Abstract
The ubiquitin-proteasome pathway responsible for the turnover of many cellular proteins represents an attractive target in the development of new drug therapies: In particular, modulation of the proteasome activity by specific inhibitors may represent a useful tool for the treatment of tumours. Here, we report synthesis and activity of a new series of oligopseudopeptide analogues bearing a vinyl ketone pharmacophoric unit at the C-terminal position. Some derivatives showed inhibition in the µM range of the trypsin-like (T-L) active site of the proteasome.
Introduction
The proteasome 26S, a multi-catalytic proteaseCitation1, is an essential component of the ubiquitin-proteasome pathway (UPP) that degrades many proteins in eukaryotic cells. Fundamental cellular functions are linked to an ubiquitin- and ATP-dependent degradation of proteins involved in different pathways such as stress response, cell cycle control and differentiation, apoptosis and the regulation of transcription factors generationCitation2. Proteins destined to degradation are tagged by a covalently linked polyubiquitin chain in a process involving three enzymes in a successive action E1 (Ubiquitin-activating enzyme), E2 (Ubiquitin-conjugating enzyme) and E3 (Ubiquitin-ligase)Citation3,4. Poliubiquitin chain linked to proteins represents the signal for degradation by multi-catalytic complex that contains a central barrel-like core and a 20S proteolytic chamber composed of four stacked rings capped by two 19S structuresCitation5Citation,6. The two outer rings of the 20S are composed of seven α-subunits, whereas the two inner rings are made up by seven different β-subunits, and each β-ring contains three different active sites. In particular, the β1 subunit contains a post-acidic (PGPH) active site, the β2 subunit expresses trypsin-like (T-L) activity, and a chymotrypsin-like (ChT-L) proteolytic function is carried out by the β5 subunit. All the proteolytic cavities utilize the γ-hydroxyl function as a nucleophile and the α-amine as a proton donor-acceptor of the N-terminal threonine residue in the catalytic cycleCitation7Citation,8.
A proteasome isoform can be formed in response to cytokine signalling that induces the expression of different β-subunits and regulatory cap to constitute immunoproteasome capable to generate epitopes for presentation by MHC class I moleculesCitation9.
Considering the crucial implication of the proteasome in various cellular processes, modulation of enzymatic activities is extremely interesting from a therapeutic perspectiveCitation10–12. Natural and synthetic products have been tested as inhibitors of the different multi-catalytic complex subunitsCitation13–21. In vitro and in vivo studies demonstrated that proteasome inhibitors showed anti-proliferative and pro-apoptotic activities against solid and haematologic tumours. In particular, the boron derivative PS341 (Bortezomib) was used in the treatment of multiple myelomaCitation22Citation,23. Other molecules were evaluated for their effect on many disease states, including inflammation and cancer, as well as on modulation of immune responsesCitation24.
Our studies report the development of numerous series of peptide-based proteasome inhibitors containing a different pharmacophoric moiety as a potential substrate for the catalytic threonine through a mechanism similar to that of the well-known vinyl sulphone inhibitors, and as recently reportedCitation25, to that the natural pseudopeptidic compound Syringolin A and, even if in minor way, the analogue Syringolin B that irreversibly inhibits the β2 and β5 subunits of the proteasome, using the same catalytic mechanism.
Generally, our C-terminal pharmacophoric units (arecoline, vinyl ester and α,β-unsatured N-acylpyrrole) present a carbonylic function conjugated to a double bond. These electrophilic trap at the C-terminous are capped with oligopeptides (three residues) variously functionalized at the N-terminal. The electrophilic part of the molecules represents a potential substrate for the Michael addition by catalytic threonine, whereas aminoacidic sequences and groups at N-terminous interact with binding pockets of the enzymatic subunits and determine active site selectivityCitation26–28.
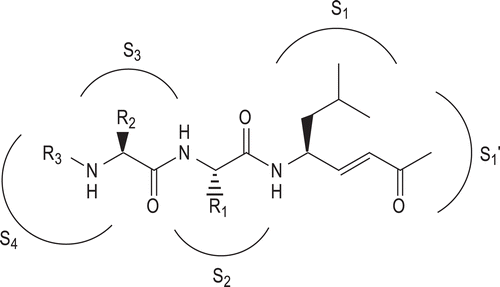
Herein, we describe the synthesis and biological activities of novel vinyl ketone-based peptide derivatives (). The aim of our work was to evaluate the capacity of the new C-terminal vinyl ketone pharmacophoric unit to interact with catalytic threonine, and the influence of the N-derivatized peptide portions on the inhibition potency and specificity. We synthesized and tested molecules containing a central tripeptidic sequence Leu-Leu-Leu (compounds 1–5) or Val-Ser-Leu (compounds 6–10) carrying 3-Hydroxy-2-methylbenzoyl (HMB), Z-protected 6-aminohexanoyl or 8-aminooctanoyl groups at the N-terminous, in accordance with the results obtained in previous series.
Figure 1. General structure of the vinyl ketone pseudotripeptides.
Matherials and methods
Chemistry-general
Amino acids, amino acid derivatives and chemicals were purchased from Bachem, Novabiochem and Fluka, respectively (Switzerland).Crude products were purified by preparative reversed-phase HPLC using a Water Delta Prep 4000 system with a Waters PrepLC 40 mm Assembly column C18 (30 × 4 cm, 300 Å, 15 μm spherical particle size column). The column was perfused at a flow rate of 30 mL/min, with a mobile phase-containing solvent A (10%, v/v, acetonitrile in 0.1% TFA), and a linear gradient from 0 to 100% of solvent B (60%, v/v, acetonitrile in 0.1% trifluoroacetic acid (TFA)); 30 min was the time adopted for elution of the compounds. HPLC analysis was performed using a Beckman System Gold with a Hypersil BDS C18 column (5 μm; 4.6 × 250 mm). Analytical determination and capacity factor (K′) of the peptides were assayed via HPLC conditions in the above solvent system (solvents A and B), programmed at flow rates of 1 mL/min, using the following linear gradients: (i) from 0 to 90% B for 25 min and (ii) from 30 to 100% B for 25 min. No pseudopeptide showed more than 1% impurity when monitored at 220 and 254 nm. The molecular weights of the compounds were determined by electrospray ionisation (ESI) (MICROMASS ZMD 2000), and the values are expressed as [MH]+. TLC was performed on pre-coated plates of silica gel F254 (Merck, Darmstadt, Germany), exploiting the following solvent systems: (iii) AcOEt/n-hexane (1:1, v/v), (iv) CH2Cl2/methanol (9.5:0.5, v/v), (v) CH2CL2/methanol (9:1, v/v) and (vi) CH2CL2/methanol/toluene (17:2:1, v/v/v). Ninhydrin (1%) or chlorine iodine spray reagents were employed to detect the peptides. Melting points were determined by a Kofler apparatus and are uncorrected. Optical rotations were determined by a Perkin–Elmer 141 polarimeter with a 10-cm water-jacketed cell. 1H NMR spectroscopy was obtained using a 400 spectrometer.
Synthesis
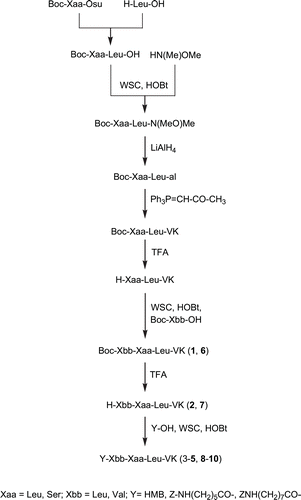
Vinyl ketone pseudotripeptides 1–10 were prepared using a C-terminal stepwise elongation. Following the strategy reported in , C-terminal dipeptide H-Xaa-Leu-VK was synthesized starting from leucine acylation by Boc-protected succinimidyl ester residue (Leu or Ser). Pharmacophoric unit was introduced by a Wittig reaction between dipeptide aldheydeCitation29, and the ylide [(methylcarbonyl)methylidene]triphenylphosphorane. Boc was removed by TFA and N-terminal residues were condensed using water soluble carbodiimide/N-hydroxybenzotriazole (WSC/HOBt) to complete the pseudotripeptide sequence 1,6 that after TFA treatment permitted to obtain the corresponding free N-terminal analogues. Finally, the other derivatives were obtained from 2 and 7, respectively by acylation with 3-hydroxy-2-methylbenzoic (3,8), Z-protected 6-aminohexanoic (4,9) or 8-aminooctanoic acids (5,10) always with WSC/HOBt as coupling reagent.
Scheme 1. Synthesis of new vinyl ketone derivatives 1–10.
All products were purified and isolated by preparative RP-HPLC, and the homogeneity of the lyophilized products was assessed by HPLC. Analytical characterization was then achieved by electrospray ionisation (ESI) mass spectrometry () and 1H NMR spectroscopy.
Table 1. Analytical data and physicochemical properties of the novel pseudotripeptides 1–10.
General synthetic procedures
TFA deprotection
Boc was removed by treating intermediates with aqueous 90% TFA (1:10, w/v) for 30–40 min. After evaporation, the residue was triturated with Et2O, centrifuged and the resulting solid was collected and dried.
Coupling with WSC/HOBt
The deprotected α-amine intermediate (1 mmol), N-methylmorpholine (NMM) (2 mmol) WSC (1 mmol) and HOBt (1 mmol) were added to a solution of carboxylic component (1 mmol) in dimethylformamide (3 mL) at 0°C. The reaction mixture was stirred for 1 h at 0°C and 18 h at rt; then the solution was diluted with AcOEt (80 mL) and washed consecutively with HCl 0.1 N, NaHCO3 and brine. The organic phase was dried (MgSO4) and evaporated to dryness. The residue was treated with Et2O and the resulting solid separated by centrifugation.
1H NMR of the selected compounds
Boc-Leu-Leu-Leu-VK (1). 1H NMR (CDCl3): δ 1.01–1.12 (m, 18H); 1.50–1.77 (m, 15H); 1.89-1.97 (m, 3H); 2.41 (s, 3H); 4.11 (m, 1H); 4.37–4.49 (m, 2H); 6.18 (d, J = 16.2, 1H); 6.87 (dd, J = 16.1, 1H); 7.36 (br s, 3H).
Z-NH-(CH2)5-CO-Leu-Leu-Leu-VK (4). 1H NMR (CDCl3): δ 0.98–1.08 (m, 18H); 1.27 (m, 2H); 1.50–1.65 (m, 6H); 1.81-1.94 (m, 7H); 2.24 (t, 2H); 2.33 (s, 3H); 2.88 (t, 2H); 4.17 (m, 1H); 4.49–4.60 (m, 2H); 5.18 (s, 2H); 6.21 (d, J = 16.4, 1H); 6.85 (dd, J = 16.3, 1H); 7.11–7.23 (m, 5H); 7.58 (br s, 4H).
HMB-Val-Ser-Leu-VK (8). 1H NMR (CDCl3): δ 1.01–1.12 (m, 12H); 1.47 (m, 2H); 1.85 (m, 1H); 2.25 (br s, 1H); 2.34 (s, 3H); 2.45 (s, 3H); 2.83 (m, 1H); 4.10–4.19 (m, 2H); 4.38 (m, 1H); 4.58-4.70 (m, 2H); 5.08 (br s, 1H); 5.97 (d, J = 16.0, 1H); 6.72 (dd, J = 16.2, 1H); 7.05–7.21 (m, 3H); 7.76 (br s, 3H).
Z-NH-(CH2)7-CO-Val-Ser-Leu-VK (10). 1H NMR (CDCl3): δ 1.04–1.13 (m, 12H); 1.31-1.42 (m, 6H); 1.55-1.64 (m, 6H); 1.87 (m, 1H); 2.15 (br s, 1H); 2.24 (t, 2H); 2.39 (s, 3H); 2.73 (m, 1H); 3.01 (t, 2H); 4.11 (m, 2H); 4.28 (m, 1H); 4.50-4.59 (m, 2H); 5.37 (s, 2H); 6.07 (d, J = 16.3, 1H); 6.92 (dd, J = 16.1, 1H); 7.09–7.23 (m, 5H); 7.83 (br s, 4H).
Biological investigation
Proteasome purification
Proteasomes were isolated from lymphoblastoid cell lines (LCL) as previously describedCitation30.
Proteasome subunit inhibition assays
Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC (Sigma) were used to measure chymotrypsin-like, trypsin-like and post-acidic proteasome activities, respectively. Substrates were incubated at 37°C for 30 min with proteasomes, untreated or pre-treated with 0.001–10 μM of test compounds, in activity buffer. Fluorescence was determined by a fluorimeter (Spectrafluor plus, Tecan, Salzburg, Austria), using an excitation of 360 nm and emission of 465 nm. Activity was evaluated in fluorescence units and the inhibitory activity of the compounds is expressed as IC50. The data were plotted as percentage control (the ratio of percentage conversion in the presence and absence of inhibitor) versus inhibitor concentration, and fitted with the equation Y = 100/1 + (X/IC50)A, where IC50 is the inhibitor concentration at 50% inhibition and A is the slope of the inhibition curve.
Enzymatic stability assays
The stability of the vinyl ketones under proteases degradation was studied in human plasma. Test compounds were incubated with plasma (0.6 mL) in a total volume of 1.5 mL of 10 mM Tris-HCl buffer, pH 7.5. Incubation was performed at 37° C for 360 min. The incubation was terminated by addition of ethanol (0.2 mL), the mixture poured at 21°C, and, after centrifugation (5000 rpm for 10 min) aliquots (20 µL) of the clear supernatant were injected into an RP-HPLC column. HPLC was performed as described in analytical determinations. The degradation half-life (T1/2) was obtained by a least-squares linear regression analysis of a plot of the logarithmic inhibitor concentration versus time, using a minimum of five points.
Results and discussion
Vinyl ketone pseudotripeptides 1–10 were synthesized following the strategy reported in .
Inhibition of β1, β2 and β5 active sites of the 20S proteasome, previously purified from lymphoblastoid cell lines, was determined using fluorogenic substrates specific for the three main proteolytic activities of the enzymatic complex. Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC were used to measure chymotrypsin-like, trypsin-like and caspase-like proteasome activities, respectively. Substrates were incubated, at 37°C for 30 min, with the proteasome, pre-treated with incremented concentrations (from 0.001 to 10 µM) of the new vinyl ketone derivatives in activity buffer. Activity was evaluated in fluorescence units, and the inhibitory activity of the compounds is expressed here as IC50.
General analysis of the activity profile shows that the new compounds have a low capacity to inhibit the proteasome activities suggesting that the C-terminal new pharmacophoric group is not a good substrate for the catalytic threonine. Indeed, all compounds were less active compared to previously described inhibitorsCitation26–28.
Chymotrypsin-like activity was in a µM range for analogues presenting N-terminal linear amino acids with a long Z-protected chain on the terminal amine group. Likewise, inhibition of the trypsin-like was relatively pronounced, with IC50 values in the order of 3–10 µM for compounds 4, 5, 9 and 10. Furthermore, 3-Hydroxy-2-methylbenzoyl N-functionalized derivative showed mild inhibitory capacity of the trypsin-like activity of the proteasome. Generally, the biological response was independent from the central tripeptide sequence but it correlated to N-terminal substituents; in particular, compounds 5 and 10 with the more bulky groups resulted the best analogues of the series. All compounds were unable to inhibit post-acidic (PGPH) activity.
The susceptibility of five selected vinyl ketone derivatives to enzymatic hydrolysis was determined by incubation at 37°C in human plasma. The pseudopeptides, according to terminal modifications of the peptide chain, showed great stability to plasma proteases ().
Table 2. Inhibition of proteasome subunits and metabolic stability of vinyl ketone derivatives.
Conclusions
The UPP plays an important role in many cellular processes. Considering the high therapeutic potential of inhibitors selective and specific for the catalytic subunits of the 20S proteasome, we synthesized and tested new peptide-based compounds with new C-terminal pharmacophoric units. Clinical trials indicate that inhibitors exhibit toxic effects during prolonged drug treatment, therefore the availability of new potent and selective molecules without side-effects is ever required. In this optic, we prepared a new series of peptide-based compounds containing a vinyl ketone pharmacophoric unit at the C-terminal as potential substrates of the catalytic γ-hydroxy threonine side-chain in Michael addition. Generally, inhibition of active subsites of the proteasome is detectable in a µM range only for some derivatives. The biological response is function of the N-terminal substituent and not dependent by physicochemical properties of the central tripeptidic sequence. Finally, vinyl ketone is not a favourable electrophilic functionality for the primary interaction with the proteasome catalytic subunits.
Declaration of interest
Financial support of this work was provided by the University of Ferrara, the Ministero dell’Università e della Ricerca Scientifica e Tecnologica (MURST).
References
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell 1994;79:13–21.
- Reed SI. The ubiquitin-proteasome pathway in cell cycle control. Results Probl Cell Differ 2006;42:147–181.
- Brannigan JA, Dodson G, Duggleby HJ, Moody PC, Smith JL, Tomchick DR et al. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 1995;378:416–419.
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 1996;65:801–847.
- Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995;268:533–539.
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997;386:463–471.
- Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Catalytic mechanism and assembly of the proteasome. Chem Rev 2009;109:1509–1536.
- Kisselev AF, Songyang Z, Goldberg AL. Why does threonine, and not serine, function as the active site nucleophile in proteasomes? J Biol Chem 2000;275:14831–14837.
- Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol 1999;17:739–779.
- Testa U. Proteasome inhibitors in cancer therapy. Curr Drug Targets 2009;10:968–981.
- Ludwig A, Fechner M, Wilck N, Meiners S, Grimbo N, Baumann G et al. Potent anti-inflammatory effects of low-dose proteasome inhibition in the vascular system. J Mol Med 2009;87:793–802.
- Orlowski RZ. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ 1999;6:303–313.
- Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev 2001;21:245–273.
- Moore BS, Eustáquio AS, McGlinchey RP. Advances in and applications of proteasome inhibitors. Curr Opin Chem Biol 2008;12:434–440.
- Groll M, Götz M, Kaiser M, Weyher E, Moroder L. TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chem Biol 2006;13:607–614.
- Nazif T, Bogyo M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA 2001;98:2967–2972.
- Furet P, Imbach P, Noorani M, Koeppler J, Laumen K, Lang M et al. Entry into a new class of potent proteasome inhibitors having high antiproliferative activity by structure-based design. J Med Chem 2004;47:4810–4813.
- Hogan PC, Corey EJ. Proteasome inhibition by a totally synthetic β-lactam related to salinosporamide A and omuralide. J Am Chem Soc 2005;127:15386–15387.
- Momose I, Umezawa Y, Hirosawa S, Iinuma H, Ikeda D. Structure-based design of derivatives of tyropeptin A as the potent and selective inhibitors of mammalian 20S proteasome. Bioorg Med Chem Lett 2005;15:1867–1871.
- Kaiser M, Groll M, Renner C, Huber R, Moroder L. The core structure of TMC-95A is a promising lead for reversible proteasome inhibition. Angew Chem Int Ed Engl 2002;41:780–783.
- Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, Schellenberg B et al. Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc Natl Acad Sci USA 2009;106:6507–6512.
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004;5:417–421.
- Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer 2004;4:349–360.
- Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. J Cell Commun Signal 2011;5:101–110.
- Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H. Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc Natl Acad Sci USA 1997;94:6629–6634.
- Marastoni M, Baldisserotto A, Canella A, Gavioli R, Risi CD, Pollini GP et al. Arecoline tripeptide inhibitors of proteasome. J Med Chem 2004;47:1587–1590.
- Marastoni M, Baldisserotto A, Trapella C, Gavioli R, Tomatis R. P3 and P4 position analysis of vinyl ester pseudopeptide proteasome inhibitors. Bioorg Med Chem Lett 2006;16:3125–3130.
- Baldisserotto A, Ferretti V, Destro F, Franceschini C, Marastoni M, Gavioli R et al. α,β-unsaturated N-acylpyrrole peptidyl derivatives: new proteasome inhibitors. J Med Chem 2010;53:6511–6515.
- Fehrentz JA, Pothion C, Califano JC, Loffet A, Martinez J. Synthesis of chiral N-protected α-amino aldehydes by reduction of N-protected N-carboxyanhydrides (UNCAs). Tetrahedron Lett 1994;35:9031–9034.
- Gavioli R, Vertuani S, Masucci MG. Proteasome inhibitors reconstitute the presentation of cytotoxic T-cell epitopes in Epstein-Barr virus-associated tumors. Int J Cancer 2002;101:532–538.