Abstract
NADPH oxidase isoform-2 (NOX2) generates reactive oxygen species (ROS) that contribute to neurodegenerative and cardiovascular pathologies. However, validation of NOX2 as a pharmacotherapeutic target has been hampered by a lack of mechanistically-defined inhibitors. Using cellular and biochemical assays, we explored previously reported inhibitors of ROS production (perhexiline, suramin, VAS2870 and two Shionogi patent compounds) as direct NOX2 inhibitors. All but suramin, which presumably lacks cell penetrance, inhibit cellular ROS production. However, only perhexiline and suramin inhibit biochemical NOX2 activity. Indeed, our data suggest that NOX2 inhibition by perhexiline may contribute significantly to its demonstrated cardioprotective effects. Inhibition of protein kinase CβII explains the cellular activity of the Shionogi compounds, whereas VAS2870 inhibits by an as-yet unidentified mechanism unrelated to direct NOX2 function or subunit assembly. These data delineate the mechanisms of action of these compounds and highlight their strengths and limitations for use in future target validation studies.
Introduction
Small-molecule inhibitors are valuable probes for defining the role of a particular enzyme in a given biological process. Application of these compounds to relevant pathophysiological cellular and animal models enables validation of the catalytic activity of that enzyme as a target for pharmacotherapy. However, a thorough interpretation of such studies requires an adequate understanding of the mechanisms of action of these tools. For complex enzymatic systems with multiple modes of regulation and activation, this mechanistic description is even more critical, given the numerous possibilities for small-molecule interactionCitation1.
The NADPH oxidase (NOX) family of enzymes represents one such complex enzyme system. These are multisubunit, membrane-bound enzymes that generate superoxide anion from NADPH and molecular oxygenCitation2. NADPH oxidase isoform-2 (NOX2), the phagocytic isoform of NOX and best-characterized member of this family, initiates production of intraphagosomal reactive oxygen species (ROS) for microbial killingCitation3. However, expression of NOX2 is not limited to phagocytic cell types, as expression in a variety of tissues, including vascular smooth muscle cells, adventitial fibroblasts and cardiomyocytes, has been reportedCitation3,Citation4. NOX2 is activated by phosphorylation of the cytosolic subunit p47phox, which stimulates translocation and assembly of a cytosolic complex of subunits (p47phox, p67phox, p40phox and Rac2) with the membrane-bound, cytochrome-containing components (gp91phox and p22phox)Citation2. The interaction of cytosolic and membrane components permits binding and oxidation of NADPH on gp91phox and subsequent electron transfer through its flavin site and dual heme domains to the oxygen substrate to yield superoxide anion.
Although NOX2 and other ROS-producing enzymes serve beneficial functions, dysregulation of the generation and/or removal of ROS upsets the balance between oxidative and antioxidant processes. Ultimately, cellular damage from persistent oxidative stress leads to impaired tissue function and the onset and progression of a disease state, as has been implicated for a number of diseases, including Alzheimer’s disease, arthritis, hypertension, stroke, heart failure and atherosclerosisCitation5–8. Indeed, the improved functional endpoints noted in cardiovascular disease models for mice lacking various subunits of the NOX2 enzyme complexCitation9–12 strongly suggest a pathophysiological role for NOX2. Emerging data also implicate the NOX1 and NOX4 isoforms in various aspects of cardiovascular disease, although these enzymes are not as well-characterized and reagents for detailed analysis are more limitedCitation13.
Many inhibitors of NOX enzymes have been claimed in the literature, but few have been shown to be specific to NOX or to act directly on the enzymeCitation14–16. As examples, in vitro pharmacological inhibition of NOX2 by covalent modifiers such as apocynin, diphenyleneiodonium (DPI) and a peptidic inhibitor of NOX2 assembly, gp91ds-tat, has been described, inclusive of demonstrated inhibition of cellular ROS productionCitation17–22. Furthermore, apocynin and gp91ds-tat have been utilized in vivo with reports of cardiovascular benefitCitation23–26, in support of the hypothesis that NOX2 inhibition would be therapeutic in cardiovascular disease. However, a direct association of cellular and in vivo effects of these compounds must be viewed cautiously, given their potential for multiple off-target cellular effectsCitation27.
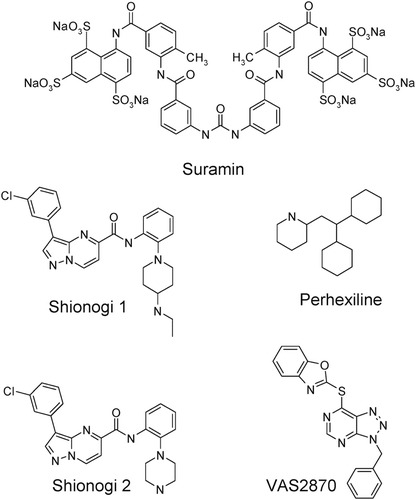
The complexity of the activation cascade for NOX2 reveals several potential mechanisms for pharmacological disruption: e.g. by inhibiting an initiating kinase, inhibiting oxidase assembly, competing with the substrate NADPH or scavenging the superoxide product. Recently, several new compounds have been described as inhibitors of ROS formation in cellular or tissue preparations (structures shown in ): perhexiline, an antianginal agent prescribed in New Zealand and AustraliaCitation28,Citation29; suramin, a purinergic receptor antagonist used as an antiparasitic agentCitation30–32; two compounds described by Shionogi, Ltd. as inhibitors of cellular ROS generation (US patent US20060089362A1); and VAS2870, from the company VasopharmCitation33,Citation34. Furthermore, several of these have been suggested to act as NOX inhibitors, but of unclear mechanism. The purpose of the current study was to provide an in vitro characterization of these compounds, primarily whether any directly inhibit NOX2 and could serve as better-defined tool molecules for further validating NOX2 as a target for therapeutic intervention.
Figure 1. Structures of inhibitors of ROS production, proposed to act at NOX.
Methods
General
Structures of perhexiline, suramin, Shionogi compounds 1 and 2 and VAS2870 are shown in . Perhexiline was purchased from Sigma Chemical Co. (St Louis, MO). Suramin was purchased from Biomol International (Plymouth Meeting, PA). Apocynin was purchased from Calbiochem (Gibbstown, NJ). Shionogi compound 1 [3-(3-chlorophenyl)-N-{2-[4-(ethylamino)-1-piperidinyl]phenyl}pyrazolo[1,5-a]pyrimidine-5-carboxamide] and Shionogi compound 2 [3-(3-chlorophenyl)-N-[2-(1-piperazinyl)phenyl] pyrazolo[1,5-a]pyrimidine-5-carboxamide] were prepared according to the procedures described in US patent US20060089362A1. VAS2870 [7-(1,3-benzoxazol-2-ylthio)-3-(phenylmethyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidine] was purchased from Butt Park, Ltd. (Okehampton, Devon, UK). All other chemicals were purchased from Sigma Chemical Co (St. Louis, MO), and were of the highest analytical grade.
Isolation of human neutrophils and neutrophil membrane fractions
Human biological samples were sourced ethically and their research use was in accord with the terms of the informed consents. Blood was collected from healthy human donors using 50 mM EDTA as the anticoagulant. Blood was diluted 1:1 in Dulbecco’s phosphate buffered saline without calcium or magnesium (DPBS) and layered over Ficoll-Paque™ PLUS, followed by centrifugation (400× g for 30 min at room temperature). The resultant pellet was resuspended in DPBS to twice the pellet volume and 6% dextran was added to a final concentration of 1%. Following 1 h incubation at room temperature to sediment erythrocytes, supernatants were removed and further treated with BD Pharm Lyse™ buffer (BD Biosciences, San Jose, CA) as per manufacturer’s instructions to lyse remaining erythrocytes. The supernatants were then centrifuged at 1000x g for 10 min and the resultant pellet (neutrophils) was resuspended in either Hank’s balanced salt solution (HBSS, 1.3 mM CaCl2, 5.4 mM KCl, 0.4 mM KH2PO4, 0.5 mM MgCl2, 0.4 mM MgSO4, 137 mM NaCl, 0.4 mM NaHCO3, 0.3 mM Na2HPO4, 5.6 mM glucose) for cellular NADPH oxidase activity experiments, or in ice-cold sonication buffer [11% sucrose, 50 mM potassium phosphate pH 7.0, 120 mM NaCl, 5 mM EGTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/mL leupeptin, 1 µM pepstatin, 1% protease inhibitor cocktail and 2 mM diisopropyl fluorophosphate] for isolation of membrane fractions.
To isolate neutrophil membrane fractions, cells were sonicated in sonication buffer and centrifuged at 1000× g for 15 min at 4°C. Supernatants were layered onto a 20/40% (w/v) discontinuous sucrose gradient and centrifuged at 150,000× g for 40 min. Membrane fractions were collected from the 20/40% sucrose interface and protein concentrations were determined using the bicinchoninic acid method (Pierce, Rockford, IL). Membrane fractions were snap-frozen in liquid nitrogen and stored at −80°C.
Recombinant His-tagged p47phox, p67phox and Rac2
Recombinant p47phox, p67phox and Rac2 were prepared as described by Rotrosen, et al.Citation35. Briefly, DNA encoding the NOX2 cytosolic components (p47phox, p67phox and Rac2) were amplified from human neutrophil cDNA using the polymerase chain reaction with the following primer sequences: p47phox forward = 5′-GGAATT CACCATGC ATCATCACCAT CACCAT GGGGAC ACCTTCAT CCGT-3′, reverse = 5′-GGGGTACCT CAGACGGC AGACGCCAGC-3′; p67phox forward = 5′-GGAATT CACCATGCA TCATCACCA TCACCATTC CCTGGTGGAG GCCATC-3′,reverse = 5′-GGGGTACC TAGACTTCTC TCCGAGTGC-3′; Rac2 forward = 5′-GGAATT CACCATG CATCATC ACCATCA CCATCA GGCCATCAAG TGTGTG-3′,reverse = 5′ -GGGGTAC CTAGAG GAGGCTGC AGGC-3′. For all primer sets, an EcoRI site, a Kozak sequence and a (His)6 tag were included in the forward primer and a KpnI site was included in the reverse primer. Open reading frames for each subunit were subcloned into pFastBac-1 (Invitrogen, Carlsbad, CA), and transfected into Sf9 insect cells for expression of proteins according to Fornwald, et al.Citation36. Proteins were purified using nickel-nitrilotriacetic acid (nickel-NTA) columns according to manufacturer’s instruction (Qiagen, Valencia, CA).
Immunoblotting
Protein samples were subjected to SDS-PAGE using 4–20% polyacrylamide gels and then transferred to PVDF membranes. Membranes were blocked with 5% nonfat dry milk in tris-buffered saline containing 0.2% Tween-20 (TBST), then probed with primary antibody in 2% nonfat dry milk in TBST, followed by either goat anti-mouse or anti-rabbit IRDye-labeled IgG secondary antibody (LI-COR Biosciences, Lincoln, NE; diluted to 1:10,000, v/v) in 2% nonfat dry milk in TBST. The following primary antibodies were used: mouse anti-p47phox (BD Biosciences, San Jose, CA; diluted to 1:2,000, v/v), mouse anti-p67phox (BD Biosciences; diluted to 1:2,000, v/v) and mouse anti-Rac2 (Abnova Corp., Walnut, CA; diluted to 1:2,000, v/v). Blots were imaged using the LI-COR Odyssey infrared imaging system, and, when necessary, bands were quantified with the ImageQuant software package (GE Healthcare, Piscataway, NJ).
Enzyme activities
Cellular NOX2 activity was determined by monitoring the reduction of exogenously-added cytochrome c by stimulated primary neutrophils. Cytochrome c from horse heart (100 µM; Sigma Chemical Co., St. Louis, MO) was added to neutrophils suspended in HBSS at a density of 2 × 10Citation6 cells/mL. In a 96 well plate, cells (200,000 cells/well) were incubated with vehicle (0.1% DMSO) or compounds for 20 min at 37°C before stimulation with 1 µM phorbol 12-myristate 13-acetate (PMA). In pilot experiments, this concentration of PMA was shown to stimulate superoxide release to 80% of the maximum level (EC80). The rate of cytochrome c reduction was determined by measuring the change in absorbance at 550 nm for 30 min after PMA stimulation. Superoxide production was determined by subtracting the absorbance of reaction wells containing an excess of superoxide dismutase (SOD, 50 units/100 μL) from those containing no SOD. The change in absorbance at 550 nm was converted to concentration of cytochrome c reduced using an extinction coefficient of 2.1 × 10Citation4 M−1 cm−137. Experiments were performed in triplicate.
For analysis of semi-recombinant NOX2 activity, 1 µg of human neutrophil membrane was combined with purified recombinant cytosolic subunits of NOX2 (2 ng of p47phox, 2 ng of p67phox and 1 ng of Rac2) and added to wells containing assay buffer [final concentrations: 50 mM potassium phosphate (pH 7), 1 mM EGTA, 0.01 mM FAD, 4 mM MgCl2, 1 µM GTPγS, 40 µM arachidonic acid, 100 µM cytochrome c and 1% DMSO]. These concentrations of protein subunits and cofactors were determined in pilot experiments to provide the maximal activity of the enzyme when stimulated with NADPH. Reactions were run at room temperature and initiated by addition of 50 µM NADPH, which was determined in pilot experiments to be the Km level. The rate of cytochrome c reduction was determined for 10 min after addition of NADPH using the method described above. Experiments were performed in triplicate.
Steady-state inhibition mechanisms, relative to NADPH substrate, were determined for the direct inhibitors of semi-recombinant NOX2 by measuring reaction velocities as a function of varied concentrations of both NADPH and inhibitor. All other reaction components and conditions were as described above. For each inhibitor, entire data sets were globally fit to standard inhibition models for assignment of best-fit mechanism, as described below.
The effects of the test compounds on the neutrophil-abundant isoform of protein kinase C (PKC), PKCβII, were assayed using a commercially available kit (IMAP™ FP kit, Molecular Devices, Sunnyvale, CA) and recombinant human PKCβII (Upstate/Millipore, Billerica, MA), according to manufacturer’s instructions.
To assess xanthine oxidase activity, compounds or vehicle (0.1% DMSO) were incubated with xanthine oxidase (from bovine milk, EMD Biosciences, Carlsbad, CA; 10 milliunits/well in HBSS containing 100 µM cytochrome c) for 20 min prior to stimulation with 50 µM hypoxanthine. Experiments were performed in triplicate wells, with the third well containing an excess of SOD (40 units/100 μL). Cytochrome c reduction was measured by the change in absorbance at 550 nm. Superoxide production was determined by subtracting the absorbance of the SOD-containing wells from the duplicate wells containing no SOD.
NOX2 assembly (p47phox translocation)
Isolated human neutrophils were incubated with vehicle (0.1% DMSO), 1 µM PMA alone or 1 µM PMA in the presence of compound (10 µM) or apocynin (100 µM), a known inhibitor of NOX2 assemblyCitation18. Neutrophils were preincubated with inhibitors or vehicle for 30 min at 37°C prior to stimulation with PMA. After 10 min stimulation with PMA, cells were pelleted by low-speed centrifugation, membrane fractions were recovered, and protein concentrations were determined as described above. Protein from membrane fractions (20 µg) was used for immunoblot analysis as described above using either mouse anti-p47phox (BD Biosciences, San Jose, CA; diluted to 1:1,000, v/v) or rabbit anti-gp91phox (Upstate/Millipore, Billerica, MA, diluted to 1:1,000, v/v) as the primary antibodies.
Data analysis
All data are reported as mean ± standard error of the mean. For kinetic evaluations, nonlinear regression analyses were performed using the GraphPad Prism 5 software package (GraphPad Software, Inc., San Diego, CA). Concentration response curves were fit to the following equation to determine IC50 values:
where [I] represents the concentration of compound and h refers to the Hill coefficient. Given the reduced solubility of the Shionogi compounds (observed as precipitation) at concentrations of 10 µM and above, data obtained at or above this limit were excluded from further analysis. To determine inhibition modality with respect to NADPH substrate, initial velocities obtained at multiple NADPH and inhibitor concentrations were globally fit to competitive, uncompetitive and noncompetitive models, represented, respectively, by the following equations:
The best-fit model was identified using the extra sum-of-squares F-test as implemented by the GraphPad Prism software package (GraphPad Software, Inc., San Diego, CA).
For the Western analysis of p47phox translocation, statistical comparisons between each treatment group and the PMA-treated control group (n = 3 biological replicates) were made using one-way ANOVA with Dunnett’s multiple comparisons post-tests (**, p < 0.01; ***, p < 0.001). One outlier from the apocynin-treated samples was identified using the Grubb’s test and excluded from data analysis.
Results
Inhibition of cellular and semi-recombinant NOX2 activity by tool compounds
Human neutrophils were used both to assess the specific effects of compounds on NOX2 in an intact cellular assay format and as a source of enzyme to examine the effects of compounds on NOX2 in a biochemical, cell-free format. Namely, these cells selectively express NOX2 over the other NOX isoforms (NOX1, NOX3, NOX4 and NOX5), as reported previouslyCitation38 and confirmed by us (data not shown). Furthermore, deletion of the gp91phox subunit of NOX2 leads to loss of the respiratory burst and undetectable superoxide production in neutrophilsCitation39.
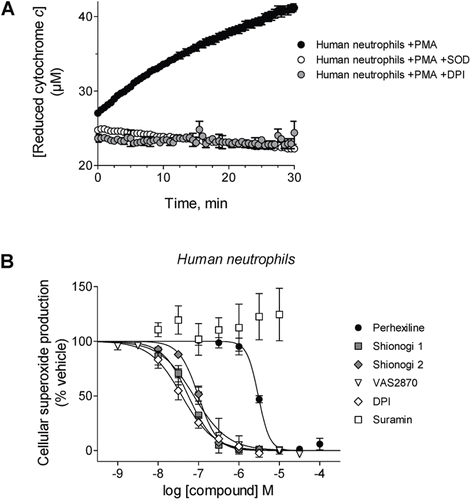
Extracellular superoxide production by intact, PMA-stimulated human neutrophils was assessed by the spectroscopic detection of cytochrome c reduction ()Citation40. Perhexiline, VAS2870, and DPI, a positive control, inhibited cellular superoxide formation with IC50 values of 3.0 ± 0.2 µM, 77 ± 14 nM and 39 ± 5 nM, respectively (). Shionogi 1 and Shionogi 2 inhibited superoxide formation with IC50 values of 56 ± 3 nM and 99 ± 6 nM, respectively. Suramin did not inhibit neutrophil superoxide formation at concentrations ranging from 10 nM to 10 µM.
Figure 2. Effects of compounds on cellular NOX activity. (A) Representative kinetic traces of cytochrome c reduction by PMA-stimulated human neutrophils alone (black circles) or in the presence of either SOD (white circles) or 100 µM DPI (gray circles). (B) Prior to stimulation with phorbol ester, human neutrophils were incubated in the presence of perhexiline (IC50 = 3.0 ± 0.2 µM, Hill slope = −3.1 ± 0.9), Shionogi 1 (IC50 = 56 ± 3 nM, Hill slope = −1.4 ± 0.1), Shionogi 2 (IC50 = 99 ± 6 nM, Hill slope = −1.8 ± 0.2), VAS2870 (IC50 = 77 ± 14 nM, Hill slope = −1.0 ± 0.2), DPI (IC50 = 39 ± 5 nM, Hill slope = −1.1 ± 0.1), or suramin (inactive). NOX activity was determined using detection of SOD-inhibitable cytochrome c reduction as described in “Methods”.
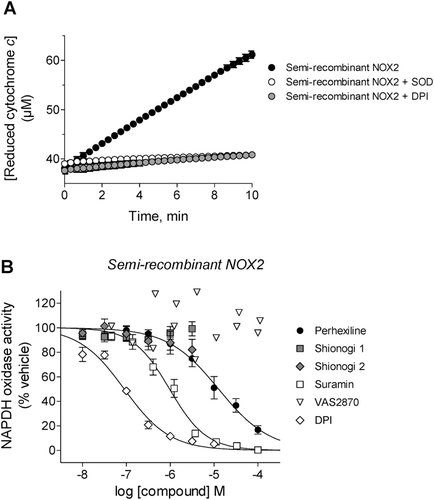
The activity of NOX2 in a cell-free environment was assessed using a semi-recombinant system in which neutrophil membranes were combined with purified recombinant p47phox, p67phox and Rac2. Confirmation of gp91phox in these membranes and the purity of the recombinant proteins were obtained by electrophoresis and immunoblotting (). Enzymatic activity was determined using the same cytochrome c-based assay format as was used for intact neutrophils (). As shown in , perhexiline and suramin inhibited semi-recombinant NOX2 activity (IC50 of 13 ± 2 µM and 1.0 ± 0.1 µM, respectively), when assayed with NADPH at its Km value (50 µM, as determined in pilot experiments). Neither VAS2870 nor the Shionogi compounds inhibited semi-recombinant NOX2 activity at concentrations ranging from 10 nM to 100 µM (for VAS2870) or 3 µM (for Shionogi 1 or 2). DPI, the positive control, inhibited semi-recombinant NOX2 activity with an IC50 of 90 ± 10 nM.
Figure 3. Protein components of the semi-recombinant NOX2 assay. (A) Representative anti-gp91phox immunoblot analysis of neutrophil membranes used in the semi-recombinant assay. A characteristic broad band centered at ~91 kD was observed when the membranes were denatured at 70°C for 5 min (left), consistent with literature reports for glycosylated gp91phox (see, for example, ref 35. Denaturation at 100°C for 10 min elicited a single band at ~60 kD (right), consistent with deglycosylation of the protein (theroretical MW = 65.3 kD) as a result of sample preparation. (B) Representative gel images of purified, recombinant, His-tagged cytosolic proteins p47phox (left), p67phox (middle) and Rac2 (right). For each protein, the Coomassie-stained gel (2 µg of protein) and the corresponding immunoblot (40 ng of protein) are shown.
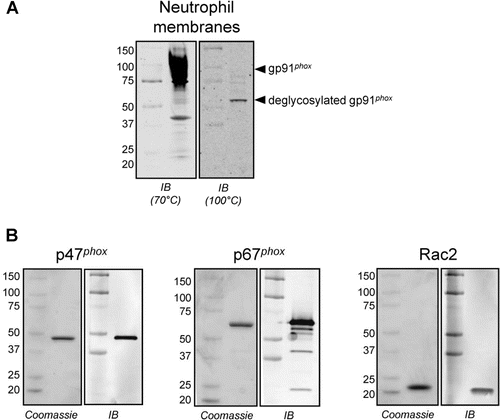
Figure 4. Effects of compounds on purified semi-recombinant NOX2 activity. (A) Representative kinetic traces of cytochrome c reduction by NADPH-stimulated semi-recombinant NOX2 alone (black circles) or in the presence of either SOD (white circles) or 2 µM DPI (gray circles). (B) Prior to reaction initiation with NADPH, neutrophil membranes and recombinant cytosolic components were added to perhexiline (IC50 = 13 ± 2 µM, Hill slope = −0.75 ± 0.08), Shionogi 1 (inactive), Shionogi 2 (inactive), suramin (IC50 = 1.0 ± 0.1 µM, Hill slope = −1.0 ± 0.1), VAS2870 (inactive), or DPI (IC50 = 90 ± 10 nM, Hill slope = −0.84 ± 0.07). NOX2 activity was determined using SOD-inhibitable cytochrome c reduction as described in “Methods”.
Determination of inhibition mechanisms of compounds
Given its activity in both NOX2 assay formats, perhexiline was also tested against an alternate superoxide generator, xanthine oxidase (XO), in order to rule out the possibility that this compound acts as a general superoxide scavenger. At concentrations ranging from 100 nM to 100 µM, perhexiline showed no effect on XO activity, whereas the known XO inhibitor luteolinCitation41 showed characteristic inhibition ().
Figure 5. Testing for inhibitory effects of compounds by NOX2-independent mechanisms. (A) Inhibition of xanthine oxidase activity by perhexiline (inactive) and luteolin (a positive control) was measured using SOD-inhibitable cytochrome c reduction as described in “Methods”. (B) Inhibition of PKCβII activity by cellular-active inhibitors Shionogi 1 (IC50 = 4.6 ± 0.8 nM, Hill slope = −1.0 ± 0.1), Shionogi 2 (IC50 = 9.4 ± 1.0 nM, Hill slope = −1.0 ± 0.1), perhexiline (inactive) and VAS2870 (inactive) as well as sangivamycin (a positive control, IC50 = 6.8 ± 2.0 µM, Hill slope = −0.7 ± 0.1), was determined using a fluorescence polarization IMAP™ kit as described in “Methods”.
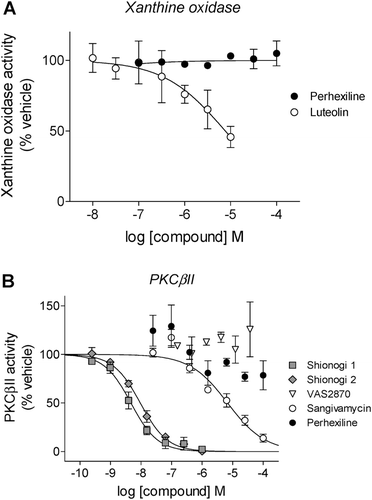
Neutrophils contain an abundance of the βII isoform of protein kinase C (PKCβII), which is critical for the activation of NOX2 by phorbol estersCitation42,Citation43 such as PMA, the stimulus used in our isolated neutrophil activity assay. Those compounds which inhibited superoxide formation in intact cells, but not semi-recombinant NOX2 − VAS2870, Shionogi 1 and Shionogi 2 − were tested for inhibition of PKCβII. VAS2870 did not inhibit PKCβII activity, whereas both Shionogi compounds displayed potent inhibition (IC50 = 4.6 ± 0.8 nM and 9.4 ± 1.0 nM, respectively, for Shionogi 1 and Shionogi 2) (). Consistent with its action as a direct inhibitor of NOX2, perhexiline did not inhibit PKCβII activity. As a positive control, the PKC inhibitor sangivamycinCitation44 displayed characteristic inhibition of PKCβII, with an IC50 of 6.8 ± 2.0 µM ().
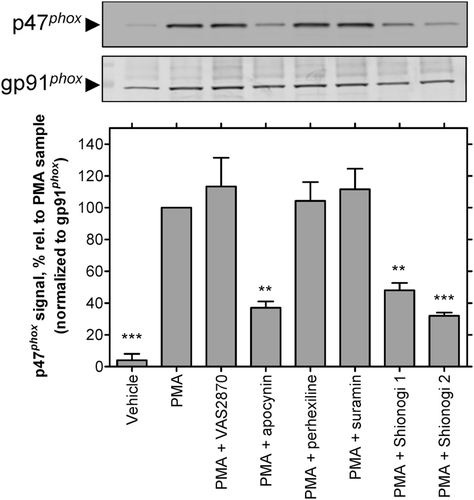
The ability of the compounds to affect translocation of p47phox to the membrane for assembly of NOX2 was examined using Western blotting of membrane fractions from PMA-stimulated neutrophils, as previously describedCitation45. As shown in , apocynin, a known inhibitor of this processCitation18,Citation20, blocked the appearance of p47phox in membrane fractions from stimulated cells. Shionogi 1 and 2 also blocked p47phox translocation, as expected from the role of PKCβII in this process and its inhibition by these compounds (see above). Perhexiline and suramin, which directly inhibited the purified semi-recombinant enzyme (see above), did not affect PMA-stimulated complex assembly. Consistent with a prior reportCitation34, VAS2870 also did not inhibit this response, suggesting that this compound does not interfere with NOX2 assembly or activation (e.g. by an activating kinase other than PKCβII).
Figure 6. Inhibition of p47phox translocation in PMA-stimulated neutrophils: representative immunoblots (top) and observed intensities of the p47phox bands (normalized to the intensity of the corresponding band for the integral membrane component of NOX2, gp91phox), relative to the PMA-treated samples from corresponding gels (bottom). Neutrophils were treated with vehicle, PMA (1 µM), or PMA following pretreatment with apocynin, VAS2870, perhexiline, suramin, Shionogi 1, or Shionogi 2, then membrane fractions were isolated and analyzed by Western analysis as described in “Methods”. With the exception of apocynin (100 µM), all compounds were tested at 10 µM. The order of sample lanes (top) correspond to the order of intensity bars (bottom).
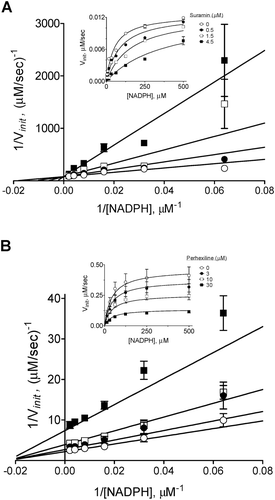
The inhibition modalities of perhexiline and suramin with respect to NADPH were determined in the semi-recombinant NOX2 assay. As shown in , suramin exhibited NADPH-competitive inhibition of NOX2 with a Ki of 0.7 ± 0.1 µM. In contrast, perhexiline () yielded a noncompetitive pattern with respect to NADPH (Ki = 13 ± 2 µM).
Figure 7. Mode of NOX2 inhibition by suramin (A) and perhexiline (B). Inhibition modalities were determined by global nonlinear fitting and model comparison as described in “Methods”. Lineweaver-Burk plots (foreground) were generated by reciprocal transformation of the best nonlinear fits (inset). Solid lines represent the best-fit global trends. (A) Suramin exhibits a competitive pattern of inhibition with respect to NADPH (best-fit parameters: Ki = 0.7 ± 0.1 µM, Km = 52 ± 8 µM, Vmax = 0.013 ± 0.001 µM/s). (B) Perhexiline exhibits a noncompetitive pattern (best-fit parameters: Ki = 13 ± 2 µM, Km = 44 ± 8 µM, Vmax = 0.46 ± 0.03 µM/s). Data shown are representative; differences in Vmax values between the two data sets reflect the typical variability observed with neutrophil membrane preparations.
Discussion
The data presented in this paper address mechanisms of action of reported inhibitors of cellular ROS production to determine whether any represent tools which could be used to validate NOX2 inhibition as a pharmacological approach for therapeutic intervention. We envisioned several distinct, readily-testable mechanisms by which these compounds might inhibit ROS production through the NOX2 pathway: direct inhibition of NOX2 enzymatic activity (perhaps by competition with NADPH substrate), general antioxidant activity, inhibition of PKC and inhibition of NOX2 assembly. As described below, our approach of evaluating inhibition both within the intact cellular milieu and directly against purified, semi-recombinant NOX2 enabled discrimination between compounds with respect to mechanisms of action and targets of effect. Furthermore, this approach underscored the limitations of these molecules as potential tool molecules to report on direct effects of NOX2 inhibition in cellular studies.
Of the compounds tested in the current study, only suramin and perhexiline directly inhibit NOX2 activity in vitro. Suramin inhibits NOX2 via competitive inhibition of NADPH. This is the first report of an NADPH-competitive inhibitor of NOX, and is consistent with its demonstration as a purinergic receptor antagonistCitation30 and competitive inhibitor of NAD+-dependent sirtuins such as SIRT5Citation46. The lack of cellular activity of suramin is consistent with its lack of membrane permeability, as previously suggestedCitation47, and indicates that further work to improve its membrane permeability would be necessary to exploit suramin as a tool for validating NOX2 as a therapeutic target. By contrast, we have yet to determine the precise mechanism of the observed noncompetitive inhibition of NOX2 by perhexiline. However, we interpret the similar potencies between biochemical and cellular activities of this compound as evidence that direct enzymatic inhibition is a major cause of cellular functionality.
Of all the compounds examined, only perhexiline inhibits NOX2 activity of both intact neutrophils and the purified enzyme. The use of perhexiline clinically and its efficacy in heart failure patients have been attributed to its inhibition of carnitine palmitoyl transferase-1 (CPT-1Citation48). Inhibition of CPT-1 shifts myocardial energy usage from fatty acid to glucose metabolism, which lowers oxygen demandCitation49. Interestingly, perhexiline has also been reported to inhibit superoxide formation in cardiovascular tissues and neutrophilsCitation28. Because our results also rule out antioxidant or kinase-dependent mechanisms, the inhibition of myocardial oxidative stress via direct NOX2 inhibition could be a major contributor to the beneficial effects of perhexiline, above and beyond its effects on cardiac energetics.
VAS2870 and both Shionogi compounds are extremely potent inhibitors of cellular superoxide formation, but not semi-recombinant NOX2 activity. Consistent with other studies of VAS2870 and a close analog, VAS3497Citation50, we have shown that the mechanism of VAS2870, as well as the Shionogi compounds, cannot include generalized antioxidant activity, as this would present as inhibition in both cellular and purified enzyme assays. Therefore, these compounds must act upstream of the activated NOX2 enzyme complex. Such a conclusion appears inconsistent with prior findings from ten Freyhaus, et al.Citation34, who reported inhibition of NOX2 by VAS2870 in whole cell lysates. However, this inhibition appeared to be dependent on the dynamics of complex assembly and order of stimulus addition (unpublished observation in reference 50) that could reflect events upstream of complex activation not recapitulated by our semi-purified enzyme preparation. As such, their observations with this compound likely reflect events more complex than direct inhibition of NOX2 catalysis.
Because intact neutrophils were stimulated with phorbol ester to activate PKCβII and trigger p47phox phosphorylation and subsequent translocation to membrane-associated gp91phox, a possible mechanism for cell-active compounds is inhibition of PKCβII. Both Shionogi compounds inhibit PKCβII with IC50 values similar to those seen in intact cells. Thus, PKCβII inhibition is responsible for the observed inhibition of cellular superoxide production by these compounds.
On the other hand, VAS2870 is not an inhibitor of PKCβII. The lack of effect of VAS2870 on PKCβII or as an antioxidant left open the possibility that VAS2870 might inhibit cellular NOX activity by blocking some other aspect of oxidase assembly. For example, apocynin, following peroxidase-dependent dimerization, inhibits NOX via covalent modification of p47phox and thereby blocking translocation of the subunit to the membraneCitation18. Indeed, apocynin inhibited the appearance of p47phox in PMA-stimulated neutrophil membrane fractions in our experiments, but VAS2870 had no effect at concentrations two orders of magnitude above its IC50 in intact cells, supportive of previous results with this compoundCitation34. Thus, the precise mode of action of VAS2870 against NOX is undetermined, but distinct from the mechanisms examined in the current study.
Conclusions
In this study, we profiled several emerging tool compounds as possible inhibitors of NOX2 and sought to probe their mechanisms by monitoring effects on direct activity of the purified enzyme and in intact cells. VAS2870 appears to be the most potent cellular inhibitor of NOX2 reported, but its precise mechanism of action remains to be determined. Our definition of whether VAS2870 is a direct inhibitor of NOX2 contrasts with that given by Wind et al.Citation50, which probably reflects differences in our assay formats. However, we reach a similar conclusion that the cellular inhibition of ROS generation by this class of compounds is not yet defined and may furthermore involve unrecognized elements of NOX2 regulation. The demonstration of NADPH-competitive inhibition of NOX2 by suramin is intriguing, and perhaps represents a route to identify other inhibitors of the enzyme. However, its activity at purinergic receptors and its absent or poor membrane penetrance render suramin, per se, an unsuitable tool for cellular studies of NOX2 inhibition. The identification of perhexiline as a direct inhibitor of NOX2 may represent a novel “add-on” for this compound, which is already used as a therapeutic for heart failureCitation29. For example, the β-adrenergic receptor antagonist carvedilol has shown added cardiovascular benefit over drugs in the same class due to added beneficial mechanisms of action, including antioxidant activityCitation51.
Additional work is required to identify a direct NOX2 inhibitor of sufficiently-defined mechanism for use as a tool for in vivo pharmacological target validation. However, our analysis has highlighted the various aspects of the NOX2 activation pathway that can be used to improve our understanding of the role of NOX2 in disease.
Acknowledgments
This work is dedicated to the memory of our friend and colleague Stephen A. Douglas. We are grateful to David J. Behm for discussions and critical review of this paper.
Declaration of interest
All authors are either currently employees of GlaxoSmithKline or were employed by GlaxoSmithKline at the time that this work was performed.
References
- Copeland RA, Harpel MR, Tummino PJ. Targeting enzyme inhibitors in drug discovery. Expert Opin Ther Targets 2007;11:967–978.
- Babior BM. NADPH oxidase. Curr Opin Immunol 2004;16:42–47.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev 2007;87:245–313.
- Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 2006;8:691–728.
- Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 2003;91:7A–11A.
- Sirker A, Zhang M, Murdoch C, Shah AM. Involvement of NADPH oxidases in cardiac remodelling and heart failure. Am J Nephrol 2007;27:649–660.
- Paravicini TM, Drummond GR, Sobey CG. Reactive oxygen species in the cerebral circulation: Physiological roles and therapeutic implications for hypertension and stroke. Drugs 2004;64:2143–2157.
- Lambeth JD. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic Biol Med 2007;43:332–347.
- Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002;105:293–296.
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H et al. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002;40:511–515.
- Thomas M, Gavrila D, McCormick ML, Miller FJ Jr, Daugherty A, Cassis LA et al. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 2006;114:404–413.
- Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM et al. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 2007;100:894–903.
- Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008;31 Suppl 2:S170–S180.
- Cross AR, Segal AW. The NADPH oxidase of professional phagocytes–prototype of the NOX electron transport chain systems. Biochim Biophys Acta 2004;1657:1–22.
- Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal 2009;11:2535–2552.
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011;10:453–471.
- O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J 1993;290 (Pt 1):41–49.
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 1994;11:95–102.
- DeLeo FR, Nauseef WM, Jesaitis AJ, Burritt JB, Clark RA, Quinn MT. A domain of p47phox that interacts with human neutrophil flavocytochrome b558. J Biol Chem 1995;270:26246–26251.
- Johnson DK, Schillinger KJ, Kwait DM, Hughes CV, McNamara EJ, Ishmael F et al. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium 2002;9:191–203.
- Ximenes VF, Kanegae MP, Rissato SR, Galhiane MS. The oxidation of apocynin catalyzed by myeloperoxidase: Proposal for NADPH oxidase inhibition. Arch Biochem Biophys 2007;457:134–141.
- Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: Molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther 2008;120:254–291.
- Beswick RA, Dorrance AM, Leite R, Webb RC. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001;38:1107–1111.
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res 2001;89:408–414.
- Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res 2004;95:587–594.
- Park YM, Park MY, Suh YL, Park JB. NAD(P)H oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem Biophys Res Commun 2004;313:812–817.
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I et al. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab 2008;9:686–696.
- Kennedy JA, Beck-Oldach K, McFadden-Lewis K, Murphy GA, Wong YW, Zhang Y et al. Effect of the anti-anginal agent, perhexiline, on neutrophil, valvular and vascular superoxide formation. Eur J Pharmacol 2006;531:13–19.
- Killalea SM, Krum H. Systematic review of the efficacy and safety of perhexiline in the treatment of ischemic heart disease. Am J Cardiovasc Drugs 2001;1:193–204.
- Voogd TE, Vansterkenburg EL, Wilting J, Janssen LH. Recent research on the biological activity of suramin. Pharmacol Rev 1993;45:177–203.
- Heyneman RA. Inhibition by suramin of the NADPH oxidase from horse polymorphonuclear leukocytes. Vet Res Commun 1987;11:149–157.
- Sobey C, Chrissobolis S, Hickey H, Drummond G. Suramin inhibits NADPH oxidase activity in cerebral arteries after subarachnoid hemorrhage. FASEB J 2006;20:A725.
- Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH et al. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun 2006;344:200–205.
- ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Bäumer AT, Vantler M et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 2006;71:331–341.
- Rotrosen D, Yeung CL, Katkin JP. Production of recombinant cytochrome b558 allows reconstitution of the phagocyte NADPH oxidase solely from recombinant proteins. J Biol Chem 1993;268:14256–14260.
- Fornwald JA, Lu Q, Wang D, Ames RS. Gene expression in mammalian cells using BacMam, a modified baculovirus system. Methods Mol Biol 2007;388:95–114.
- Abo A, Segal AW. Reconstitution of cell-free NADPH oxidase activity by purified components. Meth Enzymol 1995;256:268–278.
- Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001;269:131–140.
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 1995;9:202–209.
- Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol 2004;286:R431–R444.
- Nagao A, Seki M, Kobayashi H. Inhibition of xanthine oxidase by flavonoids. Biosci Biotechnol Biochem 1999;63:1787–1790.
- Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J et al. Protein kinase C-β contributes to NADPH oxidase activation in neutrophils. Biochem J 2000;347 Pt 1:285–289.
- Korchak HM, Kilpatrick LE. Roles for β II-protein kinase C and RACK1 in positive and negative signaling for superoxide anion generation in differentiated HL60 cells. J Biol Chem 2001;276:8910–8917.
- Loomis CR, Bell RM. Sangivamycin, a nucleoside analogue, is a potent inhibitor of protein kinase C. J Biol Chem 1988;263:1682–1692.
- Kim C, Dinauer MC. Impaired NADPH oxidase activity in Rac2-deficient murine neutrophils does not result from defective translocation of p47phox and p67phox and can be rescued by exogenous arachidonic acid. J Leukoc Biol 2006;79:223–234.
- Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A et al. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 2007;15:377–389.
- Stein CA. Suramin: A novel antineoplastic agent with multiple potential mechanisms of action. Cancer Res 1993;53:2239–2248.
- Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol 1996;52:273–280.
- Lopaschuk GD, Spafford M. Response of isolated working hearts to fatty acids and carnitine palmitoyltransferase I inhibition during reduction of coronary flow in acutely and chronically diabetic rats. Circ Res 1989;65:378–387.
- Wind S, Beuerlein K, Eucker T, Müller H, Scheurer P, Armitage ME et al. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 2010;161:885–898.
- Lysko PG, Webb CL, Gu JL, Ohlstein EH, Ruffolo RR Jr, Yue TL. A comparison of carvedilol and metoprolol antioxidant activities in vitro. J Cardiovasc Pharmacol 2000;36:277–281.