Abstract
In this study, eight non-natural peptides and peptoids incorporating the pentacycloundecane (PCU) lactam were designed and synthesized as potential inhibitors of the wild type C-SA HIV-protease. Five of these inhibitors gave IC50 values ranging from 0.5 up to 0.75 µM against the resistance-prone wild type C-South African HIV-protease. NMR EASY-ROESY studies enabled us to describe the secondary structure of three of these compounds in solution. The 3D structures of the selected cage peptides were also modelled in solution using QM/MM/MD simulations. Satisfactory agreement between the NMR observations and the low energy calculated structures exists. Only one of these inhibitors (11 peptoid), which showed the best IC50(0.5 µM), exhibited a definable 3-D structure in solution. Autodock4 and AutodockVina were used to model the potential interaction between these inhibitors and the HIV-PR. It appears that the docking results are too crude to be correlated with the relative narrow range of experimental IC50 values (0.5–10 µM). The PCU-peptides and peptoides were several orders less toxic (145 μM for 11 and 102 μM for 11 peptoid) to human MT-4 cells than lopinavir (0.025 μM). This is the first example of a polycyclic cage framework to be employed as an HIV-PR transition state analogue inhibitor and can potentially be utilized for other diseases related proteases.
Graphical abstract: PCU-diastereomers peptide and peptoid inhibitors with arrows showing the observed EASY-ROESY correlations.
| Abbreviations | ||
| CNS, | = | central nervous system |
| HIV-PR, | = | HIV-protease |
| PCU, | = | pentacycloundecane |
| PIs, | = | protease inhibitors |
Introduction
The human immunodeficiency virus (HIV) encodes an aspartic protease (PR) that is essential for the formation of mature and infectious virionsCitation1. The HIV-PR is an established target in the chemotherapy of acquired immunodeficiency syndrome (AIDS)Citation2. At present, several protease inhibitors (PIs) have been approved by the Food and Drug Administration (FDA) as commercial HIV drugs, and several others are now in clinical trialsCitation3–5. However, the binding affinity of these drugs to the African prevalent HIV-protease strains A and C is lower; since, they were developed for the type B strain prevalent in Northern America and Western EuropeCitation6. Thus, there is anobvious demand for the development of type A and C PIs.
In this study, we designed cage derived inhibitors and assessed them against the wild type C-SA HIV-1 protease. This follows the successful application of the same cage lactamCitation7–10 as a new transition state analogueCitation11 attached to short peptides. These cage peptide inhibitorsCitation12 showed much less toxicity (between 6000 and 8500 times) against human cells than lopinavir. The pentacycloundecane (PCU) cage moiety was chosen since it has been reported that the incorporation of polycyclic bulky hydrophobic cage frameworks into biologically active molecules often enhances their activityCitation13–16 and retards biodegradation thereofCitation17,Citation18. Various cage systems were also shown to have antiviral activityCitation14,Citation16,Citation19–21.
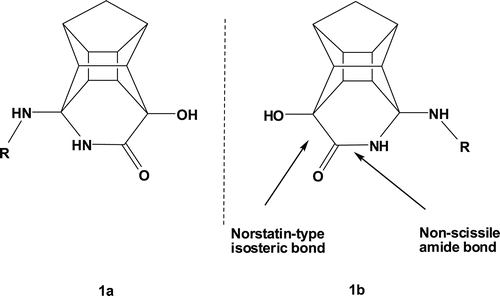
Biodegradation is one of the major limiting factors of peptide drugs. Considerable effort has gone into improving the pharmacological properties of peptides, mainly by increasing their enzymatic stability while preserving the chemical moieties required for functionalityCitation21,Citation22. Peptoids represent a class of oligomeric compounds that closely mimic the natural structure of peptides and possess increased enzymatic stability as compared with homologous peptides. Peptoids are polymeric compounds composed of N-substituted glycine (NSG) residues. The advances in the synthetic strategies used to prepare peptoidoligomers make them an attractive class of peptidomimeticsCitation11,Citation23–25. Further variations of the peptide chain and combination of the cage lactam with peptoids could potentially reveal important information about the HIV-PR inhibition characteristics of the PCU-lactam derivatives. The cage lactamCitation7 serves as a non-cleavable peptide bond under proteolytic conditions and the lactam hydroxyl group a norstatin type isostere transition state mimicCitation26 ().
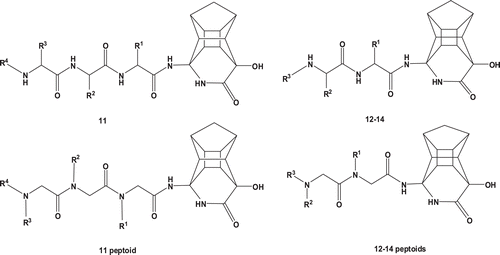
Figure 1. Structure of non-cleavable PCU-lactam peptides with a norstatin type isostere. The PCU-lactam is synthesized as a racemate 7–9 (R = peptide).
In our previous reportCitation11,Citation12, the most active inhibitor was PCU-Glu-Ala-Ile-Ser (IC50 of 0.078 µM) and we wanted to see whether variation of the peptide sequence could further improve the HIV-PR activity. This paper also investigates the effect of protection of the terminal amino acid with groups of different solubilities. The benzyloxy carbonyl (Cbz) and 2-pyrimidinyl-ethane-thioic acid (2-Peta) groups are used for this purpose.
Phenylalanine is considered to be the most common substituent at the S1 subsite of the HIV-PR and it is expressed in 40% of gag-pol gene sequences of the virusCitation27,Citation28. The S1/S1′ subsites accommodate hydrophobic substituents whereas the S2/S2′ and S3/S3′ subsites accommodate relatively less hydrophobic substituents such as valine and alanineCitation28. It was postulated that inhibitors must have peptidic character with the scissile bond of the substrate being replaced by a non-cleavable bondCitation29–32. The inhibitor specificity is determined by the substituent located close to the scissile bond (i.e. P2-P2′) with the P1 and P1′ residues being responsible for recognition and the remaining side chains are particularly important for the binding affinity to the enzymeCitation32.
With this in mind, it was decided to synthesize PCU-Phe-Val-Ala, PCU-Val-Ala, PCU-Val-Ala-Cbz (Cbz = Benzyloxycarbonyl), PCU-Val-Ala-2-Peta (Peta = Pyrimidinyl-ethane-thioic acid) and PCU-Val-Ala peptides and the corresponding peptoid analogues. This should enable us to validate if the theory for substrates in the S1 and S2 subsites is valid for our PCU-transition state analogue and whether the proposed increase in biostability of cage peptoids will have a positive effect on the inhibition of HIV-PR.
Material and methods
Experimental
PCU-peptide and peptoid synthesis
The peptides were synthesized via solid phase peptide synthesis (SPPS) on a 2-chlorotrityl chloride resin. Upon activation of the resin, the first amino acid was loaded onto the resin in the presence of N,N-diisopropylethylamine (DIPEA). The remaining amino acids were coupled to the loaded resin using microwave assistance on an automated peptide synthesizer (see the supporting information for more details).
The completed peptides were cleaved from the resin with a 5% trifluoroacetic acid/dichloromethane (TFA/DCM) mixture. In most cases, the peptides/peptoids were of sufficient purity to be coupled to the lactam after lyophilization without further purification.
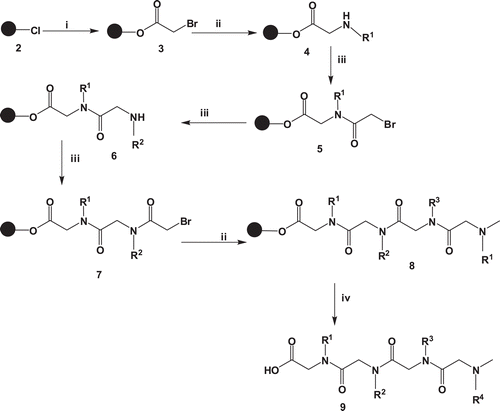
The peptoidoligomers were synthesized via solid phase synthesis on an activated 2-chlorotrityl chloride resin 2 by attaching bromoacetic acid in the presence of DIPEA in dry DCM affording compound 3. Upon filtration, the amine solution in DMF was added and mixed under a stream of nitrogen gas to yield compound 4. The chain was elongated by coupling bromoacetic acid in the presence of N, N′-diisopropylcarbodiimide (DIC). Similarly the corresponding amine groups were coupled to the extended chain followed by bromoacetic acid to give compounds 5–9. The peptoids were cleaved from the resin using 5% TFA/DCM as illustrated in .
Scheme 1. General procedure for the synthesis of peptoids. i = bromoacetic acid, DIPEA, dry DCM ii = amine, DMF iii = bromoacetic acid, DIC, DMF iv = 5% TFA/DCM.
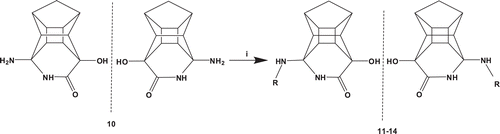
The PCU-lactam 14 was synthesized as beforeCitation7–10. The peptides and peptoids were coupled to the amino lactam with 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) and DIPEA in dimethylformamide (DMF) at ambient temperature as shown in . The compounds were purified via semi-preparative high-performance liquid chromatography (HPLC) and obtained as non-separable diastereomeric mixtures for the peptides and enantiomeric mixtures for the corresponding peptoids.
Scheme 2. Synthesis of the PCU-lactam peptide and peptoid analogues. i = HATU, DIPEA, DMF, RT, R = peptides or peptoids.
NMR studies
The EASY-ROESYCitation33 measurements required for structural analysis were recorded on a Bruker AVANCE III 600 MHz spectrometer using a BBO probe with z-gradient at a transmitter frequency of 600.1 MHz (spectral width, 10.0143 Hz; acquisition time, 0.1704436 s; 90° pulse width, 11.02 µs; scans, 8; relaxation delay, 2.0 s, mixing time 0.12 s, 2048 points in F2 dimensions). Processing and assignments were carried out using the Topspin 2.3 software from Bruker Karlsruhe. As observed previouslyCitation11,Citation12, the spectra of the inhibitor in DMSO-d6or D2O gave the same ROESY spectra. DMSO-d6 was used for the rest of the NMR samples.
In vitro HIV-1 protease activity
The catalytic activities of the HIV-1 protease was determined by monitoring the enzymatic hydrolysis of the chromogenic peptide substrate His-Lys-Ala-Arg-Val-Leu-Phe(p-NO2)-Glu-Ala-Nle-Ser in the presence of the inhibitors (). This substrate mimics the conserved KARVL/AEAM cleavage siteCitation6,Citation34 between the capsid protein and nucleocapsid (CA-p2) in the gag poly protein precursor and its use for the determination of HIV-1 PIs efficacy has been establishedCitation35,Citation36. Atazanavir and lopinavir were used as the positive control compounds.
Figure 2. PCU-lactampeptide and peptoid derivatives 11–14 tested for inhibition of wild type C-SA HIV-1 protease.
To determine the concentration of the inhibitors that resulted in 50% inhibition (IC50) of HIV-1 protease enzyme activity, the enzyme (100 nM) and chromogenic substrate (50 µM) were added into 120 µL microcuvettes containing increasing concentrations of inhibitor in a pH 5.0 buffer (50 mM sodium acetate and 0.1 M NaCl). Protease hydrolytic activity was measured by monitoring the relative decrease in absorbance at 300 nm with an Analytik Jena AG Specord 210 spectrophotometer.
The in vitro toxicology screening assay for 11 peptoid was performed as reported in our previous workCitation12. The method is based on the activity of mitochondrial enzymes of viable human MT-4 cells which cleave the tetrazolium ring of XTT yielding orange formazan crystals.
Docking and molecular modelling
To investigate the molecular behaviour of the synthesized pentacycloundecane lactam-peptide and peptoid inhibitors, we performed a two prong computational investigation. First, hybrid quantum mechanical/molecular mechanical and molecular dynamics (QM/MM/MD) simulationCitation37 were executed to determine the prominent low energy conformations for selectedCitation1 PCU-lactam peptides and peptoids in aqueous solution. Second, these peptides and peptoids were subjected to molecular docking studies. The binding energies of the best-docked inhibitor-enzyme complex were calculated. Since the peptides were synthesized as diastereomeric mixtures, both diastereomers were modelled. 1Only the peptides/peptoids that have shown a specific solution structures from EASY-ROESY data were modelled.
QM/MM/MD simulations of PCU-peptide/peptoid inhibitors in solution
The selected structures of the cage diastereoisomeric peptides, namely, Ala and racemic peptoids, were constructed using Avogadro softwareCitation38 with the Amber force field.subjected to a hybrid QM/MM/MD simulationCitation37 to determine the low energy conformations of the inhibitors in aqueous solution.
The gas phase AM1 optimized structures of the PCU-Val-Ala-Cbz (12), PCU-Val-Ala (13) and PCU-Ac-N(i-prop)-Ac-N(Me)-Cbz (12 peptoid) inhibitors (and their corresponding diasteromers/enantiomers) were individually enveloped in a cubic box of TIP3P waterCitation39 of side-length 34.5 Å. The whole system was energy minimized to a residual gradient of less than 0.001 kJ mol−1 Å−1 with a QM/MM method using the DYNAMO packageCitation40. In the QM/MM model, the inhibitors were included in the QM region which was described by the AM1 semi-empirical HamiltonianCitation41, the solvent was treated at the molecular mechanical (MM) level of theory using the OPLS-AACitation42 force field. The whole system was then equilibrated without any constraints under hybrid QM/MM Langevin-Verlet molecular dynamics (MD) conditions at 300 K for 50 ps, using the NVT ensembleCitation43. This was followed by 1 ns of separate production MD runs at 400, 500 and 600 K.
Molecular docking and MD simulations of the inhibitor-enzyme complex
A detailed computational simulation scenario adopted in this work was reported in our previous reportCitation44; however, a summarized procedure is presented herein.
Preparation of inhibitors
The 3-D structures of the fully optimized and equilibrated PCU peptide/peptoid inhibitor, resulted from the QM/MM/MD simulations, were subjected to docking and MD studies.
C-SA HIV-PR enzyme model system
Since the X-ray structure of the South African HIV-1 protease subtype C (C-SA) has not yet been reported, the initial 3-D structure of the enzyme was taken from the reported X-ray data of subtype B HIV-PR (PDB accession code 1HXW)Citation6. C-SA HIVs-PR differs from subtype B at eight positions; T12S, I15V, L19I, M36I, R41K, H69K, L89M and I93LCitation34. Mutations at the eight positions were manually induced. Throughout this work, the modelled C-SA HIV-PR was used for docking and MD simulations. This constructed enzyme was successfully used in our previous study on cage lactam peptidesCitation36,Citation37.
Docking studies were performed using AutodockCitation45. Docking results from Autodock were compared to the more accurate results from AutodockVina softwareCitation46 employing a standard set of parameters. The Cartesian coordinates of the docked structures with the best binding energies are available with the supplementary information.
MD simulations were performed for the inhibitor-enzyme complex of the most active compound 11a using the Amber10 suiteCitation47,Citation48.
Results and discussion
The inhibition activity (IC50) of the PCU-lactam peptides 11-14 and peptoid derivatives against wild type C-SA HIV-protease is listed in . All the cage peptides gave similar activities, whereas the three of the four cage peptoids exhibited much reduced activity (ten to twenty times).
Table 1. Inhibitory concentration (IC50) values for the inhibition of wild type C-SA HIV-1 protease by PCU-lactam peptide and peptoid derivatives 11-14.
Peptide 11 (PCU-Phe-Val-Ala-Cbz) showed slightly weaker inhibition in comparison to pepdide 12 (PCU-Val-Ala-Cbz) in which phenylalanine was removed as the first amino acid. This could be attributed to the phenylalanine of 11 occupying the S2 subsite instead of the S1 subsite potentially due to the PCU-cage filling this space. As reported before, the S2 subsite prefers less bulky molecules such as valine; thus, phenylalanine would not have maximum interaction with this subsiteCitation26. We therefore, postulate that for 12 (PCU-Val-Ala-Cbz), the cage norstatine moiety interacts with the active site while the PCU-cage occupies the S1 subsite which can accommodate bulky groups. The valine and alanine segments will then fit into the S2 and S3 subsite, respectively. The Cbz group was removed from 12 to increase aqueous solubility but this led to a decrease in activity for 13 (PCU-Val-Ala) with an IC50 = 0.75 µM. In another attempt to increase the aqueous solubility, the Cbz group was replaced with 2-pyrimidinyl-ethane-thioic acidCitation35 and this led to an increased inhibitory activity (14 with an IC50 = 0.5 µM).
Peptoid 11 consisting of an aromatic moiety showed the best inhibitory activity when compared to the other PCU-lactam peptoid analogs (12–14 peptoids). This observation can be attributed to the conformational difference reported by Fowler et al.Citation36 between peptoids consisting of an aromatic and/or negatively charged side chain and those only with an alkyl chain. According to their report, aromatic groups on the side chains cause charge-charge repulsion with backbone carbonyl groups that assist in peptoid folding. This folding can be advantageous since it gives the peptoid a well-defined secondary structure, which are lacking in most peptoids. Peptoids are highly flexible because of the lack of hydrogen bonding and the tertiary nature of the amide bonds that allows them to isomerize between cis and trans conformations far more readily than secondary amides in peptidesCitation36. The decrease in activity observed for three of the four PCU-peptoids (12–14 peptoids in comparison to the corresponding PCU-peptides) could be attributed to the lack of a defined structure.
The cytotoxicity results revealed that the PCU-peptides (145 μM for 11) and PCU-peptoids (102 μM for 11 peptoid) are significantly less toxic to human MT-4 cells than lopinavir (0.025 μM). These results clearly confirm the potential bioactive contribution of the cage, with a non-cleavable norstatine-type bond and the importance of the appropriate peptide and peptoide sequences for intensifying specific binding to the active site.
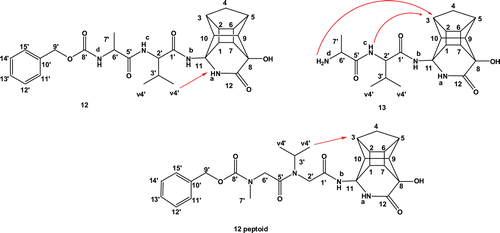
As was the case in our previous reportCitation11, it was again clear from NMR observations that some of the diastereomeric peptides exhibit different conformational preferences. The ROESY spectra of compound 12 (PCU-Val-Ala-Cbz) and the corresponding peptoid confirmed the presence of more than one conformation. Based on the 2D spectra, it was concluded that the diastereomeric mixture of 12 exhibited fewer conformations than the corresponding peptoid (see and in the supplementary material). The HSQC spectrum of peptoid 12 revealed three different chemical shifts for each of the methylene protons (H-2′ and H-6′) of the peptoid chain. According to the ROESY spectra, the most prevalent conformations in both inhibitors are those in which the valine methyl protons H-4′ interact with the cage protons H-1 and H-3 (). In the less prevalent conformation, the H-4′ protons interact with the cage lactam proton NHa. It is also clear from the NMR data that the peptoids exhibit much more structural flexibility than their peptide analogues.
Table 2. Potential EASY-ROESY correlations measured for the ten lowest energy (LE) conformations* of inhibitor 12 (PCU-Val-Ala-Cbz) extracted from the QM/MM/MD simulation at 400 K.
Figure 3. Inhibitor 12 (PCU-Val-Ala-Cbz), 13 (PCU-Val-Ala) and 12 peptoid (PCU-Ac-N(i-prop)-Ac-N(Me)-Cbz) with arrows showing the observed EASY-ROESY correlations.
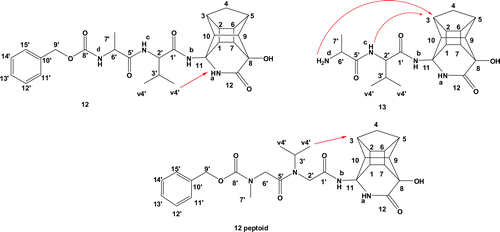
The ROESY spectrum of compound 13 (PCU-Val-Ala) showed an interaction between the alanine amine NHd proton and the cage protons H-1 and H-3 and the valine amine NHc proton with H-3 (). This suggests a sharp turn conformation, which could potentially not be complimentary to the active site of the protease. Most of the clinically available inhibitors extend to either the S2 or S3 pocket of the enzyme for maximum bindingCitation26,Citation28.
QM/MM/MD simulations of synthesized inhibitors
The inhibitors that revealed specific ROESY interactions were modelled in aqueous solution. The MD simulations for both diastereomers of 12 (PCU-Val-Ala-Cbz), 13 (PCU-Val-Ala) and both enantiomers of 12 peptoid PCU-Ac-N(i-prop)-Ac-N(Me)-Cbz were executed at 400, 500 and 600 K. Our previous reportsCitation11,Citation12,Citation17,Citation49 showed that 300 K is insufficient to explore the complete conformational space of cage peptides.
The ten lowest energy conformations of each inhibitor were extracted from the trajectory of the hybrid QM/MM/MD simulations using the VMDCitation50 software. The results for peptides 12 and 13 are presented in and .
Table 3. EASY-ROESY correlations measured for the ten lowest conformations* of inhibitor 13 (PCU-Val-Ala) at 400 K.
Clear conformational differences exist at 400 K. At higher temperatures, the peptides exhibit more extended conformations, where these interactions are largely absent. According to calculated distances, a considerable distinction in terms of potential NOESY interaction distances between the two diasteriomeric forms (a and b) is evident and these measured values correlate to the observed EASY-ROESY interactions. In the case of 12 (PCU-Val-Ala-Cbz), the interaction between the hydrogen atoms of the methyl group of Valine (v4′ in ) with the NHa part of lactam cage for 12a is more evident than for 12b (see ).
The calculated MD distances between hydrogen atoms of the cage and NHc and NHd for the ten lowest conformations of 13 (PCU-Val-Ala) from the MD reveal that the distances for H-3…NHc (Å) of 13b is closer to the observed NMR EASY-ROESY correlations (see ). These observations confirm our previous findingsCitation11,Citation12 that the chirality of the PCU moiety plays an important role to induce different side chain conformations.
The corresponding analysis for 12 peptoid at 300 and 400 K did not exhibit any definite structural features. It is clear from the MD results that the peptoids display much more conformational flexibility compared to the corresponding peptides.
Docking and MD studies
Docking studies are used at different stages in drug discovery such as in the prediction of ligand-receptor complex structures and also to rank the ligand molecules based upon the binding energies of the corresponding ligand-enzyme complexes. The objective of our docking study was to elucidate the potential interaction mode of the PCU-peptide/peptoid derivatives with C-SA HIV-PR and to see whether a correlation between the binding energies and the observed IC50 results exists.
Both diasteromeric peptides (12a, 12b and 13a, 13b) and racemic peptoids (12a peptoid and 12b peptoid) were considered in the docking study. The affinity energies (EA) of the best-docked inhibitor-enzyme complex are depicted in .
Table 4. The docked* binding energies of the inhibitors and the corresponding IC50 values against the wild type C-SA HIV-1 protease.
The docking results (binding energies) suggest that the docking methods are too crude to correlate with such a narrow range of experimentally obtained IC50 inhibition results. However, the docking results are useful to determine the possible orientation of inhibitors in the active site of C-SA HIV-1 PR; these results may be used to design new more potent inhibitors of HIV-1 protease.
The competitive inhibition of HIV-1 protease depends on the interaction between the inhibitor and the aspartyl dyad (Asp25 and Asp25′)Citation51. shows the distances (Å) between the inhibitors and aspartyl dyad in the active site of wild type C-SA HIV-1 protease.
Table 5. Distances (Å) between the inhibitors and key residues in the active site of wild type C-SA protease obtained from molecular docking with AutodockVina.
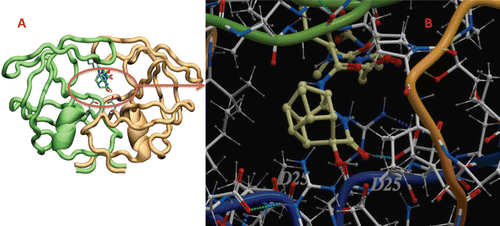
A general conclusion derived from these docking results is that the side chain of the aspartyl dyad Asp25 or Asp25′ may form a hydrogen bond with the cage lactam of all inhibitors studied here (). The nature of these hydrogen bonds involves an interaction between the aspartyl dyad (OD1 or OD2) and the carbonyl oxygen or the hydroxyl-group of the PCU-lactam (). Interestingly, this interaction is stronger for 11 peptoid, which is also the peptoid that exhibits better activity against C-SA protease (). As can be observed from docking studies (), the hydroxyl-group of the PCU-lactam of 11a peptoid interacts through a hydrogen bond with Asp25, whereas 12a peptoid interacts through hydrogen bond with Asp25′. Another residue that appears to interact through hydrogen bond with the hydroxyl-group of the PCU-lactam is the Gly27 residue ().
Finally, it is clear from the docking results that the low energy solution structures of the inhibitors are not prevalent in the resulting inhibitor-enzyme complex. This suggests that an induced fit binding mechanism could be applicable to these inhibitors.
However, the docking calculations provided us with a general static picture of the most energetically favourable binding orientation of inhibitors to the enzyme (). To obtain further insight into the dynamic changes of the docked inhibitors within the active site of the enzymatic pocket over time, the lowest energy docked complex of the most active inhibitor, 11a, was subjected to MD simulations (1 ns).
Figure 4. (A) Lowest energy docked structure for the most active compound 11a peptoid with C-SA HIV-PR. (B) A closer view showing the binding mode of compound 11a inside the C-SA HIV-PR active site. The two PR monomers are coloured as the light brown and green ribbon. The inhibitor and the two Asp25 residues are presented as coloured sticks.
The MD simulations clearly show that the inhibitor easily fits into the active enzyme pocket. To assess the quality of our MD simulations, energetic and structural properties are monitored along the entire 1 ns MD trajectory of the complex (). The 1 ns averaged backbone Root-mean-square deviations (RMSDs) for the 11a enzyme complex is less than 1 Å and this is an indication that the stability of the generated MD trajectory of the complex is quite satisfactory.
Figure 5 (A) The potential energy of 11a C-SA HIV-PR complex observed in MD simulation as a function of time.(B) Root-mean-square deviations (RMSD) observed in MD simulations as a function of time. (C) Hydrogen bond distance between the hydroxyl-group located at the cage and the Asp25 carboxyl group.
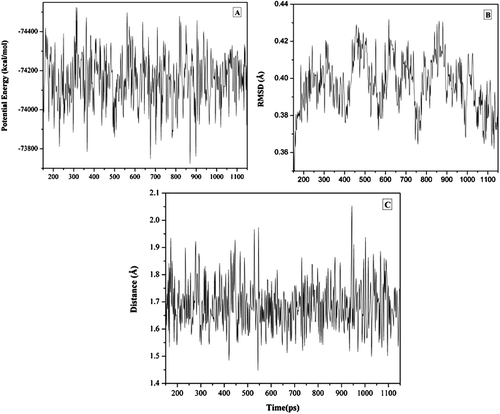
As evident from the MD trajectory analysis, we found that such inhibitors fit quite well in the active site and no abrupt change in the orientation as obtained from docking simulations. Most interestingly, a strong hydrogen bond formed between the hydroxyl-group on the cage skeleton and Asp5 residue was found to be stable over the whole MD run which we assume that such hydrogen bond maintains the inhibitor anchored in the active site ( and is essential for inhibitory activity.
Conclusion
The coupling of the PCU-lactam to a series of peptide side chains suggests again that the HIV-PR activity is largely due to the presence of the cage lactam; since, variation of the peptide side chain has little effect on the activity. Removal of the cage was previously shownCitation11,Citation12 to result in a complete loss of activity. It also appears that the incorporation of a peptoid side chain does not contribute to an increase in activity. The PCU-peptides and peptoids are several orders of magnitude less toxic to human MT-4 cells than lopinavir. MD simulations of the cage peptides are able to provide the low energy conformations of the peptides in solution, which agree with the experimental NMR observations. It is also clear from the MD simulations that the peptoids exhibit much more conformational flexibility than the corresponding cage peptides. Docking results suggest that the cage hydroxyl-group is interacting with the Asp25 residues of the HIV-PR as observed beforeCitation11,Citation12. The docking results are too crude to correlate well with the narrow range of experimentally observed IC50 values (0.5–10 µM). Nevertheless, these results provide insight into the potential interaction of inhibitors inside the active site of C-SA HIV-1. This knowledge will be useful for the design of more potent inhibitors for HIV-1 protease.
Supplementary Material
Download PDF (3.3 MB)Acknowledgements.
We thank Aspen Pharmacare, University of KwaZulu-Natal for financial support and the CHPC (HYPERLINK “http://www.chpc.ac.za” www.chpc.ac.za) for computational resources. We also thank Mr. Dilip Jagjivan (UKZN, South Africa) for his assistance with the NMR experiments. YS acknowledges the South African National Research Foundation (Thuthuka) for financial support.
Declaration of interest
This research was supported by NRF (SA) TG (GUN: 66319), KP (GUN: 69728), HGK and PIA (SA-Sweden bilateral grant), GEMM and CNA (IBSA). Financial support was provided by Aspen Pharmacare, University of KwaZulu-Natal and the computational resources by CHPC (HYPERLINK “http://www.chpc.ac.za” www.chpc.ac.za”).
Related Research Data
References
- West ML, Fairlie DP. Targeting HIV-1 protease: A test of drug-design methodologies. Trends Pharmacol Sci 1995;16:67–75.
- Martinez-Cajas JL, Wainberg MA. Protease inhibitor resistance in HIV-infected patients: Molecular and clinical perspectives. Antiviral Res 2007;76:203–221.
- Jacobsen H, Yasargil K, Winslow DL, Craig JC, Kröhn A, Duncan IB et al. Characterization of human immunodeficiency virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31-8959. Virology 1995;206:527–534.
- Sylvia L-H, Huang PL, Dawei Z, Wook LJ, Ju B, Yongtao S et al Discovery of small-molecule HIV-1 fusion and integrase inhibitors oleuropein and hydroxytyrosol: Part I. Integrase inhibition. Biochem Biophys Res Commun 2007;354:872–878.
- Hammer SM. Clinical practice. Management of newly diagnosed HIV infection. N Engl J Med 2005;353:1702–1710.
- Velázquez-Campoy A, Vega S, Fleming E, Bacha U, Sayed Y, Dirr HW et al. Protease inhibition in African subtypes of HIV-1. AIDS Rev 2003;5:165–171.
- Kruger HG, Martins FJ, Viljoen AM. Interconvertions between delta-lactam and delta-lactone derivatives initiated by unique transannular interactions of the rigid cyclohexane boat structure in pentacycloundecane. J Org Chem 2004;69:4863–4866.
- Martins FJC, Viljoen AM, Kruger HG, Joubert JA, Wessels PL.. Synthesis of δ-Lactams from. Tetrahedron 1994;50:10783–10790.
- Kruger HG, Martins FJC, Viljoen AM, Boeyens JCA, Cook LM, Levendis DC. Structure and conformation of the hydrate of 8,11-dihydroxypentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-carbolactam. Acta Crystallogr 1996;52:838–841.
- Boyle GA, Govender T, Kruger HG, Ndlovu IN 8-Amino-11-hydroxypentacycloundecane-8,11-lactam. Acta Crystallogr 2007;63:O3906–U3914.
- Makatini MM, Petzold K, Sriharsha SN, Ndlovu N, Soliman ME, Honarparvar B et al. Synthesis and structural studies of pentacycloundecane-based HIV-1 PR inhibitors: A hybrid 2D NMR and docking/QM/MM/MD approach. Eur J Med Chem 2011;46:3976–3985.
- Makatini MM, Petzold K, Sriharsha SN, Soliman ME, Honarparvar B, Arvidsson PI et al. Pentacycloundecane-based inhibitors of wild-type C-South African HIV-protease. Bioorg Med Chem Lett 2011;21:2274–2277.
- James B, Viji S, Mathew S, Nair MS, Lakshmanan D, Kumar RA. Synthesis of novel highly functionalized biologically active polycyclic caged amides.Tetrahedron Lett 2007:48:6204–6208.
- Geldenhuys WJ, Malan SF, Bloomquist JR, Marchand AP, Van der Schyf CJ. Pharmacology and structure-activity relationships of bioactive polycyclic cage compounds: A focus on pentacycloundecane derivatives. Med Res Rev 2005;25:21–48.
- Oliver DW, Malan SF. Medicinal chemistry of polycyclic cage compounds in drug discovery research. Med Chem Res 2008;17:137–151.
- Oliver DW, Dekker TG, Snyckers FO, Fourie TG. Synthesis and biological activity of D3-trishomocubyl-4-amines. J Med Chem 1991;34:851–854.
- Bisetty K, Corcho FJ, Canto J, Kruger HG, Perez JJ. A theoretical study of pentacyclo-undecane cage peptides of the type [Ac-X-Y-NHMe]. J Pept Sci 2006;12:92–105.
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y et al.. Synthesis and biological evaluation of rigid polycyclic derivatives of the Diels-Alder adduct tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione. Science 2010;327:271–282.
- Inamoto Y, Aigami K, Kadono T, Nakayama H, takatsuki A, Tamura G. Biological active polycycloalkanes. 4. Phosphoric esters of trimethylenenorbornyl alcohols. J Med Chem 1977;20:1371–1374.
- Aigami K, Inamoto Y, Takaishi N, Fujikura Y. Biologically active polycycloalkanes. 2. Antiviral 4-homoisotwistane derivatives. J Med Chem 1976;19:536–540.
- Adessi C, Soto C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr Med Chem 2002;9:963–978.
- Hruby VJ, Balse PM. Conformational and topographical considerations in designing agonist peptidomimetics from peptide leads. Curr Med Chem 2000;7:945–970.
- Miller, SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH.. Proteolytic studies of homologous peptide and n-substituted glycine peptoid oligomers. Bioorg Med Chem Lett 1994;4:2657–2662.
- Zuckermann RN, Kerr JM., Kent, SBH, Moos WH. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid phase synthesis. J Am Chem Soc 1992;114:10646–10647.
- Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH.. Comparison of the proteolytic susceptibilities of homologous L-amino Acid, D-amino Acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev Res 1995;35:20–32.
- Kruger HG, Rademeyer M, Ramdhani R.. Pentacyclo[5.4.0.0- 2,6.03,10.05,9]undecane-8,11-dione ethylene acetal. Acta Crystallogr Sect E Struct Rep Online 2006;62:268–270.
- Clemente JC, Coman RM, Thiaville MM, Janka LK, Jeung JA, Nukoolkarn S et al. Analysis of HIV-1 CRF_01 A/E protease inhibitor resistance: Structural determinants for maintaining sensitivity and developing resistance to atazanavir. Biochemistry 2006;45:5468–5477.
- Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct 1998;27:249–284.
- Fairlie DP, Tyndall JD, Reid RC, Wong AK, Abbenante G, Scanlon MJ et al. Conformational selection of inhibitors and substrates by proteolytic enzymes: Implications for drug design and polypeptide processing. J Med Chem 2000;43:1271–1281.
- Wu XY, Oehrngren P, Ekegren JK, Unge J, Unge T, Wallberg H et al. Two-carbon-elongated HIV-1 protease inhibitors with a tertiary-alcohol-containing transition-state mimic J Med Chem 2008;51:1053–1057.
- Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA 1995;92:2484–2488.
- Ridky TW, Cameron CE, Cameron J, Leis J, Copeland T, Wlodawer A et al. Human immunodeficiency virus, type 1 protease substrate specificity is limited by interactions between substrate amino acids bound in adjacent enzyme subsites. J Biol Chem 1996;271:4709–4717.
- Thiele CM, Petzold K, Schleucher J. EASY ROESY: Reliable cross-peak integration in adiabatic symmetrized ROESY. Chemistry 2009;15:585–588.
- Mosebi S, Morris L, Dirr HW, Sayed Y. Active-site mutations in the South African human immunodeficiency virus type 1 subtype C protease have a significant impact on clinical inhibitor binding: Kinetic and thermodynamic study. J Virol 2008;82:11476–11479.
- Huang, L, Lee A, Ellman JA.. Identification of potent and selective mechanism-based inhibitors of the cysteine protease cruzain using solid-phase parallel synthesis pages J Med Chem 2002;45:676–684.
- Fowler SA, Blackwell HE. Structure-function relationships in peptoids: Recent advances toward deciphering the structural requirements for biological function. Org Biomol Chem 2009;7:1508–1524.
- Warshel A, Levitt M. Folding and stability of helical proteins: Carp myogen. J Mol Biol 1976;106:421–437.
- Avogadro. Version 1.0.0. Availabe at: http://avogadro.openmolecules.net/
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML.. Comparison of Simple Potential Function for Simulating Liquid Water pages. J Chem Phys 1983;79:926–935.
- Field MJ, Albe M, Bret C, Proust-De Martin F, Thomas A.. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J Comp Chem 2000;21:1088–1100.
- Dewar MJS, Zoebisch EG, Healy EF, Stewart JJPJ.. A new general purpose quantum mechanical molecular model. Am Chem Soc 1985;107:3902–3909.
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL.. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 2001;105:6474–6487.
- Zhang XQ, Schooley RT, Gerber JG. The effect of increasing α1-acid glycoprotein concentration on the antiviral efficacy of human immunodeficiency virus protease inhibitors. J Infect Dis 1999;180:1833–1837.
- Karpoormath R, Sayed Y, Govender P, Maguire GEM, Kruger HG, Govender T et al. Pentacycloundecane derived hydroxyl acid peptides: A new class of irreversible non-scissile bridged ether type isoster as potential HIV-1 wild type C-SA protease. Bioorgan Chem 2011. In Press. Available at: http://dx.doi.org/10.1016/j.bioorg.2011.08.002.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK et al. J Comp Chem 1998;19:1639.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–461.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–1174.
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr et al. The Amber biomolecular simulation programs. J Comput Chem 2005;26:1668–1688.
- Bisetty K, Gomez-Catalan J, Aleman C, Giralt E, Kruger HG, Perez JJ. Computational study of the conformational preferences of the ®-8-amino-pentacyclo[5.4.0.0(2,6).0(3,10).0(5,9)]undecane-8-carboxylic acid monopeptide. J Pept Sci 2004;10:274–284.
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph 1996;14:33–8, 27.
- Adachi M, Ohhara T, Kurihara K, Tamada T, Honjo E, Okazaki N et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc Natl Acad Sci USA 2009;106:4641–4646.