Abstract
The aspartic protease inhibitory efficiency of rBm-33, an aspin from a filarial parasite Brugia malayi was investigated. rBm-33 was found to be thermostable up to 90°C and it forms a stable ‘enzyme-product’ complex with human pepsin. Aspartic protease inhibitory activity was investigated using UV spectroscopy and isothermal titration calorimetry. Our results suggest that rBm-33 inhibits the activity of important human aspartic proteases that were examined with binding constants (Kb) values between 10.23 × 103 and 6.52 × 103 M−1. The binding reactions were enthalpy driven with ΔHb values between −50.99 and −46.07 kJ mol−1. From kinetic studies, pepsin inhibition by rBm-33 was found to be linear competitive with an inhibition constant (Ki) of 2.5 (±0.8) nM. Because of the inhibitory efficacy of Bm-33 against important human aspartic proteases which play a vital role in immune-regulation along with other functions, Bm-33 can be projected as a drug target for the filariasis.
Introduction
Lymphatic filariasis, a serious debilitating disease, affects nearly 120 million people and, an estimated 2% of the world’s populationCitation1. Wuchereria bancrofti, Brugia malayi and Brugia timori are the lymph-dwelling parasitic nematodes which cause lymphatic filariasis. These parasites have a remarkable ability to modulate the host immune responses which makes them able to survive for a long time in the hostile immune niche of the host. The parasite protease inhibitors are believed to be the key players in this phenomenonCitation2. It is also believed that these protease inhibitors are crucial for parasite survival and are perhaps the logical sites to be attacked for the control of the parasite development and migrationCitation3.
Parasites produce three types of protease inhibitors namely; serine protease inhibitors (serpins), cysteine protease inhibitors (cystatins) and aspartic protease inhibitors (aspins), Aspartic proteases are believed to play a key role in the pathogenesis of human pathogens like HIVCitation4, Plasmodium falciparum (malaria)Citation5 and fungi such as CandidaCitation6. Hence the design of specific and potent inhibitors for these aspartic proteases may help in controlling many human diseases.
Different approaches have been employed in designing the new inhibitors for these aspartic proteases, and several HIV protease inhibitors have been now licensed by the Food and Drug Administration of the USACitation7–9; however, all these approaches had only limited usage.
In contrast, only a few naturally occurring protein inhibitors of this protease family like human renin-binding proteinCitation10, aspartic proteinase inhibitors from plants such as potatoCitation11, squashCitation12 and yeastCitation13 and pepsin inhibitors from Ascaris lumbricoidesCitation14 have been identified. Among these the best characterized one is the aspin (PI-3) from the nonfilarial nematode Ascaris suum that infects the pigsCitation15. Similarly, an aspin (Bm-33) from the filarial nematode Brugia malayi has also been identified and the immunological characterizations have been reportedCitation16–18. In addition to this, the earlier studies from this laboratory reveal rBm-33 to be a human pepsin inhibitorCitation19.
Four major groups of aspartic proteases have been identified in vertebrates namely pepsin, cathepsin-D, cathepsin-E and reninCitation20. Among these, cathepsin-D and cathespin-E are widely distributed in tissues and are thought to be involved in the regulation of physical activities like, lysosomal biogenesisCitation21, protein targetingCitation22, antigen processing and presentation by degradation of proteins and peptidesCitation23–25. In order to understand the role of aspin (Bm-33) in the parasite survival and its immune evasion strategies, the study of its inhibitory efficiency against the human aspartic proteases becomes imperative, which is attempted in the present study.
Materials and methods
rBm-33 was expressed in Escherichia coli, purified from the inclusion bodies and refolded as described earlierCitation19.
Thermal stability of rBm-33 and circular dichrosim
rBm-33 was incubated in 100 mM sodium acetate buffer (pH 5.6) at different temperatures (40–95°C) for 10 min. The samples were then centrifuged at 17,091 g for 45 min and the clear supernatants were assayed for their pepsin inhibition activityCitation19. Heat treated (95°C) rBm-33 was diluted with water at a final concentration of 0.1 mg/ml at pH 6.9. CD measurements were performed using a Jasco J-810 CD spectropolarimeter at 293 K using a quartz cell of 0.5 mm path-length in the wavelength range of 190–260 nm at 293 K at a resolution of 1 nm. K2D2 web serverCitation26 was used to analyze the secondary structure composition.
Role of N-terminal end in pepsin inhibition
Equimolar quantities of rBm-33 and human pepsin were incubated together for 10 min at 37°C. A final volume of 2.5% glutaraldehyde was added to this mixture for cross-linking rBm-33 and human pepsin if they are already complexed. The reaction was stopped by adding an equal volume of 20 mM Tris buffer (pH 8.0). This nonreactive glutaraldehyde was removed by ultrafiltration and the samples were loaded on an Immobilized metal affinity chromatography (IMAC) column (Ni2+-IDA) equilibrated with a buffer containing 20 mM NaH2PO4 (pH 8.0) and 100 mM NaCl (buffer A). After a brief wash with the equilibration buffer, elution was carried out with buffer A containing increasing concentrations of imidazole (25–200 mM), and the eluted flow-through fraction was analyzed on immunoblot using anti Bm-33 antisera (1:20,000) (in-house produced) and anti his-tag antibody (1:20,000) (Amersham Pharmacia, Piscataway, NJ, USA).
Human aspartic protease inhibition assay
rBm-33 (purified), pepsin (Himedia, Mumbai, India), cathepsin-D, cathepsin-E and renin (SIGMA, St. Louis, MO, USA) were used for the assays.
UV spectroscopy
Equimolar quantities of rBm-33 and human proteases (pepsin, cathepsin-D, cathepsin-E, and renin) were mixed and incubated in 100 mM sodium acetate buffer (pH 5.6) for 10 min at 37°C separately. To measure the residual protease activity, 10 µl (1 mg/ml) of casein were added and the reaction was stopped after 5 min by adding ice cold trichloroacetic acid (TCA) (5 %) solution. After incubation at 4°C for 30 min, the mixture was centrifuged for 40 min with 17,091g. When the whole proteins get precipitated by TCA, the peptide fragments in the supernatant show UV absorption at 280 nm, which is proportional to the digestion of casein by the proteases. The whole experiment was repeated six times and the mean values obtained are graphically representedCitation27.
Kinetics of pepsin inhibition by rBm-33
Kinetics of pepsin inhibition by rBm-33 was determined by the spectroscopic methodCitation27. With a fixed quantity of pepsin (5 mM) and fixed reaction time, the rate of proteolysis in the presence of inhibitor was measured. Human pepsin (5 mM) alone and human pepsin preincubated with increasing concentration of rBm-33 (1 mM, 2.5 mM and 5 mM, respectively) for 10 min at 37°C in 100 mM sodium acetate buffer (pH 5.6) were taken. To measure the residual protease activity, 10 µl of casein (7–80 µM) was added and the reaction was stopped after 5 min by adding ice cold TCA (5%) solution. After incubation at 4°C for 30 min, the mixture was centrifuged for 40 min with 17,091g. The reaction was monitored by measuring the UV absorbance at 280 nm. The Km value for rBm-33 was determined by linear regression from plots of 1/V vs. 1/S, using substrate concentrations of 7–80 µM. Three fixed concentrations, 1 mM, 2.5 mM and 5 mM, respectively of rBm-33 were used to determine the inhibition constant (Ki) against six concentrations of casein. Assays were carried out in triplicates and the kinetic constants were determined using Graphpad Prism 2.0 (San Diego, CA, USA).
Isothermal titration calorimetry
The calorimetric titrations were performed at 298 K with a VP-ITC isothermal titration calorimeter from Microcal (Northampton, MA, USA). Small aliquot (typically 5 µl) of the protease solution (0.036 mM) was added from a rotating stirrer-syringe to a solution of r-Bm-33 (1.8 ml, 0.036 mM) in the calorimeter cell. Successive additions were separated by a 120-s interval to allow the exothermic peak resulting from the reaction to return to the baseline. Usually the first injection was found to be inaccurate; hence, a 2 µl injection was added first and the resultant point was deleted before the remaining data were analyzed. No correction was made as the heat of dilution for all the protease buffer (protein buffer) was found to be very small and negligible when compared to the heat changes corresponding to the protease–rBm-33 binding. The total heat released or absorbed at the end of the ith injection, Q (i), was then fitted by a non-linear least squares method to the total protease concentration, Xi, according to the equationCitation28:
where, n refers to the number of sites, Pt to the protein concentration, V to the volume of the cell, Kb to the binding constant and ΔHb to the binding enthalpy.
The heat released in ith injection is given by:
where, dVi to the volume of the protease added to the solutionCitation29. The thermodynamic parameters like ΔGb° and ΔSb° are calculated according to equations 3 and 4:
Results
Thermal stability of rBm-33 and circular dichrosim
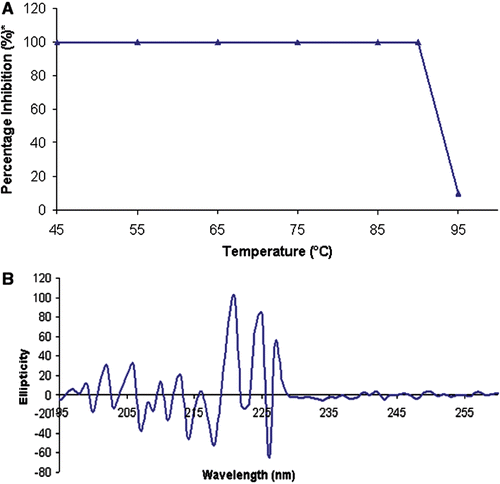
Thermal stability of rBm-33 was studied by incubating at different temperatures (40°C–95°C) for 10 min, followed by pepsin inhibition activity of the heat treated protein. The results obtained are shown in (). The CD spectrum (at 190–260 nm) of heat treated (95°C) rBm-33 showed a complete collapse of the secondary structure pattern ().
Figure 1. Thermal stability expressed through the CD spectrum of rBm-33 heat treated to (95°C). Thermal stability of rBm-33 on pepsin inhibition activity.*The percentage activity was calculated assuming the maximum activity at 37°C is 100% (A). CD spectrum of heat treated (95°C) rBm-33 diluted with water at final concentration of 0.1 mg/ml at pH 6.9 (B).
Role of N-terminal end in pepsin inhibition
When the flow-through fraction from the IMAC experiment was analyzed, a 66 kDa (corresponding to rBm-33-pepsin complex) was found to be probed with anti-Bm-33 antisera and no other fractions showed the reactivity. Probing with anti his-tag antibody failed to show any immunoreactivity ().
Figure 2. Immunoblot to show the removal of N-terminal his-tag from rBm-33 after the incubation with pepsin. M: protein molecular-weight marker; Lane 1: flow-through of IMAC (Ni2+-IDA) experiment containing glutaraldehyde cross linked rBm-33-pepsin complex probed with anti Bm-33 antisera; Lane 2: same flow-through probed with anti his-tag antibody.

Human aspartic protease inhibition assay
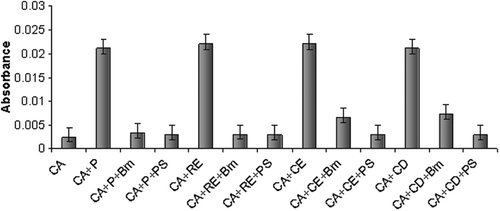
The action of rBm-33 on the human aspartic proteases was examined (). When assayed for the inhibition of casein digestion, rBm-33 showed inhibition against the four important human aspartic proteases that were examined.
Figure 3. Inhibition activity of rBm-33 against human aspartic proteases using UV spectroscopy. CA: only casein, P: pepsin, RE: renin, CE: cathepsin-E, CD: cathepsin-D, PS: pepstatin Bm: rBm-33. All the values represent the mean of six different experiments ± SD
Kinetics of pepsin inhibition by rBm-33
The substrate concentration vs. velocity curve () and the corresponding double inverse Lineweaver-Burk plot are depicted (). While the saturating concentration of substrate (Vmax) seems to be the same, the rate of the proteolysis decreased with the increasing rBm-33 concentrations. Inhibition constant (Ki) for pepsin inhibition of rBm-33 was found to be 2.5 (± 0.8) nM.
Figure 4. Kinetics of pepsin inhibition by rBm-33. The assay was performed with the fixed quantity of pepsin (5 mm) and varying concentrations of rBm-33 in absence and in the presence of (1 mM, 2.5 mM and 5 mM, respectively). Substrate-velocity curve (A) and Lineweaver-Burk plot (1/V vs. 1/S) (B) are shown indicating competitive inhibition of pepsin by rBm-33 (Ki = 2.5 (± 0.8) nM). P: pepsin (5 mM), P+ [B 1 mM]: pepsin+rBm-33 (1 mM), P+ [B 2.5 mM]: pepsin+rBm-33 (2.5 mM), P+ [B 5 mM]: pepsin+rBm-33 (5 mM). Assay was carried out in triplicates and the kinetic constants were determined using Graphpad Prism 2.0 (San Diego, CA, USA).
![Figure 4. Kinetics of pepsin inhibition by rBm-33. The assay was performed with the fixed quantity of pepsin (5 mm) and varying concentrations of rBm-33 in absence and in the presence of (1 mM, 2.5 mM and 5 mM, respectively). Substrate-velocity curve (A) and Lineweaver-Burk plot (1/V vs. 1/S) (B) are shown indicating competitive inhibition of pepsin by rBm-33 (Ki = 2.5 (± 0.8) nM). P: pepsin (5 mM), P+ [B 1 mM]: pepsin+rBm-33 (1 mM), P+ [B 2.5 mM]: pepsin+rBm-33 (2.5 mM), P+ [B 5 mM]: pepsin+rBm-33 (5 mM). Assay was carried out in triplicates and the kinetic constants were determined using Graphpad Prism 2.0 (San Diego, CA, USA).](/cms/asset/33d26180-ae45-4d04-9cdf-15c0a90d0f38/ienz_a_710849_f0004_b.gif)
Isothermal titration calorimetry
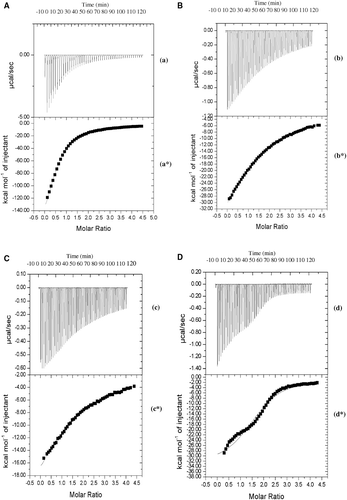
The experimental values of the calorimetric titration experiment for the binding of human aspartic proteases to rBm-33 at 298 K are shown in , , and 5D. From these figures, it is evident that “exothermic heat of binding” decreases monotonically with successive injections until saturation is achieved. This behaviour is found to be consistent with all the proteases (sub-legends a, b, c and d). A plot of incremental heat as a function of all the four proteases/rBm-33 ratio was obtained with non-linear least squares fit of the data to equation 1. The titration data could be fitted satisfactorily for a model with ‘single type of binding site’ and the fit is shown as a solid line in (*, 5b*, 5c* and 5d*). The values of various parameters for binding of rBm-33 with the important human aspartic proteases that were examined are presented in ().
Table 1. Association constants (Kb) and thermodynamic parameters for the binding of important human aspartic proteases to rBm-33.
Figure 5. Iso-thermal calorimetric titration of rBm-33 (refolded) with cathepsin-E (C). Raw data obtained from 50 automatic injections of 5 µl aliquots of 0.036 mM cathepsin-E into 0.036 mM of rBm-33 (refolded) at 298 K (c). Non-linear least squares fit for the cathepsin-E and rBm-33 (refolded) (c*). Iso-thermal calorimetric titration of rBm-33 (refolded) with cathepsin-D (D). Raw data obtained from 50 automatic injections of 5 µl aliquots of 0.036 mM cathepsin-D into 0.036 mM of rBm-33 (refolded) at 298 K (d). Non-linear least squares fit for the cathepsin-D and rBm-33 (refolded) (d*).
Discussion
rBm-33 is an aspartic protease inhibitor secreted by the human filarial parasite Brugia malayi and is believed to play a key role in host pathogenesis and immune modulationCitation18. It was purified under denaturing conditions and refolded by step wise dialysis using buffers of pH ranging from 11 to 7. It was characterized with respect to its human pepsin inhibition activityCitation19.
In continuation of our earlier workCitation19, we have characterized the thermal stability of rBm-33 and investigated the role of N-terminal end in the pepsin inhibition. Further, we have also studied the kinetics of pepsin inhibition, inhibitory effect and binding characteristics of rBm-33 with the other important human aspartic proteases using UV absorption and iso thermal titration calorimetry.
The thermal stability experiment clearly shows that the pepsin inhibition activity of rBm-33 is unaffected up to 90°C. The presence of two disulfide bonds in rBm-33 could have given this high-thermal stabilityCitation30 similar to PI-3 (homologous aspin from Ascaris suum) which had three disulfide bondsCitation31. However, the activity decreases rather steeply at 95°C. This loss of protease inhibition activity can be correlated with CD spectroscopy results, which indicate distorted secondary structural pattern of the protein when incubated at 95°C for 10 min.
As the N-terminal β strand insertion of PI-3 (from Ascaris suum) into the pepsin was demonstrated in the case of PI-3 – pepsin crystal structureCitation15, N-terminal his-tag removal from rBm-33 for pepsin inhibition was hypothesized. The presence of N-terminal his-tag in rBm-33 prior to the incubation with pepsin and its removal after incubation was demonstrated by immunoblotting. This is further supported by the IMAC experiment. When the glutraldehyde cross linked rBm-33-pepsin complex was added to IMAC resin, the complex was not retained on the column. The presence of a 66 kDa band corresponding to rBm-33-pepsin complex, only in the flow-through which could be probed with anti-Bm-33 antisera but not with anti-his-tag monoclonal antibody reveals the removal of his-tag from rBm-33 by pepsin. The presence of a pepsin cleavage site at the 11th amino acid position (N-terminus) in rBm-33 can be hypothesized as a contributing factor for the N-terminal his-tag removal by pepsin. Hence when the rBm-33 is incubated with pepsin for 10 min at 37°C it forms a stable ‘enzyme-product’ complex suggesting the insertion of N-ter strand after altering the rBm-33 N-terminus.
The effect of rBm-33 on the various human aspartic proteases was examined. Unlike PI-3, where the inhibitor was found to be ineffective against renin and cathepsin-DCitation31, an appreciable influence on all the four aspartic proteases was evident with rBm-33. In all cases, addition of equimolar amounts of the inhibitor (rBm-33) resulted in almost complete inhibition of the proteolytic activity of all the proteases that were examined. The difference in the nature of N-terminal composition from that of PI-3 (data not shown) or the excess of about 66 amino acid residues (not as a single stretch) present in rBm-33 compared to PI-3 may be attributed to its universal inhibition. Among all the proteases that were examined, rBm-33 was found to be equally effective against pepsin and renin whereas few folds less are effective towards cathepsin-E and D. UV based kinetic experiments were performed with rBm-33 – pepsin to identify its mode of inhibition. These experiments suggest almost a linear competitive inhibition towards pepsin. Such mode of inhibition was comparable to that of pepsin inhibition by pepstatin suggesting that mode of inhibition of rBm-33 is similar to that of pepstatin inhibitionCitation32.
Isothermal titration calorimetric experiment values in indicate that the rBm-33 has one binding site (1:1 stoichiometry) for pepsin, cathepsin-E and renin whereas more than one site (1:1.7 stoichiometry) with cathepsin D. It is further seen that binding constant (Kb) for human cathepsin-E and D are 1.8 and 3.6 times less specific than that of pepsin and renin. A slight preference towards pepsin over renin is also observed. The thermodynamic data indicate that aspartic protease binding to rBm-33 is governed primarily by enthalpic forces, with negative contribution from the enthalpy of binding for the aspartic proteases.
Biological implications in aspartic protease inhibition
Considering the findings of pronounced inhibitory activity of rBm-33 against the important human aspartic proteases, it is interesting to speculate Bm-33 as an important drug target for future drug designing attempts. Cathepsin E and D have shown to be involved in the processing of antigens and thereby construction of Complementarity determining regions (CDRs.)Citation33,Citation34. Renin is believed to be an important component of blood plasma, lymph and intestinal fluid to maintain arterial blood pressure of the human bodyCitation35. Though the filarial worm does not encounter the host digestive system, pepsin is believed to be one of the principal enzymes that play a key role in immunoregulationCitation36,Citation37. Hence the inhibition of these aspartic proteases by Bm-33 might assist the parasite by inhibiting important proteases of the host resulting in the invasion and survival of the filarial parasite.
Conclusion
The physicochemical properties and aspartic protease inhibition efficiency of rBm-33 have been investigated. These studies reveal that rBm-33 is active up to 90°C. The formation of a stable ‘enzyme-product’ complex is evident by N-terminal his-tag removal from rBm-33 when incubated with pepsin. These findings are similar to those of PI-3-pepsin complex suggesting that the binding mode of Bm-33 with aspartic protease may be a β strand insertionCitation15. Unlike in PI-3, UV absorption and isothermal titration calorimetry results clearly suggest that rBm-33 inhibits cathepsin-D and renin. The reason for rBm-33 inhibition of all the important aspartic proteases can be attributed to its different N-terminal composition to that of PI-3 which may allow interactions with primary/secondary interaction sites of all the human aspartic proteases that were examined. In addition to this, the presence of excess 66 amino acid residues in Bm-33 was compared to PI-3 which may induce a conformational change on the proteases. This study sheds light on the inhibition of important human aspartic proteases by rBm-33, which opens up the possibility of exploring rBm-33 as a potential drug target for human lymphatic filariasis.
Acknowledgements
The authors wish to thank Dr. Sadasivan at Kannur University, Kerala, India for allowing us to carry out the isothermal titration calorimetric experiments.
Declaration of interest
NRSK and SSR thank University Grants Commission, India. RBN thanks CSIR, India, for financial assistance. The authors report no conflict of interest.
References
- Molyneux DH, Bradley M, Hoerauf A, Kyelem D, Taylor MJ. Mass drug treatment for lymphatic filariasis and onchocerciasis. Trends Parasitol 2003;19:516–522.
- Martzen MR, Geise GL, Hogan BJ, Peanasky RJ. Ascaris suum: localization by immunochemical and fluorescent probes of host proteases and parasite proteinase inhibitors in cross-sections. Exp Parasitol 1985;60:139–149.
- Peanasky RJ, Martzen MR, Homandberg GA, Cash JM, Babin, DR, Litweiler B. (1987). Molecular paradigms for eradicating helminthic parasites. New York: Alan R. Liss, 349–366.
- Craig C, Race E, Sheldon J, Whittaker L, Gilbert S, Moffatt A et al. HIV protease genotype and viral sensitivity to HIV protease inhibitors following saquinavir therapy. AIDS 1998;12:1611–1618.
- Dame JB, Reddy GR, Yowell CA, Dunn BM, Kay J, Berry C. Sequence, expression and modeled structure of an aspartic proteinase from the human malaria parasite Plasmodium falciparum. Mol Biochem Parasitol 1994;64:177–190.
- Monod M, Togni G, Hube B, Sanglard D. Multiplicity of genes encoding secreted aspartic proteinases in Candida species. Mol Microbiol 1994;13:357–368.
- Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct 1998;27:249–284.
- Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C et al. Expression and characterisation of plasmepsin I from Plasmodium falciparum. Eur J Biochem 1997;244:552–560.
- Roberts NA, Craig JC, Sheldon J. Resistance and cross-resistance with saquinavir and other HIV protease inhibitors: theory and practice. AIDS 1998;12:453–460.
- Takahashi S, Takahashi K, Kaneko T, Ogasawara H, Shindo S, Kobayashi M. Human renin-binding protein is the enzyme N-acetyl-D-glucosamine 2-epimerase. J Biochem 1999;125:348–353.
- Kreft S, Ravnikar M, Mesko P, Pungercar J, Umek A, Kregar I et al. Jasmonic acid inducible aspartic proteinase inhibitors from potato. Phytochemistry 1997;44:1001–1006.
- Christeller JT, Farley PC, Ramsay RJ, Sullivan PA, Laing WA. Purification, characterization and cloning of an aspartic proteinase inhibitor from squash phloem exudate. Eur J Biochem 1998;254:160–167.
- Saheki T, Matsuda Y, Holzer H. Urification and characterization of macromolecular inhibitors of proteinase A from yeast. Eur J Biochem 1974;47:325–332.
- Abu-Erreish GM, Peanasky RJ. Pepsin inhibitors from Ascaris lumbricoides. Isolation, purification, and some properties. J Biol Chem 1974;249:1558–1565.
- Petersen W, Cherney MM, Garen C, Zalatoris JJ, Rao-Naik C, Dunn BM et al. Structural basis for the inhibition of porcine pepsin by Ascaris pepsin inhibitor-3. Nat Struct Biol 2000;7:653–657.
- Dissanayake S, Xu M, Nkenfou C, Piessens WF. Molecular cloning and serological characterization of a Brugia malayi pepsin inhibitor homolog. Mol Biochem Parasitol 1993;62:143–146.
- Krushna NS, Shiny C, Dharanya S, Sindhu A, Aishwarya S, Narayanan RB. Immunolocalization and serum antibody responses to Brugia malayi pepsin inhibitor homolog (Bm-33). Microbiol Immunol 2009;53:173–183.
- Krushna NS, Shiny C, Manokaran G, Elango S, Babu S, Narayanan RB. Immune responses to recombinant Brugia malayi pepsin inhibitor homolog (Bm-33) in patients with human lymphatic filariaisis. Parasitol Res 2011;108:407–415.
- Krishna NR, Krushna NS, Narayanan RB, Rajan SS, Gunasekaran K. Expression, purification and characterization of refolded rBm-33 (pepsin inhibitor homolog) from Brugia malayi: a human Lymphatic Filarial parasite. Protein Expr Purif 2011;79:245–250.
- Takahashi K. (1995). Aspartic proteinases: structure, function, biology and biomedical implications. New York: Plenum Press, 581–587.
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol 1989;5:483–525.
- von Figura K, Hasilik A. Lysosomal enzymes and their receptors. Annu Rev Biochem 1986;55:167–193.
- Lapresle C, Barrett AJ, Dingle J. (1971). Tissue proteinases. Amsterdam: North-Holland Publishing Co, 135–155.
- Barrett AJ. (1977). Proteinases in mammalian cells and tissues. Amsterdam: Elsevier/North-Holland Bio-medical Press, 209–248.
- Kageyama T, Ichinose M, Tsukada S, Miki K, Kurokawa K, Koiwai O et al. Gastric procathepsin E and progastricsin from guinea pig. Purification, molecular cloning of cDNAs, and characterization of enzymatic properties, with special reference to procathepsin E. J Biol Chem 1992;267:16450–16459.
- Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol 2008;8:25.
- Anson ML. The estimation of pepsin, trypsin, papain, and cathepsin with hemoglobin. J Gen Physiol 1938;22:79–89.
- Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem 1989;179:131–137.
- ITC data analysis in Origin”, Tutorial guide 7.0, Microcal#. Northampton, MA, 2002, 99.
- Wetzel R, Perry LJ, Baase WA, Becktel WJ. Disulfide bonds and thermal stability in T4 lysozyme. Proc Natl Acad Sci USA 1988;85:401–405.
- Kageyama T. Molecular cloning, expression and characterization of an Ascaris inhibitor for pepsin and cathepsin E. Eur J Biochem 1998;253:804–809.
- Rich DH, Sun ET. Mechanism of inhibition of pepsin by pepstatin. Effect of inhibitor structure on dissociation constant and time-dependent inhibition. Biochem Pharmacol 1980;29:2205–2212.
- Bennett K, Levine T, Ellis JS, Peanasky RJ, Samloff IM, Kay J et al. Antigen processing for presentation by class II major histocompatibility complex requires cleavage by cathepsin E. Eur J Immunol 1992;22:1519–1524.
- Kageyama T, Yonezawa S, Ichinose M, Miki K, Moriyama A. Potential sites for processing of the human invariant chain by cathepsins D and E. Biochem Biophys Res Commun 1996;223:549–553.
- Imai T, Miyazaki H, Hirose S, Hori H, Hayashi T, Kageyama R et al. Cloning and sequence analysis of cDNA for human renin precursor. Proc Natl Acad Sci USA 1983;80:7405–7409.
- Ohnshi H. Pepsin-immunoregulation hypothesis. Med Hypotheses 1984;13:189-198.
- Ohnshi H. Pepsin like enzyme in macrophages and its role in the immune system. Clin Exp Immunol 1984;58:206–212.