
Abstract
A practical protocol has been used for the synthesis of benzimidazoles. The reaction of iminoester hydrochlorides of phenylacetic with 4,5-dichloro-1,2-phenylenediamine under microwave irradiation leads to the benzimidazole derivatives with good yields and in short reaction times. After the synthesis of benzimidazoles, we synthesized ester and hydrazide derivatives under microwave irradiation with good yields. All compounds were evaluated with regard to pancreatic lipase activity and 3b, 3c, 5a and 6a showed lipase inhibition at various concentrations.
Introduction
Benzimidazoles are an important group of heterocyclic compounds in the field of medicinal chemistryCitation1. Some benzimidazole derivatives with different biological activity such as anticancerCitation2, antihelminticCitation3, antimicrobialCitation4, antifungalCitation5, antitubercularCitation6, antiallergicCitation7, antioxidantCitation8, antihistaminicCitation9, antitumorCitation10, anti-inflammatory and analgesic activitiesCitation11 have been revealed in the literature. Benzimidazole derivatives exhibit significant activity against several viruses such as HIVCitation12, herpes (HSV-1)Citation13 and human cytomegalovirusCitation12. In addition, the treatment potency of benzimidazoles in diseases such as ischemia–reperfusion injuryCitation14, hypertensionCitation15, obesityCitation16 has been recently reported. Because of these reasons, benzimidazole derivatives have been studied by numerous scientists. Obesity is widely recognized as a major public health problem which is caused by an imbalance between energy intake and expenditure. Obesity can be a cause to different serious diseases, including hypertension, hyperlipidemia, arteriosclerosis and type II diabetesCitation17. Pancreatic lipase plays a key role for fat digestion. Moreover, pancreatic lipase inhibitors, such as Orlistat, are used as therapeutic agent for curing obesityCitation18.
In the literature, there are many synthetic routes that are common to the preparation of benzimidazoles. Generally, the condensation of ortho-phenylenediamines and carboxylic acids (or their derivatives such as nitriles, imidates and orthoesters) had been widely used for the synthesis of benzimidazoleCitation19,Citation20. However, many of these procedures are associated with several drawbacks such as expensive reagents, harsh reaction conditions, extended reaction times, the occurrence of side products, unsatisfactory yields and complicated experimental procedures. Also, microwave technology has been used to synthesize benzimidazole derivatives, and important changes have been seen on yield and reaction timeCitation1,Citation21,Citation22. In this study, synthesis of benzimidazoles has shown that iminoester hydrochlorides could be a useful intermediate in the reaction with 4,5-dichloro-1,2-phenylenediamine under microwave irradiation in methanol and solvent-free conditions. This method can provide a convenient way of synthesizing potential bioactive benzimidazoles. Furthermore, a practical method has been developed for the synthesis of bis-benzimidazoles. Iminoester hydrochlorides of phenylacetic acids were used as intermediates in the reaction with 3,3′-diaminobenzidine under microwave irradiation, leading to the products with good yields and short reaction times. This method can be a general technique for the synthesis of bis-benzimidazoles. The synthetic path of the target compounds is shown in .
Scheme 1. Synthetic path of the target compounds.
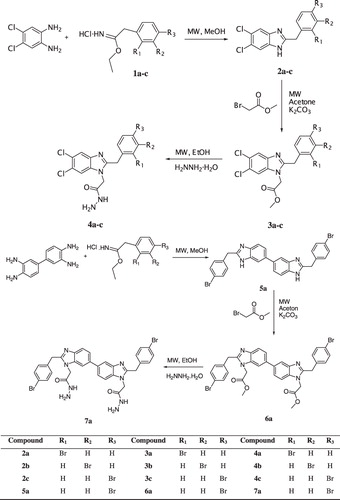
Figure 1. Dose-dependent inhibitory effect of the compounds 3b, 3c, 5a and 6a. Orlistat was used positive control. All compounds and Orlistat were measured at final concentrations of 0.625, 1.25 and 6.25 µg/mL. Residual activities of compounds are expressed as the mean ± SD in triplicate.
Results and discussion
Iminoester hydrochloride could be a useful intermediate in the reaction with o-phenylenediamine under microwave irradiation in methanol for the synthesis of benzimidazole derivatives. In this method, we obtained products within short reaction times and with high yields. In addition, the reaction was carried out catalyst-free under mild conditions. Compounds (2a–c) were also obtained using conventional heating in methanolCitation23. We also synthesized bis-benzimidazole from iminoester hydrochloride reaction of the 3,3′-diaminobenzidine under microwave irradiation, leading to the product with good yield. After the synthesis of benzimidazoles, we synthesized esters (3a–c, 6a) and hydrazides (4a–c, 7a) under microwave irradiation with good yields. Simple alkylation reaction was applied to 2a–c, 6a with methyl α-bromoacetate in acetone and then, the MeO group was displayed with NH2NH2·H2O in EtOH. The structures of new compounds were confirmed by FT-IR (infrared), 1H-NMR, Citation13C-NMR spectroscopy and mass spectrometry. All the synthesized compounds were screened for their anti-lipase activities.
Anti-lipase activity results
All the compounds were evaluated with regard for pancreatic lipase activity and different concentrations of 3b, 3c, 5a and 6a showed anti-lipase activities at various concentrations (). No inhibitory effect was observed in other compounds. Dose-dependent pancreatic lipase activity is shown in . Among the compounds tested, 3b showed the best anti-lipase activity. The compound inhibited pancreatic lipase activity by 84%, 97% and 98% at concentrations of 0.625, 1.25 and 6.25 µg/mL, respectively. Orlistat (Xenical; Hoffmann-La Roche, Segrate, Italy), known pancreatic lipase inhibitor used as anti-obesity drug, showed inhibitory effect by 98%, 99% and 100% at the same concentrations. IC50 values of Orlistat and compound 3b were calculated as 0.04 and 0.17 µg/mL, respectively. Orlistat is the only approved anti-obesity medicationCitation18 but it has some side effects, such as fecal incontinence, flatulence and steatorrheaCitation24,Citation25. The synthesized compounds such as 3b and 6a can be a good alternative to Orlistat.
Table 1. Residual lipase activity of chemical compounds.
Experimental
Melting points were determined in open capillaries on Büchi (Essen, Germany) digital melting point apparatus and were uncorrected. IR spectra were recorded in KBr pellets on a Perkin-Elmer 100 FT-IR spectrophotometer (California). 1H- and Citation13C-NMR spectra were measured on a Varian 400 spectrometer (Varian, Darmstadt, Germany) using DMSO-d6 as solvent and TMS as internal standard. Chemical shifts are given in parts per million (ppm), coupling constants J in hertz (Hz). Mass spectra (MS) were taken in H-ESI mode on Thermo Quantum Mars (Thermo-scientific, Florida). A monomode CEM-Discover microwave (Linfort, Germany) was used in the standard configuration as delivered, including proprietary software. All experiments were carried out in microwave process vials (30 mL) with control of the temperature by IR detection temperature sensor. The temperature was computer monitored and maintained constant by a discrete modulation of the delivered microwave power. After completion of the reaction, the vial was cooled to 60 °C by air jet cooling.
Synthesis of hydrochloride of substitute benzeneimidic acid ethyl ester (1a–c)
To an ice-cooled solution of the substitute benzonitrile (1 mol) in dry EtOH (1.1 mol), dry hydrogen chloride was added until 1.1 mol had been absorbed. The resulting solution was then allowed to stand at 0 °C in the refrigerator for 12 h, after which cold Et2O was added. The precipitated crystals were filtered off immediately, washed with cold Et2O and dried in a dessicator.
General procedure for the synthesis of 5,6-dichloro-2-(substitutedbenzyl)-1H-benzimidazole (2a–c) under microwave irradiation
A mixture of 4,5-dichloro-1,2-phenylenediamine (0.010 mol) and iminoester hydrochlorides (0.013 mol; 1a–c) in dry methanol (15 mL) was irradiated in closed vessels with the pressure control at 65 °C for 10 min (hold time) at 300 W maximum power. After the completion of the reaction, (monitored by TLC, ethylacetate:hexane, 3:1), the mixture was poured onto water. The precipitate formed was filtered and recrystallized from ethanol–water (1:3) to give pure 2a–c.
5,6-Dichloro-2-(2-bromobenzyl)-1H-benzimidazole (2a). Yield (90%); m.p. 228–229 °C; IR (νmax, cm−1): 3422 (NH), 1629 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.31 (s, 2H, CH2), 7.28–7.77 (m, 6H, Ar–H) and 12.63 (s, 1H, NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 32.65 (CH2), 119.62, 123.79, 124.26, 127.21, 128.77, 129.10, 131.50, 133.23, 134.58 (Ar–C) and 155.24 (C=N). Anal. Calcd for C14H9Cl2BrN2: C, 47.23; H, 2.55 and N, 7.87; found: C, 47.20; H, 2.56 and N, 7.89. ESI-MS m/z calculated for C14H9Cl2BrN2 [M]+ 356.10 and 358.10; found: 356.70 and 358.65.
5,6-Dichloro-2-(3-bromobenzyl)-1H-benzimidazole (2b). Yield (91%); m.p. 208–209 °C; IR (νmax, cm−1): 3200 (NH), 1627 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.21 (s, 2H, CH2), 7.10–7.95 (m, 6H, Ar–H) and 12.64 (s, 1H, NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 34.89 (CH2), 113.47, 120.32, 122.39 (2C), 124.48 (2C), 128.76, 130.30, 131.37, 132.32, 132.83, 140.39 (Ar–C) and 156.61 (C=N). Anal. Calcd for C14H9Cl2BrN2: C, 47.23; H, 2.55 and N, 7.87; found: C, 47.21; H, 2.59 and N, 7.85. ESI-MS m/z calculated for C14H9Cl2BrN2 [M+H]+ 356.10 and 358.10; found: 356.78 and 358.67.
5,6-Dichloro-2-(4-bromobenzyl)-1H-benzimidazole (2c). Yield (95%); m.p. 234–235 °C; IR (νmax, cm−1): 3208 (NH), 1579 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.10 (s, 2H, CH2), 7.10–7.71 (m, 6H, Ar–H) and 12.64 (s, 1H, NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 34.77 (CH2), 113.25, 120.20, 120.61, 124.58, 131.84 (2C), 132.07 (2C), 134.69, 137.09 (2C), 143.84 (Ar–C) and 156.74 (C = N). Anal. Calcd for C14H9Cl2BrN2: C, 47.23; H, 2.55 and N, 7.87; found: C, 47.22; H, 2.58 and N, 7.87. ESI-MS m/z calculated for C14H9Cl2BrN2 [M+H]+ 357.10 and 359.10; found: 357.78 and 359.10.
General procedure for the synthesis of methyl [5,6-dichloro-2-(substitutedbenzyl)-1H-benzimidazole-1-yl] acetate (3a–c)
A mixture of compounds 2a–c (0.01 mol), methyl-α-bromoacetate (0.01 mol) and K2CO3 (0.025 mol) in acetone (10 mL) was irradiated in closed vessels with the pressure control at 85 °C for 7 min (hold time) at 300 W maximum power. After the completion of the reaction, (monitored by TLC, ethylacetate:hexane, 3:1), the mixture was poured onto water. The precipitate formed was filtered and recrystallized from acetone:water (1:3) to give pure 3a–c.
Methyl [5,6-dichloro-2-(2-bromobenzyl)-1H-benzimidazole-1-yl] acetate (3a). Yield (90%); m.p. 162–163 °C; IR (νmax, cm−1): 1738 (C=O), 1618 (C=N) and 1250 (C–O); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 3.48 (s, 3H, OCH3), 4.31 (s, 2H, CH2), 5.30 (s, 2H, N–CH2) and 7.19–7.89 (m, 6H, Ar-H); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 30.75 (CH2), 44.48 (N–CH2), 52.27 (O–CH3), 112.84, 119.68, 124.10, 124.41, 127.11, 128.56, 129.03, 131.39, 133.19, 133.25, 135.02, 141.42 (Ar–C), 155.44 (C=N) and 167.67 (C=O). Anal. Calcd for C17H13Cl2BrN2O2: C, 47.69; H, 3.06 and N, 6.54; found: C, 47.65; H, 3.11 and N, 6.53. ESI-MS m/z calculated for C17H13Cl2BrN2O2 [M]+ 428.10 and 430.10; found: 428.80 and 430.88.
Methyl [5,6-dichloro-2-(3-bromobenzyl)-1H-benzimidazole-1-yl] acetate (3b). Yield (93%); m.p. 193–194 °C; IR (νmax, cm−1): 1731 (C=O), 1594 (C=N) and 1227 (C–O); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 3.55 (s, 3H, OCH3), 4.28 (s, 2H, CH2), 5.29 (s, 2H, N–CH2) and 7.24–7.95 (m, 6H, Ar–H); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 32.57 (CH2), 45.12 (N–CH2), 52.79 (O–CH3), 112.75, 120.37, 122.06, 124.83, 128.67, 130.06, 131.03, 132.17, 135.24, 135.75, 139.06, 142.06 (Ar–C), 156.56 (C=N) and 168.39 (C=O). Anal. Calcd for C17H13Cl2BrN2O2: C, 47.69; H, 3.06 and N, 6.54; found: C, 47.65; H, 3.11 and N, 6.55. ESI-MS m/z calculated for C17H13Cl2BrN2O2 [M]+ 428.10 and 430.10; found: 428.80 and 430.91.
Methyl [5,6-dichloro-2-(4-bromobenzyl)-1H-benzimidazole-1-yl] acetate (3c). Yield (97%); m.p. 152–153 °C; IR (υmax, cm−1): 1738 (C=O), 1591 (C=N) and 1226 (C–O); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 3.64 (s, 3H, OCH3), 4.23 (s, 2H, CH2), 4.66 (s, 2H, N–CH2) and 7.09--7.85 (m, 6H, Ar–H); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 29.11 (CH2), 40.28 (N–CH2), 48.25 (O–CH3), 105.73, 116.31, 116.65, 121.93, 122.41, 125.57 (2C), 127.34 (2C), 129.10, 130.09, 137.02 (Ar–C), 149.80 (C=N) and 162.07 (C=O). Anal. Calcd for C17H13Cl2BrN2O2: C, 47.69; H, 3.06 and N, 6.54; found: C, 47.64; H, 3.10 and N, 6.57. ESI-MS m/z calculated for C17H13Cl2BrN2O2 [M]+ 428.10 and 430.10; found: 428.81 and 430.84.
General procedure for the synthesis of 2-[5,6-dichloro-2-(substitutedbenzyl)-1H-benzimidazole-1-yl] acetohydrazide (4a–c)
A mixture of compounds 3a–c (0.01 mol) and hydrazine hydrate (0.025 mol) in absolute ethanol (10 mL) was irradiated in closed vessels with the pressure control at 120 °C for 5 min (hold time) at 300 W maximum power. After the completion of the reaction, (monitored by TLC, ethylacetate:hexane, 3:1), the mixture was cooled to room temperature. The precipitate formed was filtered, washed with excess ethanol and dried over CaCl2 to give pure 4a–c.
2-[5,6-Dichloro-2-(2-bromobenzyl)-1H-benzimidazole-1-yl] acetohydrazide (4a). Yield (80%); m.p. 248–249 °C; IR (νmax, cm−1): 3331, 3302 (NH2), 3173 (NH), 1674 (C=O) and 1588 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.33 (s, 2H, CH2), 4.46 (s, 2H, NH2), 4.83 (s, 2H, N–CH2), 7.17–7.82 (m, 6H, Ar–H) and 9.51 (s, 1H, –NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 31.63 (CH2), 45.34 (N–CH2), 112.56, 120.42, 124.49, 125.01, 127.87, 129.34, 129.83, 132.31, 134.10, 134.87, 135.91, 142.32 (Ar–C), 156.58 (C=N) and 166.19 (C=O). Anal. Calcd for C16H13Cl2BrN4O1: C, 44.89; H, 3.06 and N, 13.09; found: C, 44.86; H, 3.07 and N, 13.10. ESI-MS m/z calculated for C16H13Cl2BrN4O1 [M+H]+ 429.10 and 431.10; found: 429.89 and 431.92.
2-[5,6-Dichloro-2-(3-bromobenzyl)-1H-benzimidazole-1-yl] acetohydrazide (4b). Yield (83%); m.p. 231–232 °C; IR (νmax, cm−1): 3304, 3150 (NHNH2), 1658 (C=O) and 1550 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.27 (s, 2H, CH2), 4.35 (s, 2H, NH2), 4.91 (s, 2H, N–CH2), 7.26–7.86 (m, 6H, Arom-H) and 9.53 (s, 1H, NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 32.72 (CH2), 45.23 (N–CH2), 112.50, 120.14, 120.29, 122.07, 124.61, 124.90, 128.79, 130.04, 130.98, 132.30, 135.69, 139.49, 139.60, 142.16 (Ar–C), 156.89 (C=N) and 166.00 (C=O). Anal. Calcd for C16H13Cl2BrN4O1: C, 44.89; H, 3.06 and N, 13.09; found: C, 44.86; H, 3.08 and N, 13.11. ESI-MS m/z calculated for C16H13Cl2BrN4O1 [M+H]+ 429.10 and 431.10; found: 429.02 and 430.98.
2-[5,6-Dichloro-2-(4-bromobenzyl)-1H-benzimidazole-1-yl] acetohydrazide (4c). Yield (85%); m.p. 264-265 °C; IR (νmax, cm−1): 3310, 3190 (NHNH2), 1678 (C=O), and 1596 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.23 (s, 2H, CH2), 4.35 (s, 2H, NH2), 4.88 (s, 2H, N–CH2), 7.26 (d, 2H, Ar–H, J = 8.4 Hz), 7.51 (d, 2H, Ar–H, J = 8.4 Hz), 7.84 (s, 2H, Ar–H) and 9.50 (s, 1H, NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 32.64 (CH2), 45.19 (N–CH2), 112.48, 120.24, 120.34, 124.55, 124.85, 131.74 (2C), 131.86, 135.74, 136.13, 142.14 (Ar–C), 157.00 (C=N) and 165.97 (C=O). Anal. Calcd for C16H13Cl2BrN4O1: C, 44.89; H, 3.06 and N, 13.09; found: C, 44.85; H, 3.04 and N, 13.12. ESI-MS m/z calculated for C16H13Cl2BrN4O1 [M]+ 428.11 and 430.11; found: 428.81 and 430.91.
General procedure for the synthesis of 2,2′-bis(4-bromobenzyl)-1H,1′H-5,5′-bibenzimidazole under microwave irradiation (5a)
A mixture of 3,3′-diaminobenzidine (0.010 mol) and 1c (0.026 mol) in dry MeOH (15 mL) was irradiated with microwave at 60 °C for 10 min at 300 W maximum power. After the completion of the reaction, (monitored by TLC, AcOEt:hexane, 3:1), the mixture was poured onto H2O. The precipitate formed was filtered and recrystallized from EtOH:H2O (1:3) to give pure compounds, 5a.
2,2′-Bis(4-bromobenzyl)-1H,1′H-5,5′-bibenzimidazole (5a). Yield (90%); m.p. 312–313 °C; IR (νmax, cm−1): 3335 (NH), 1569 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.30 (s, 4H, 2CH2), 7.19–7.56 (m, 14H, Ar–H) and 12.33 (s, 2H, 2NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 34.69 (2CH2), 120.44, 121.91, 130.98, 131.15, 131.79, 132.06, 132.15, 135.99, 137.68 (Ar–C) and 154.34 (2C=N). Anal. Calcd for C28H20Br2N4: C, 58.76; H, 3.52 and N, 9.79; found: C, 58.75; H, 3.54 and N, 9.80. ESI-MS m/z calculated C28H20Br2N4 [M]+ 572.29 and 574.26; found: 572.33, 574.16 and symmetric division signal 286.14.
General procedure for the synthesis of dimethyl 2,2′-[2,2′-bis(4-bromobenzyl)-1H,1′H-5,5′-bibenzimidazole-1,1′-diyl]diacetate (6a)
A mixture of compounds 5a (0.01 mol), methyl-α-bromoacetate (0.02 mol) and K2CO3 (0.05 mol) in acetone (15 mL) was irradiated in closed vessels with the pressure control at 85 °C for 20 min (hold time) at 300 W maximum power. After the completion of the reaction, (monitored by TLC, ethylacetate:hexane, 3:1), the mixture was poured onto water. The precipitate formed was filtered and recrystallized from acetone:water (1:3) to give pure 6a.
Yield (85%); m.p. 104–105 °C; IR (νmax, cm−1): 1740 (C=O), 1622 (C=N), 1215, and 1180 (C–O); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 3.34 (s, 6H, 2OCH3), 4.22 (s, 4H, 2CH2), 5.25 (s, 4H, 2N–CH2) and 7.11–7.95 (m, 14H, Ar–H); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 34.96 (2CH2), 47.34 (2N–CH2), 55.11 (2OCH3), 113.29, 119.77, 121.61, 122.63, 124.46, 124.72, 134.13, 138.25, 138.78, 139.21, 144.11, 145.63 (Ar–C), 156.91 (2C=N) and 171.23 (2C=O). Anal. Calcd for C34H28Br2N4O4: C, 57.00; H, 3.94 and N, 7.82; found: C, 57.05; H, 3.90 and N, 7.81. ESI-MS m/z calculated C34H28Br2N4O4 [M]+ 716.42 and 718.41; found: 716.56, 718.71 and symmetric division signal 358.14.
General procedure for the synthesis of 2,2′-[2,2′-bis(4- bromobenzyl)-1H,1′H-5,5′-bibenzimidazole-1,1′-diyl]diacetohydrazide (7a)
A mixture of compounds 6a (0.01 mol) and hydrazine hydrate (0.05 mol) in absolute ethanol (10 mL) was irradiated in closed vessels with the pressure control at 125 °C for 13 min (hold time) at 300 W maximum power. After the completion of the reaction, (monitored by TLC, ethylacetate:hexane, 3:1), the mixture was cooled to room temperature. The precipitate formed was filtered, washed with excess ethanol and dried over CaCl2 to give pure 7a.
Yield (80%); m.p. 185–186 °C; IR (νmax, cm−1): 3292, 3200 (NHNH2), 1658 (C=O), 15 621 (C=N); 1H-NMR (DMSO-d6, 200 MHz) δ (ppm): 4.25 (s, 4H, 2CH2), 4.45 (bs, 4H, 2NH2), 4.85 (s, 4H, 2N–CH2), 7.16–7.85 (m, 14H, Ar–H) and 9.60 (s, 2H, 2NH); Citation13C-NMR (DMSO-d6, 50 MHz) δ (ppm): 36.20 (2CH2), 45.33 (2N–CH2), 109.27, 110.57, 121.25, 122.33, 124.16, 132.13, 132.28, 136.62, 138.10, 139.21, 141.96, 144.03 (Ar–C), 155.34 (2C=N) and 166.33 (2C = O). Anal. Calcd for C32H28Br2N8O2: C, 53.65; H, 3.94 and N, 15.64; found: C, 53.63; H, 3.95 and N, 15.65. ESI-MS m/z calculated C32H28Br2N8O2 [M]+ 716.43 and 718.43; found: 716.60, 718.53 and symmetric division signal 358.23.
Anti-lipase activity
The inhibitory effects of those compounds were evaluated against PPL (15 ng/µL). Lipase activity assay were done according to Verger and ChahinianCitation26. Microtiter plates were coated with purified tung oil TAGs. Compounds were mixed with PPL 1:2 (v/v) and incubated for 30 min. The microtiter plates containing purified tung oil, lipase solution and assay buffer (10 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl, 6 mM CaCl2, 1 mM EDTA and 3 mg/mL β-cyclodextrin) were recorded continuously for 40 min against the buffer alone using microplate reader (SpectraMax M5, Molecular Devices) at 272 nm. The inhibitory activities of those compounds and Orlistat, a positive control against pancreatic lipase, were measured at concentrations of 6.25, 1.25 and 0.625 µg/mL. Residual activities were calculated by comparing to control without inhibitor (T+). The assays were done in triplicate. The IC50 value was determined as the concentration of compound that give 50% inhibition of maximal activity.
Conclusion
In conclusion, we have developed a novel and practical method for the synthesis of benzimidazole derivatives under microwave irradiation. This method can provide a convenient way of synthesizing potential bioactive benzimidazoles. This is the first example of benzimidazoles with their inhibitory properties toward pancreatic lipase. The results could be an inspiration for further investigation of potential lipase inhibitors within heterocycles.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Hosamani KM, Seetharamareddy HR, Keri RS, et al. Microwave assisted, one-pot synthesis of 5-nitro- 2-aryl substituted-1H-benzimidazole libraries: screening in vitro for antimicrobial activity. J Enzyme Inhib Med Chem 2009;24:1095–100
- Demirayak S, Kayagil I, Yurttas L. Microwave supported synthesis of some novel 1,3-diarylpyrazino[1,2-a]benzimidazole derivatives and investigation of their anticancer activities. Eur J Med Chem 2011;46:411–16
- Tuncbilek M, Kiper T, Altanlar N. Synthesis and in vitro antimicrobial activity of some novel substituted benzimidazole derivatives having potent activity against MRSA. Eur J Med Chem 2009;44:1024–33
- Desai KG, Desai KR. Green route for the heterocyclization of 2-mercaptobenzimidazole into betalactum segment derivatives containing –CONH– bridge with benzimidazole. Screening in vitro antimicrobial activity with various microorganisms. Bioorg Med Chem 2006;14:8271–9
- Mobinikhaledi A, Foroughifar N, Kalhor M, Mirabolfathy M. Synthesis and antifungal activity of novel 2-benzimidazolylimino-5-arylidene-4-thiazolidinones. J Heterocyclic Chem 2010;47:77–80
- Kılcıgil GA, Tuncbilek M, Altanlar N, Göker H. Synthesis and antimicrobial activity of some new benzimidazole carboxylates and carboxamides. Farmaco 1999;54:562–5
- Nakano H, Inoue T, Kawasaki N, et al. Synthesis and biological activities of novel antiallergic agents with 5-lipoxygenase inhibiting action. Bioorg Med Chem 2000;8:373–80
- Eke BC, Puskullu MO, Buyukbingol E, Iscan M. A study on the antioxidant capacities of some benzimidazoles in rat tissues. Chem Biol Interact 1998;113:65–77
- Kohler P. The biochemical basis of anthelmintic action and resistance. Int J Parasitol 2001;31:336–9
- Refaat HM. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur J Med Chem 2010;45:2949–56
- Kavitha CS, Hosamani KM, Seetharamareddy HR. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur J Med Chem 2010;45:2048–54
- Roth T, Morningstar ML, Boyer PL, et al. Synthesis and biological activity of novel nonnucleoside inhibitors of HIV-1 reverse transcriptase. 2-Aryl-substituted benzimidazoles. J Med Chem 1997;40:4199–207
- Migawa MT, Girardet JL, Walker JA, et al. Design, synthesis: antiviral activity of alpha-nucleosides: d- and l-isomers of lyxofuranosyl- and (5-deoxylyxofuranosyl)benzimidazoles. J Med Chem 1998;41:1242–51
- Zhu GD, Gandhi VB, Gong J, et al. Synthesis and SAR of novel, potent and orally bioavailable benzimidazole inhibitors sf poly(ADP-ribose) polymerase (PARP) with a quaternary methylene-amino substituent. Bioorg Med Chem Lett 2008;18:3955–8
- Ogino Y, Ohtake N, Nagae Y, et al. Design, syntheses, and structure–activity relationships of novel NPY Y5 receptor antagonists: 2-{3-oxospiro[isobenzofuran-1(3H),4′-piperidin]-1′-yl}benzimidazole derivatives. Bioorg Med Chem Lett 2008;18:5010–14
- Petersen AK, Olesen PH, Christiansen LB, et al. 2-(2-Hydroxyphenyl)benzimidazoles useful for the treating obesity and diabetes, US Patent. Patent no: US 7.915.299B2. 2011
- van Gaal LF, Mertens IL, de Block CE. Mechanisms linking obesity with cardiovascular disease. Nature 2006;444:875–80
- Jandacek RJ, Woods SC. Pharmaceutical approaches to the treatment of obesity. Drug Discov Today 2004;15:874–80
- Grimmet MR, Katritzky AR, Rees CW, Potts KT. İmidazole and their benzoderivatives. Compr Heterocyc Chem 1984;5:345–72
- Dudd LM, Venardou E, Garcia Verdugo E, et al. Synthesis of benzimidazoles in high-temperature water. Green Chem 2003;5:187–92
- Lu J, Yang B, Bai Y. Microwave irradiation synthesis of 2-substituted benzimidazoles using PPA as a catalyst under solvent-free conditions. Synthetic Commun 2002;32:3703–9
- Algul O, Kaessler A, Apcin Y, et al. Comparative studies on conventional and microwave synthesis of some benzimidazole, benzothiazole and indole derivatives and testing on inhibition of hyaluronidase. Molecules 2008;13:736–48
- Hunger A, Kebrle J, Rossi A, Hoffmann K. Benzimidazol-derivate und verwandte heterocyclen VI. Synthese von phenyl-[1-aminoalkyl-benzimidazolyl-(2)]-essigsäure-estern und -amiden. Helv Chim Acta 1960;43:1727–33
- Birari RB, Bhutani KK. Pancreatic lipase inhibitors from natural sources: unexplored potential. Drug Discov Today 2007;12:879–89
- Weigle DS. Pharmacological therapy of obesity: past, present, and future. J Clin Endocr Metab 2003;88:2462–9
- Verger RC, Chahinian H. Method for the high-speed detection and/or measurement of a lipase or phospholipase activity, PCT Int. Appl. WO 2006/085009. 2006