A better inhibitory activity has been observed with compounds 3, 4 for the inhibition of the rapid cytosolic isozyme hCA II at low micromolar concentrations (). Two derivatives, 5, 6, showed weak hCA II inhibitory activity with Ki-s in the range of 143–470 μM (). The best hCA II inhibitor in this series of derivatives was the bulky, compound 4, which with a Ki of 0.381 μM, is better inhibitor than acetazolamide, a clinically used sulfonamide. It must be stressed that Ki-s measured with the esterase method are always in the micromolar range because hCA I and II are weak esterasesCitation30.
(iii) 6,7-ADTN 3 and its congener 5,6-ADTN 4 are quite effective inhibitors of the secreted isozyme hCA VI, with Ki-s of 1.152 and 0.968 μM. However, derivatives 5 and 6 show a higher affinity for this isozyme, with inhibition constants in the range of 0.782–0.578 μM ().
(iv) The data in indicate that the ubiquitous cytosolic isozymes hCA I and hCA II are activated by 2-Amino-5,6-dimethoxyindan hydrochloride 7. The aromatic compound 7, serotonin 9 and tryptophan 10, acted as weak hCA I and II activators, with KA-s in the range of 27.3–45 μMCitation31. Medium activating properties against hCA I and II were previously observed on the other hand for dopamine 1, which showed KA in the range of 13.5 μM. The most effective CA activators are generally incorporated both amino and carboxy moieties, as well as a heterocyclic ring (imidazole, indole, etc.) or a substitutedaromatic one (3,4-dihydroxyphenyl, present in dopamineCitation32). The compound 7 is much more efficient hCA I and II activator as compared to serotonin 9 and tryptophan 10, and is quite effective against hCA VI.
Table 2. hCA I, II and VI activation data with compounds 1 and 7–9Citation31
Many molecules have been tested so far as CA inhibitors or activatorsCitation33–36. It is critically important to explore further classes of potent CAIs in order to detect compounds with different inhibition profiles as compared to the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes. It is also important to determine CA activators for pharmacological applications.
Conclusions
Dopaminergic compounds 3–6 inhibited the activity of these three CA isozymes investigated here, i.e., hCA I, II and VI. Our findings indicate another class of possible CAIs of interest, in addition to the well-known ones (sulfonamides, sulfamates, coumarins, etc) although the compounds investigated here exhibited very different inhibition profiles against these isoenzymes. Compound 7 was found to behave as a weak activator against CA I and II, but it was a moderate activator of CA VI. These findings point out that substituted dopaminergic compounds might be used as leads for generating interesting CAIs or CAAs.
Acknowledgement
This study was financed by Turkish Republic Prime Ministry State Planning Organization (DPT), (project no: 2010K120440) and Ağrı İbrahim Çeçen University Scientific Research Council, (project no: Ağrı BAP-2010/K-10) for (MS), and by an FP7 EU grant (Metoxia), for CTS.
Declaration of interest
The authors report no declarations of interest.
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Supuran CT. Carbonic anhydrases—an overview. Curr Pharm Des 2008;14:603–614.
- Supuran CT. Diuretics: from classical carbonic anhydrase inhibitors to novel applications of the sulfonamides. Curr Pharm Des 2008;14:641–648.
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem 1995;64:375–401.
- Ozensoy O, Arslan O, Sinan SO. A new method for purification of carbonic anhydrase isozymes by affinity chromatography. Biochemistry Mosc 2004;69:216–219.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–9105.
- Sentürk M, Talaz O, Ekinci D, Cavdar H, Küfrevioglu OI. In vitro inhibition of human erythrocyte glutathione reductase by some new organic nitrates. Bioorg Med Chem Lett 2009;19:3661–3663.
- Parkkila S, Parkkila AK. Carbonic anhydrase in the alimentary tract. Roles of the different isozymes and salivary factors in the maintenance of optimal conditions in the gastrointestinal canal. Scand J Gastroenterol 1996;31:305–317.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–3660.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–1587.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Inhibition of human mitochondrial carbonic anhydrases VA and VB with para-(4-phenyltriazole-1-yl)-benzenesulfonamide derivatives Bioorg Med Chem 2008;16:7424–7428.
- Innocenti A, Hilvo M, Scozzafava A, Parkkila S, Supuran CT. Carbonic anhydrase inhibitors: Inhibition of the new membrane-associated isoform XV with phenols. Bioorg Med Chem Lett 2008;18:3593–3596.
- Coban TA, Beydemir S, Gulcin I, Ekinci D. The inhibitory effect of ethanol on Carbonic Anhydrase isoenzymes: An in vivo and in vitro study J Enzyme Inhib Med Chem 2008; 23:266–270.
- Ekinci D, Ceyhun SB, Sentürk M, Erdem D, Küfrevioglu OI, Supuran CT. Characterization and anions inhibition studies of an a-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–748.
- Hilvo M, Tolvanen M, Clark A, Shen B, Shah GN, Waheed A et al. Characterization of CA XV, a new GPI-anchored form of carbonic anhydrase. Biochem J 2005;392:83–92.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–27809.
- Ekinci D, Beydemir S, Küfrevioglu OI. In vitro inhibitory effects of some heavy metals on human erythrocyte carbonic anhydrases. J Enzyme Inhib Med Chem 2007;22:745–750.
- Haadsma-Svensson SR, Svensson KA. PNU-99194A: a preferential dopamine D3 receptor antagonist. CNS Drug Rev 1998;4:42–57.
- Cannon JG. Dopamine agonists: structure-activity relationships. Prog Drug Res 1985;29:303–414.
- Gray AP, Reit E, Ackerly JA. Conformational requirements for direct adrenergic stimulation. J Med Chem 1973;16:1023–1027.
- Jagoe CT, Kreifels SE, Li J. Covalent binding of catechols to src family SH2 domains. Bioorg Med Chem Lett 1997;7:113–116.
- Richter H, Schenck M, German Patent 952441 1956; Chem. Abstr., 1959, 53, 2190e.
- Haadsma-Svensson SR, Cleek KA, Dinh DM, Duncan JN, Haber CL, Huff RM et al. Dopamine D(3) receptor antagonists. 1. Synthesis and structure-activity relationships of 5,6-dimethoxy-N-alkyl- and N-alkylaryl-substituted 2-aminoindans. J Med Chem 2001;44:4716–4732.
- Kivelä J, Parkkila S, Waheed A, Parkkila AK, Sly WS, Rajaniemi H. Secretory carbonic anhydrase isoenzyme (CA VI) in human serum. Clin Chem 1997;43:2318–2322.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–4229.
- Lineweaver H, Burk D. The determination of enzyme dissocation constants. J Am Chem Soc 1934;56:658–666.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Laemmli DK. Cleavage of structural proteins during in assembly of the head of Bacteriophage T4. Nature 1970;227:680–685.
- Vomasta D, Innocenti A, König B, Supuran CT. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-alpha-dithienylethene derivatives. Bioorg Med Chem Lett 2009;19:1283–1286.
- Vullo D, Innocenti A, Nishimori I, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase activators: activation of the human isoforms VII (cytosolic) and XIV (transmembrane) with amino acids and amines. Bioorg Med Chem Lett 2007;17:4107–4112.
- Ilies M, Scozzafava A, Supuran CT. Carbonic anhydrase activators. In Carbonic anhydrase – Its inhibitors and activators; Supuran CT, Scozzafava A, Conway J, Eds.; CRC Press: Boca Raton (FL), USA, 2004; p 317.
- Innocenti A, Maresca A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: thioxolone versus sulfonamides for obtaining isozyme-selective inhibitors? Bioorg Med Chem Lett 2008;18:3938–3941.
- Hisar O, Beydemir S, Gülçin I, Küfrevioglu OI, Supuran CT. Effects of low molecular weight plasma inhibitors of rainbow trout (Oncorhynchus mykiss) on human erythrocyte carbonic anhydrase-II isozyme activity in vitro and rat erythrocytes in vivo. J Enzyme Inhib Med Chem 2005;20:35–39.
- Abdülkadir Coban T, Beydemir S, Gülcin I, Gücin I, Ekinci D, Innocenti A et al. Sildenafil is a strong activator of mammalian carbonic anhydrase isoforms I-XIV. Bioorg Med Chem 2009;17:5791–5795.
- Ekinci D, Beydemir S, Alim Z. Some drugs inhibit in vitro hydratase and esterase activities of human carbonic anhydrase-I and II. Pharmacol Rep 2007;59:580–587.