Abstract
The analysis of the structural similarity between Candida albicans Sap2 and HIV-1 aspartic proteases by molecular modeling gave insight into the common requirements for inhibition of both targets. Structure superimposition of Sap2 and HIV-1 protease confirmed the similarity between their active sites and flap regions. HIV-1 protease inhibitors herein investigated can fit the active site of Sap2, adopting very similar ligand-backbone conformations. In particular, key anchoring sites consisting of Gly85 in Sap2 and Ile50 in HIV-1 protease, both belonging to their corresponding flap regions, were found as elements of a similar binding-mode interaction. The knowledge of the molecular basis for binding to both Sap2 and HIV-1 proteases may ultimately lead to the development of single inhibitor acting on both targets.
Introduction
Candida albicans is a polymorphic opportunistic fungal pathogen and it is responsible for the majority of candidiasis in humans. It usually resides as a commensal in the human mucosal surface such as the oral cavity. In the presence of a compromised host immune system, C. albicans is able to cause a wide spectrum of infections ranging from mucosal to life-threatening disseminated candidiasis. Oralpharyngeal candidiasis is recognized as one of the most frequent opportunistic infectious diseases among patients suffering from HIV infection. The appearance of this pathology is considered a prognostic indicator for the development of AIDS in HIV-positive patients, and it has been reported to occur in 54–93% of patients with AIDSCitation1,Citation2. Otherwise, Candida vaginitis occurrence seems to be equally frequent in HIV-positive patients, as well as in non HIV-infected onesCitation3. Upon the introduction of antiretroviral therapy (ART), including HIV protease inhibitors, for the treatment of HIV infection, it was observed that this type of treatment favorably influenced the frequency of mucosal candidiasis. Patients undergoing ART regimens displayed a reduction of clinically apparent oral candidiasis occurrence than patients not receiving ART, and this beneficial effect might be not completely due to a reconstitution of the immune status, but rather due to a direct effect on C. albicans. This hypothesis was confirmed in 1998 by Hoegl et al., who reported a case of a patient treated with antiretroviral drugs who recovered from fungal infection maintaining a low CD4+ T-cell countsCitation4. Effects on oral candidiasis appeared in HIV patients treated with antiretroviral agents soon after therapy and before CD4+ cell recovery. Subsequently, in 1999 the first demonstration of a direct inhibition of proteases from C. albicans with HIV protease inhibitors was reportedCitation5,Citation6. Indeed, C. albicans possesses ten distinct genes (SAP1–10) encoding secreted aspartic protease (Sap) enzymes, which appear to play a central role in infections caused by this fungus, allowing to adhere and invade host tissuesCitation7. The role of secreted aspartic proteases has been well characterized during last years by several groups, and all these studies indicated these enzymes contributing to Candida virulence and pathogenesis in human infectionsCitation8. Saps are expressed at different stages and in distinct types of the infection process, and on the basis of these characteristics they can be divided into two different subgroups. Sap1–3 seem to have a crucial role in adherence and tissue damage of localized infection, whereas Sap4–6 are involved in systemic diseasesCitation9–11. Sap9 and Sap10 are glycosyl-phosphatidyl-inositol (GPI)-anchored membrane-bound proteins, whereas the role of Sap7 and Sap8 is still unknownCitation12,Citation13. Structural studies of C. albicans proteases family have been concentrated mainly on Sap2, which is the most abundant protein expressed in vitro when grown in the presence of protein as the sole source of nitrogen, and it has been recognised as a crucial virulence factor for vaginal infectionsCitation14. Thus, this family of enzymes offers a potential target for drug intervention in fungal infections, especially when current treatments fail due to phenomena of drug resistance to available antifungal agentsCitation15. Considering the concomitant recurrence of HIV- and C. albicans-derived infectious diseases and that their major virulence factors, namely HIV-1 protease and C. albicans Sap2, respectively, belong to the same superfamily, the development of new “dual” inhibitors able to interact with both targets represents an enormous challenge and a matter of high relevance. Although the evidence of a direct inhibition of proteases from C. albicans with HIV protease inhibitors is known since last decade, to our knowledge no detailed in silico studies of the binding mode of such drugs on the two proteases has been reported, yet. In a recent work, Řezáčová et al. reported the crystallographic structure of protease Sapp1p from Candida parapsilosis in complex with HIV protease inhibitor ritonavir, thus giving a first evidence of specific binding of this class of HIV drugs to Sap enzymes, and supporting the importance of examining more in detail the common structural determinants for inhibition of HIV protease and Sap2Citation16.
In the present work, we investigated the structural similarity between these two tightly correlated targets, and the key structural features required for inhibition by a three-dimensional structural superimposition of Sap2 and HIV-1 proteases and by molecular modeling. Specifically, we simulated the interaction between two selected FDA-approved HIV-1 protease inhibitors as model compounds and Sap2 in order to analyze the molecular basis for Sap2 susceptibility to such HIV-protease inhibitors.
Methods
Tridimensional structural alignment
The Swiss PDB viewer (SPDBV) program (4.0.1 version, Swiss Institute of Bioinformatics) was used to superimpose three dimensional (3D) structures of HIV-1 aspartic protease (PDB code: 3OXC) and Secreted aspartic protease 2 from C. albicans (PDB code: 1EAG)Citation17,Citation18. Alpha-carbon atoms of the highly conserved DTG motif (e.g. Asp25, Thr26 and Gly27 for HIV-1 protease) were initially superimposed using the “fit molecule from selection” option. Then, using the “improve fit” option, SPDBV was asked to minimize the root-mean square distance (RMSD) between the corresponding atoms using a least square algorithm. The calculation was extended to neighbors until the maximum number of aligned atoms with the lowest RMSD was obtained. Structural alignment was generated after protein superimposition. Residues of the superimposed protein that were spatially close to residues of the static one were aligned. Appropriate gaps were inserted in the sequences to indicate a lack of structural correspondence.
Molecular modeling calculations
Docking analysis were performed using GOLD (Genetic Optimization for Ligand Docking) software package 5.1 version (CCDC, Cambridge, UK)Citation19. The structures of the ligands were constructed using Schrödinger MAESTRO interface, and then energy-minimized with Spartan. The equilibrium geometry was calculated through the semi-empirical method AM1. The coordinates of Sap2 enzyme were retrieved from the Protein Data Bank (PDB code:1EAG). The ligand-protein complex was unmerged for achieving free enzyme structure, water molecules were removed and hydrogen atoms were added to the enzyme. The binding site was defined as all residues of the protein located within 10 Å from the β-carbon of Asp218. ChemPLP (Piecewise Linear Potential) was chosen as the fitness function, and the standard default settings were used in all the calculationsCitation20. Docking poses were then rescored using GOLDscore fitness function. For each of 50 independent genetic algorithm runs, a maximum of 1,000,000 genetic operations were carried out using default operator weights and a population size of 200 chromosomes. Results differing by less than 1.5 Å in ligand-all atom RMSD were clustered together. The binding mode analysis of docked conformations was carried out using PyMol software version 0.99Citation21. Best-scoring docked conformations were compared with their crystal structure in complex with HIV-1 protease. Specifically, docked ligands were superimposed to the corresponding ligand structure in complex with HIV-1 protease using the same procedure as described above for 3D-structural superimposition (PDB code: 3OXC and 1HXW for saquinavir and ritonavir, respectively).
Pairwise sequence alignment
Primary sequences of HIV-1 aspartic protease and Sap2 from C. albicans were retrieved from the PDB in FASTA format. EMBOSS matcher version 6.3.1 was used to carry out local alignments of the two proteins, by identifying local similarities in the two protein sequence using an algorithm based on Bill Pearson’s lalign application, version 2.0u4 (February 1994). EBLOSUM-62 substitution matrix was used and penalties of −14 and −4 were applied for gap opening and extension, respectively.
Results and discussion
Structural features of C. albicans Sap2 and HIV-1 protease
Sap2 possesses typical features of eukaryotic aspartic proteases. Indeed, it has a monomeric structure constituted by N- and C-terminal domains, each possessing an active site region. It displays a bi-lobed shape characterized by the presence of a central binding cleft able to accommodate up to nine residues of peptide substrates ().
Figure 1. Ribbon representation of the crystal structure of C. albicans Secreted aspartic protease 2, Sap2, (PDB code: 1EAG). Residues in contact with A70450 are shown in cyan.
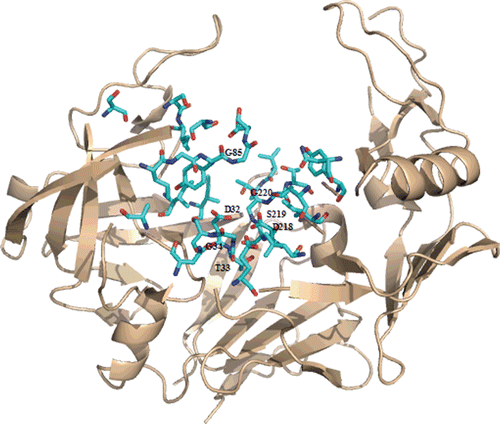
Similarly to other fungal proteases, Sap2 presents a Asp-Thr-Gly (DTG) motif in the N-terminal domain, and a Asp-Ser-Gly (DGS) motif in the C-terminal domain, whereas the majority of mammalian aspartic proteases display a DTG motif in both domainsCitation22. A highly conserved feature is represented by a β-hairpin loop, named “flap region,” which shields the active site from bulk solvent and interacts centrally with the substrate/inhibitor. In Sap2 this region encloses residues Lys81-Gln91. Sap2 displays broad substrate specificity, being able to degrade many extracellular matrix components such as keratin, collagen, mucin and proteins related to immunological defenses (i.e. IgA and lactoferrin), as extensively investigated by Koelsch et al.Citation23 At P1 site all Saps display preferentially large hydrophobic residues, and among them Sap2 displays preferentially in the order Phe>Leu>Tyr. P1′ specificities are broader than those observed for P1, and Sap2 generally prefers Tyr in such position.
HIV-1 aspartic protease inhibitors saquinavir and ritonavir inhibit Sap2, as well as other components of its subgroup, whereas they do not show inhibition potency against Saps4–6. Nevertheless, Sap2 inhibition by these protease inhibitors occurs at higher concentration as compared to that required for HIV-1 protease inhibition (). We selected the first generation protease inhibitors saquinavir and ritonavir as model compounds for our structural investigation ().
Table 1. Inhibition potency of FDA HIV-1 aspartic protease inhibitors towards Sap2 and HIV-1 protease.
HIV-1 aspartic protease, which belongs to the retropepsin family, is less than half the size of mammalian aspartic proteases. It is active as a symmetric dimer with a single active site, formed by residues facing two identical monomers (), and each monomer consists of 99 amino acids. This architecture is unique among enzymes, with no other examples of active sites formed in a similar mannerCitation27. HIV-1 protease displays the highly conserved active site sequence Asp-Thr-Gly typical of all aspartic proteases. This sequence is located in a loop whose structure is stabilized by a network of hydrogen-bonds called “fireman’s grip”.
Figure 2. Ribbon representation of the crystal structure of HIV-1 protease (PDB code: 3OXC). Residues in contact with saquinavir are shown in cyan.
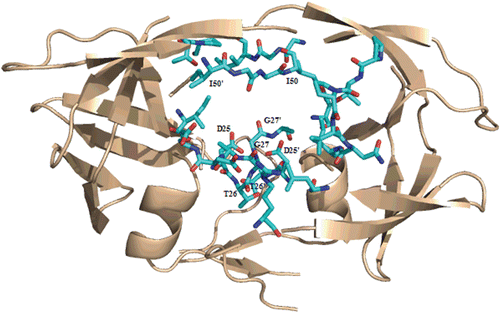
HIV-1 protease possesses two flexible flap regions, which participate on binding with the substrate/inhibitor, and differs to eukaryotic aspartic protease, which possess only one hairpin loop in that regionCitation28. This enzyme cleaves peptide bonds in polyprotein precursors of structural proteins and viral-encoded enzymes during the maturation and assembly of infectious viral particles. As regarding to substrate specificity, HIV-1 protease shows a wide variety of cleavage preferences, similarly to Sap2. Specifically, at P1 site it shows preference toward hydrophobic residues, as well as at P1′ position, where the enzyme can accommodate also a Pro residue.
Structural comparison between Sap2 and HIV-1 protease
Sequence comparison between Saps, mammalian aspartic proteases and HIV-1 protease in the active site and flap region demonstrated that, though belonging to the pepsin family, Saps are the eukaryotic proteases that most resemble the HIV-1 protease structureCitation29–31. Despite differences in both size and overall amino acid sequence between Sap2 from C. albicans and HIV-1 protease, significant homologies exist between their active sites. Similarities in the overall shape and secondary structure are also present. In order to compare the primary structures of Sap2 and HIV-1 protease, we performed a local sequence alignment using the EMBOSS matcher algorithmCitation32, and high similarity was found in the active site regions of Sap2 and HIV-1 protease (). Indeed, sequence alignment of active sites discloses a similarity of 57.1% and identity of 21.4% between Sap2 N-terminal domain (18–45 residues) and HIV-1 protease active site region (11–38 residues), and higher similarity with Sap2 C-terminal domain (66.7% of similarity and 33.3% of identity). Similarities between Sap2 and HIV-1 protease also emerge from 3D-structural superimposition performed with Swiss PDB viewer (SPDBV) software (4.0.1 version, Swiss Institute of Bioinformatics), and from the structure-based sequence alignment. The 3D-structural superimposition (RMS on alpha-carbon of 1.44 Å) highlighted the two proteins displaying structural similarities in the binding pocket and in the flap region, which interact centrally with bound inhibitor/substrate (). The two enzymes show similarities in the conserved residues flanking catalytic aspartic acid residues common in all aspartic proteases. Upstream residues DTG (Asp32-Thr33-Gly34) motif in the N-terminal domain of Sap2 possess an aliphatic nature (Val31, Ile30, Val29), and this feature is shared also by HIV-1 protease, showing two leucine residues (Leu23, Leu24) and an alanine (Ala22) in same positions.
Figure 3. Sequence comparison between Sap2 (1EAG) and HIV-1 aspartic protease (3OXC) in the active site region performed using EMBOSS matcher pairwise sequence alignment: Sap2 N-terminal domain, top; C-terminal domain, bottom.

Figure 4. Top: superimposition of HIV-1 protease (PDB: 1OXC in light pink) and C.albicans Sap2 (PDB: 1EAG in green) obtained with SPDBV. Down: binding pockets superimposition.
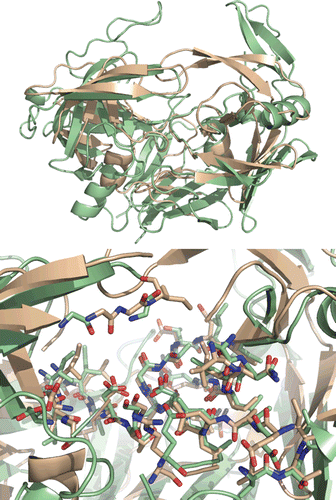
In both cases, such aliphatic sequence terminates with a polar residue (Asn28 and Glu21 in Sap2 and HIV-1 protease, respectively), and superimposition of this amino acid pattern is characterized by a RMS value of 0.789 Å. Similarly, in other domains the two proteins display the same Leu–Leu pattern (Leu216–217 in Sap2 and Leu23′–24′ in HIV-1 protease), followed by an aliphatic (Val215 in Sap2 and Ala22′ in HIV-1 protease) and an acidic residue (Asp214 and Glu21′). Even in this case, the amino acid orientation is very similar, and the superimposition displays an RMS value of 0.654 Å. An interesting structural feature shown by both enzymes is a hairpin loop called flap region projecting over the active site, which plays an important role in binding to substrate/inhibitor. The existence of two flaps in HIV-1 protease versus a single flap for Sap2 determines a more intimate embracing of the substrate by the retropepsin enzyme. Superimposition analysis showed these loops overlapping at residues directly involved in the interaction with substrate/inhibitor, namely Gly85 and Asp86 in Sap2, and Gly49 and Ile50 in HIV-1 protease. Flap regions of the two enzymes enclose ten residues, and in both cases the presence of glycine residues stands out. The amount of glycines in the flap region seems to reflect the need of conformational flexibility of this element. Residues 197–215 of Sap2, which belong to a subdomain of about 100 residues within the C-terminal domain, display structural similarities with residues 12′–22′ of HIV-1 aspartic protease (RMS 1.030 Å). Residues 298–300 and 303–310 from Sap2 are structurally related to 75′–77′ and 82′–89′ residues of HIV-1 protease with an RMS of 1.768 Å and 0.767 Å, respectively. In this position, Sap2 displays a shorter loop connecting Phe298 to Ile305, as compared to other mammalian aspartic proteases, which allows for some residues interacting with the substrate/inhibitor in the active site. In particular Asn301, Ala303 and Ile305 constitute a short “specificity ridge” unique to this fungal enzyme. Indeed, variations of these three residues could affect substrate specificityCitation33, and the superimposition showed that such feature is shared also by HIV-1 aspartic protease, as Val82′ and Ile84′ superimpose nicely with Ala303 and Ile305, respectively, which belong to this specificity ridge.
Molecular modeling studies
Two HIV protease inhibitors were selected as model compounds, ritonavir and saquinavir, for which inhibition data against Sap2 are reported in the literature, and were docked into Sap2 structure, using the genetic algorithm GOLD (Genetic Optimization for Ligand Docking, CCDC, Cambrige, UK)Citation5,Citation6,Citation19. Best-scoring docked conformations resulting from docking of each ligand were clustered and visually inspected for enzyme-ligand interactions. Furthermore, docked ligands were compared and superimposed to their corresponding crystal structure in complex with HIV-1 protease using the Swiss PDB viewer (SPDBV) software (PDB code: 3OXC for saquinavir and 1HXW for ritonavir). The two inhibitors taken into account in the present study are peptide-like inhibitors, consisting of main-chain amide bond isosteres and hydrophobic groups corresponding to side-chains. The interaction between these inhibitors and HIV-1 protease follows a common pattern characterized by the presence of hydrogen-bonds mainly formed between main-chain atoms of both protease and inhibitors. In the inhibitor, the scissile bond isostere is located near the carboxylic groups of Asp25/Asp25′ interacting through hydrogen-bonds. Also, a double hydrogen-bond system is displayed between inhibitors and a water molecule, which bridges the inhibitor carbonyl groups and Ile50/Ile50′ amide protons. Specifically, in saquinavir/HIV-1 protease complex, the hydroxyl function of hydroxyethylene isostere establishes hydrogen-bonds with side chain carboxylate oxygen atoms of both catalytic Asp25/Asp25′, and two water molecules establish three more hydrogen-bonds with saquinavir and the enzymeCitation34. The interaction between the inhibitor and flap residues is mediated by a water molecule interacting with the carbonyl oxygen of Gly27. Finally, saquinavir also interacts with Asp29 through hydrogen-bonds.
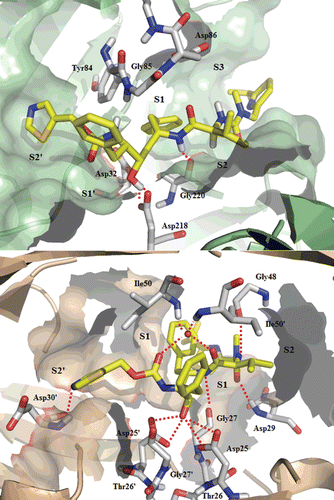
Our docking results highlighted that same polar contacts are preserved in the interaction with Sap2. In particular, saquinavir interacts with both catalytic Asp218 and Asp32, as in HIV-1 protease complex, and another hydrogen-bond is established between the main chain carbonyl moiety and Gly85 amide proton (). The latter is similar to hydrogen-bonds as established between backbone carbonyl groups and Ile50/Ile50′ located in the flap region of HIV-1 protease, although in that case the interaction is mediated by a water molecule. An amide proton of the main chain establishes a hydrogen-bond with the carbonyl oxygen of Gly34, and similarly Gly27 carbonyl moiety of HIV-1 protease is involved in an interaction with an amide proton belonging to saquinavir backbone.
Figure 5. Top: Best-scoring docked conformations resulting from the docking of saquinavir into Sap2 binding site. Down: saquinavir in complex with HIV-1 protease (PDB code: 3OXC).
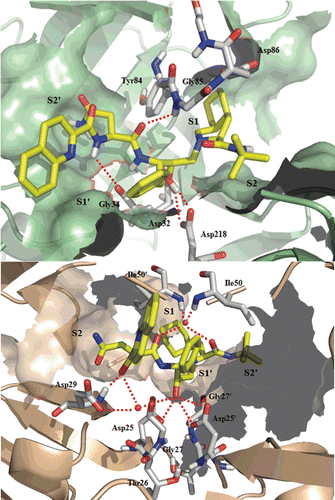
In HIV-1 protease, large hydrophobic groups at positions P3, P1 and P1′ of saquinavir occupy hydrophobic pockets S3, S1 and S1′, respectively, whereas the smaller t-butyl group and the polar asparagine side chain lie in S2′ and S2 subsites, respectively.
Docking results showed saquinavir to address S2/S2′ and S1/S1′ Sap2 binding pockets. In particular, the t-butyl function is located in S2 subsite, whereas the hydroisoquinoline in S1 subsite. The benzyl group adjacent to the transition state isostere is located in S1′ subsite, and the asparagine side chain in S2′. The isoquinoline residue, which in HIV-1 protease is located in S3 subsite, does not address a specific Sap2 subsite, and it is partially exposed to the solvent.
Ritonavir is the HIV-1 protease inhibitor which displays the strongest inhibitory effect towards Sap2 (). Docking studies revealed this drug to interact with Sap2 following the same structural arrangement, as above described for saquinavir. Unlike the first binding modes herein analyzed, ritonavir address the S3 subsite as a further binding pocket of Sap2 active site. In particular, the isopropylthiazol moiety is located in S3 subsite, and the other thiazol group in S2′. The two benzyl groups are found in S1 and S1′ subsites, whereas the isopropyl moiety occupies the S2 cavity ().
Figure 6. Top: Best-scoring docked conformations resulting from the docking of ritonavir into Sap2 binding site. Down: ritonavir in complex with HIV-1 protease (PDB code: 1HXW).
It was reported in the literature that exploiting the S3 subsite is critical for high inhibition potency, as showed for Sap2 inhibitor A-70450Citation35. Thus, optimal subsite filling can explain the higher inhibition potency of ritonavir as compared to that of saquinavir. This finding must be taken into account for the design of potential inhibitors with increasing binding strength and specificity. The main cluster of ritonavir conformations, as obtained from the docking study, is characterized by a hydrogen-bond established with Asp218 catalytic residue. Unlike the results obtained for saquinavir, ritonavir does not seem to interact with flap residues. Nevertheless, the polar contact established with the carbonyl moiety of Gly27 in the interaction with HIV-1 protease, is preserved also in the complex with Sap2, where it interacts with the corresponding carbonyl oxygen of Gly220.
Finally, our docking results are in agreement with the published crystal structure of C. albicans Sap2 in complex with ritonavir, which is a recent important entry to this topicCitation36. In fact, as showed in the crystal structure, the arrangement of ritonavir hydrophobic residues into Sap2 subsites appears similar to that obtained through molecular docking in the present study, thus these data corroborate our docking calculations.
Conclusions
In conclusion, we reported the analysis of similarity between Sap2 from C. albicans and HIV-1 aspartic protease, and we studied in detail, through molecular modeling, the binding requirements for inhibition of both targets, in order to address the clinical evidence of HIV protease inhibitors providing beneficial effects to mucosal candidiasis, as a consequence of a specific inhibition of Sap2. The tridimensional structure superimposition of Sap2 and HIV-1 protease confirmed the similarity between their active sites and flap regions. In silico experiments suggested that HIV-1 protease inhibitors herein investigated can fit the active site of Sap2, adopting very similar ligand-backbone conformations. The polar network as shown in the interaction between inhibitors and viral enzyme are also displayed in the interaction with Sap2. Nevertheless, the orientation of hydrophobic residues in Sap2 subsites does not perfectly match with the arrangements as for HIV-1 protease, in agreement with different Ki values of model inhibitors herein taken into account. The difference in the orientation of hydrophobic residues into binding pockets may be due to enlarged and more extended Sap2 subsites as compared to HIV-1 protease. Molecular modeling studies performed to investigate the molecular basis of interaction between Sap2 and two model HIV-1 protease inhibitors disclosed main common structural requirements for inhibition, which ultimately may address the generation of a “dual” inhibitor. In particular, designed molecules should display suitable groups to interact with key anchoring site consisting of Gly85 in Sap2 and Ile50 in HIV-1 protease, both belonging to the flap region, and another functional group on the opposite site, capable to form hydrogen-bonded interactions with carboxylic acid moieties of the two catalytic aspartic acid residues. Moreover, the establishment of hydrophobic contacts between potential inhibitors and S3 subsite of Sap2, which has been recognized as crucial for inhibition potency, should be considered for the design of new inhibitors. Specifically, the design of cyclic scaffolds possessing such polar functional groups, and bearing suitable hydrophobic residues in an optimal orientation to address both Sap2 and HIV-1 protease subsites, may lead to a single inhibitor acting on both targets.
Acknowledgements
Financial support received from the Fondazione Roma, MIUR, Università degli Studi di Firenze, and CINMPIS is gratefully acknowledged.
Declaration of interest
The authors have no declaration of interest
References
- Rothenberg R, Woelfel M, Stoneburner R, Milberg J, Parker R, Truman B. Survival with the acquired immunodeficiency syndrome. Experience with 5833 cases in New York City. N Engl J Med 1987;317:1297–1302.
- Schmidt-Westhausen A, Schiller RA, Pohle HD, Reichart PA. Oral Candida and Enterobacteriaceae in HIV-1 infection: correlation with clinical candidiasis and antimycotic therapy. J Oral Pathol Med 1991;20:467–472.
- Cataldo M, Tacconelli E, Sanguinetti M, Villa P, Fadda G, Cauda R. Highly active antiretroviral therapy (HAART) and probability of Candida vaginitis in HIV-infected patients. Trends Med Mycol 2003:165–169.
- Hoegl L, Thoma-Greber E, Röcken M, Korting HC. HIV protease inhibitors influence the prevalence of oral candidosis in HIV-infected patients: a 2-year study. Mycoses 1998;41:321–325.
- Korting HC, Schaller M, Eder G, Hamm G, Böhmer U, Hube B. Effects of the human immunodeficiency virus (HIV) proteinase inhibitors saquinavir and indinavir on in vitro activities of secreted aspartyl proteinases of Candida albicans isolates from HIV-infected patients. Antimicrob Agents Chemother 1999;43:2038–2042.
- Cassone A, De Bernardis F, Torosantucci A, Tacconelli E, Tumbarello M, Cauda R. In vitro and in vivo anticandidal activity of human immunodeficiency virus protease inhibitors. J Infect Dis 1999;180:448–453.
- Schaller M, Schackert C, Korting HC, Januschke E, Hube B. Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J Invest Dermatol 2000;114:712–717.
- Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 2003;67:400–28, table of contents.
- De Bernardis F, Cassone A, Sturtevant J, Calderone R. Expression of Candida albicans SAP1 and SAP2 in experimental vaginitis. Infect Immun 1995;63:1887–1892.
- Naglik JR, Newport G, White TC, Fernandes-Naglik LL, Greenspan JS, Greenspan D et al. In vivo analysis of secreted aspartyl proteinase expression in human oral candidiasis. Infect Immun 1999;67:2482–2490.
- Schaller M, Januschke E, Schackert C, Woerle B, Korting HC. Different isoforms of secreted aspartyl proteinases (Sap) are expressed by Candida albicans during oral and cutaneous candidosis in vivo. J Med Microbiol 2001;50:743–747.
- Albrecht A, Felk A, Pichova I, Naglik JR, Schaller M, de Groot P et al. Glycosylphosphatidylinositol-anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J Biol Chem 2006;281:688–694.
- Taylor BN, Hannemann H, Sehnal M, Biesemeier A, Schweizer A, Röllinghoff M et al. Induction of SAP7 correlates with virulence in an intravenous infection model of candidiasis but not in a vaginal infection model in mice. Infect Immun 2005;73:7061–7063.
- Cassone A, De Bernardis F, Mondello F, Ceddia T, Agatensi L. Evidence for a correlation between proteinase secretion and vulvovaginal candidosis. J Infect Dis 1987;156:777–783.
- Gauwerky K, Borelli C, Korting HC. Targeting virulence: a new paradigm for antifungals. Drug Discov Today 2009;14:214–222.
- Dostál J, Brynda J, Hrušková-Heidingsfeldová O, Pachl P, Pichová I, Rezácová P. The crystal structure of protease Sapp1p from Candida parapsilosis in complex with the HIV protease inhibitor ritonavir. J Enzyme Inhib Med Chem 2012;27:160–165.
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997;18:2714–2723.
- ExPAsy Proteomics Server. http://www.expasy.org
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–748.
- Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model 2009;49:84–96.
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC, San Carlos, CA, USA. http://www.pymol.org
- Cutfield SM, Dodson EJ, Anderson BF, Moody PC, Marshall CJ, Sullivan PA et al. The crystal structure of a major secreted aspartic proteinase from Candida albicans in complexes with two inhibitors. Structure 1995;3:1261–1271.
- Koelsch G, Tang J, Loy JA, Monod M, Jackson K, Foundling SI et al. Enzymic characteristics of secreted aspartic proteases of Candida albicans. Biochim Biophys Acta 2000;1480:117–131.
- Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990;248:358–361.
- Pichová I, Pavlícková L, Dostál J, Dolejsí E, Hrusková-Heidingsfeldová O, Weber J et al. Secreted aspartic proteases of Candida albicans, Candida tropicalis, Candida parapsilosis and Candida lusitaniae. Inhibition with peptidomimetic inhibitors. Eur J Biochem 2001;268:2669–2677.
- Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA 1995;92:2484–2488.
- Wlodawer A, Gustchina A. Structural and biochemical studies of retroviral proteases. Biochim Biophys Acta 2000;1477:16–34.
- Blundell TL, Lapatto R, Wilderspin AF, Hemmings AM, Hobart PM, Danley DE et al. The 3-D structure of HIV-1 proteinase and the design of antiviral agents for the treatment of AIDS. Trends Biochem Sci 1990;15:425–430.
- Gruber A, Speth C, Lukasser-Vogl E, Zangerle R, Borg-von Zepelin M, Dierich MP et al. Human immunodeficiency virus type 1 protease inhibitor attenuates Candida albicans virulence properties in vitro. Immunopharmacology 1999;41:227–234.
- Borg-von Zepelin M, Meyer I, Thomssen R, Würzner R, Sanglard D, Telenti A et al. HIV-Protease inhibitors reduce cell adherence of Candida albicans strains by inhibition of yeast secreted aspartic proteases. J Invest Dermatol 1999;113:747–751.
- Gruber A, Berlit J, Speth C, Lass-Flörl C, Kofler G, Nagl M et al. Dissimilar attenuation of Candida albicans virulence properties by human immunodeficiency virus type 1 protease inhibitors. Immunobiology 1999;201:133–144.
- EMBOSS Matcher - Pairwise Sequence Alignment from the EMBL-EBI tools website. http://www.ebi.ac.uk/Tools/psa/emboss_matcher/
- Abad-Zapatero C, Goldman R, Muchmore SW, Hutchins C, Stewart K, Navaza J et al. Structure of a secreted aspartic protease from C. albicans complexed with a potent inhibitor: implications for the design of antifungal agents. Protein Sci 1996;5:640–652.
- Tie Y, Kovalevsky AY, Boross P, Wang YF, Ghosh AK, Tozser J et al. Atomic resolution crystal structures of HIV-1 protease and mutants V82A and I84V with saquinavir. Proteins 2007;67:232–242.
- Pranav Kumar SK, Kulkarni VM. Insights into the selective inhibition of Candida albicans secreted aspartyl protease: a docking analysis study. Bioorg Med Chem 2002;10:1153–1170.
- Behnen J, Köster H, Neudert G, Craan T, Heine A, Klebe G. Experimental and computational active site mapping as a starting point to fragment-based lead discovery. ChemMedChem 2012;7:248–261.