
Abstract
Monoamine oxidase (MAO, EC 1.4.3.4) is responsible from the oxidation of a variety of amine neurotransmitters. MAO inhibitors are used for the treatment of depression or Parkinson’s disease. They also inhibit the catabolism of dietary amines. According to one hypothesis, inactivation results from the formation of a covalent adduct to a cysteine residue in the enzyme. If the adduct is stable enough, the enzyme is inhibited for a long time. After a while, enzyme can turn to its active form as a result of adduct breakdown by β-elimination. In this study, the proposed inactivation mechanism was modeled and tested by quantum chemical calculations. Eight heterocyclic methylthioamine derivatives were selected to represent the proposed covalent adducts. Activation energies related to their β-elimination reactions were calculated using ab initio and density functional theory methods. Calculated activation energies were in good agreement with the relative stabilities of the hypothetical adducts predicted in the literature by enzyme inactivation measurements.
Introduction
Monoamine oxidase (MAO) catalyzes the oxidation of various amine neurotransmitters, such as serotonin, dopamine and norepinephrine. It exists as two isozymic forms, MAO-A and MAO-BCitation1–3. These two forms display different substrate and inhibitor specificities. However, it is believed that they catalyze substrate oxidation by the same mechanism. Compounds that selectively inhibit MAO-A exhibit antidepressant activity, whereas the ones that selectively inhibit MAO-B are used in the treatment of Parkinson’s diseaseCitation4. MAO inhibitors also inhibit the catabolism of dietary amines. Other findings showed that MAO-B inhibitors have neuroprotective and antioxidant effects, as well as a role in delaying apoptotic neuronal deathCitation5,Citation6.
MAO belongs to a family of enzymes known as flavoenzymes and the flavin in MAO is covalently attached at the 8α-position to an enzyme active-site cysteine residueCitation7. It catalyzes the anaerobic oxidation of amine substrates to the corresponding imines, which are nonenzymatically hydrolyzed to the corresponding aldehydes. The flavin is reduced to the hydroquinone and converted back into the oxidized form by reaction with molecular oxygen, and the enzyme turns to its native form. Over the years, there have been extensive efforts to understand the MAO-catalyzed amine-oxidation mechanism and various mechanisms have been proposed so farCitation8–11. Previous computational resultsCitation12–14 by our group are in agreement with polar nucleophilic mechanism proposed by Miller & EdmondsonCitation15 for MAO-A. Still, the detailed mechanism of amine oxidation by MAO remains a subject for additional studies.
One approach to study the enzyme mechanisms is to investigate the inactivation reaction by mechanism-based inactivators. These inactivators are unreactive compounds that have structural similarity to the substrate or product for the target enzyme. Such compounds are converted by the normal catalytic mechanism of the enzyme into reactive species typically by covalent bond formation, and this causes inactivation of the enzyme. A mechanism-based inactivator is unreactive and selective since only the target enzyme is capable of converting it to the reactive form. Therefore, these compounds are quite useful in rational design of new drugs.
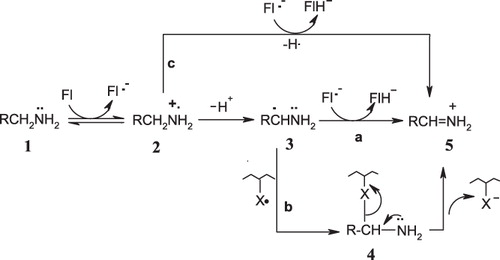
Silverman et al.Citation16–21 proposed single-electron transfer (SET) mechanisms for MAO, based on the ring-opening and SET chemistry observed in chemical model studies of mechanism-based MAO-B inactivators. Three possible paths (a, b and c) were proposed, as shown in . The present study focuses on path b. The one-electron transfer from the amine to the FAD would give the amine radical cation (2), which can lose a proton to give the carbon radical (3). The radical generated by deprotonation can undergo radical combination with an active-site radical to give a covalent adduct (4), which decomposes by β-elimination to the immonium ion (5). Silverman et al.Citation16–21 propose that enzyme activity is retarded if this hypothetical covalent adduct is much more stable than that for good substrates. Although the structure of the inactivated enzyme adduct (4) has not known, the X group in (4) could be either the flavin semiquinone formed in the first step or an amino acid radical generated from hydrogen atom transfer from the amino acid to the flavin semiquinone. Two experiments identified this amino acid to be a cysteine residueCitation22, cys365 in MAO-B being a logical candidateCitation23,Citation24. However, elucidation of the structure of human MAO-BCitation25 and MAO-ACitation26,Citation27 and of rat MAO-ACitation28 by X-ray revealed that there is no cysteine in the active site; cys365 in MAO-B and the corresponding amino acid cys374 in MAO-A are on the surface of the protein. Enzyme activity studies on cys365ACitation29 and cys374ACitation30 mutant enzymes were also conducted but could not clarify the situation. Lu et al.Citation29 and Vintém et al.Citation30 revised the proposed inactivation mechanism, suggesting that “oxidation of, at least, N-cyclopropyl-α-methylbenzylamine has to produce an activated species capable of traveling a distance from the active site prior to becoming attached to a cysteine residue on the surface of the protein”.
Figure 1. SET mechanisms proposed by Silverman et al.Citation16–21.
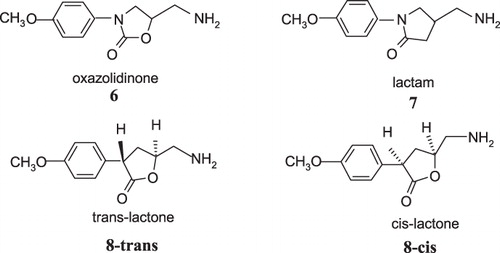
On the other hand, the data in the literature related to the influence of thiol groups on the catalytic properties of both MAO-A and MAO-B are often contradictoryCitation29–31. In addition, hypothetical covalent adducts are proposed based only on indirect measurements and chemical model reactions since it is a challenging task to trap or isolate such intermediates. Although quantum chemical modeling studies are quite useful in direct determination of the relative stabilities of highly reactive species, only two articles have been reported so far for the understanding of MAO inactivation mechanism: our previous study on oxygen containing heptylaminesCitation32 and a very recent computational study on rasagiline and selegiline by Borstnar et al.Citation33. Therefore, the aim of the present work is to investigate the relationship between inhibitory activity and the stability of the proposed covalent adduct by quantum chemical modeling of the β-elimination reaction in path b of in order to test the proposed inactivation mechanism. For this purpose, 5-(aminomethyl)-3-(4-methoxyphenyl)-2-oxazolidinone (6), 4-(aminomethyl)-1-(4-methoxyphenyl)-2-pyrrolidinone (7) and cis- and trans-5-(aminomethyl)-3-(4-methoxyphenyl)dihydrofuran-2(3H)-one (8), shown in , were utilized because they are known to be time-dependent selective inhibitors of MAO-B, and extensive chemical modeling and enzyme inactivation studies are available on these compounds and their derivativesCitation34–41.
Figure 2. MAO-B inhibitors studied by Silverman et al.Citation34–41.
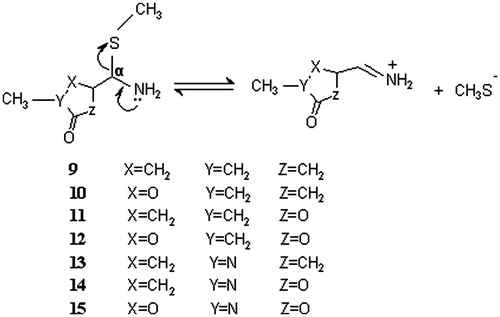
Based on enzyme inactivation and chemical model studies coupled with basic chemical reasoning, relative stability of the hypothetical covalent adducts (4) of the heterocyclic rings (oxazolidinone, pyrrolidinone and dihydrofuranone) was proposed to be the result of the electron-withdrawing ability of the heteroatoms in the heterocyclic rings. The rationale behind this approach was that the greater the electron-withdrawing effect of the heterocycle, the greater the stability of the adduct should be, and this should result in a longer half-life for reactivation. The observed half-lives of reactivation correspond to the electron-withdrawing power of the heteroatom in the ringCitation34: oxazolidinone (two heteroatoms, O, N) > dihydrofuranone (oxygen) > pyrrolidinone (nitrogen). This order is hypothesized to be parallel to the relative stability of the proposed covalent adducts. In order to test this hypothesis, we designed the model adduct molecules (9–15) and predicted their relative kinetic stabilities directly by calculating the activation energies and the rate constants of the β-elimination reaction shown in . The enzyme cysteine residue is simplified and represented by SCH3 group, which enables us to apply time-consuming ab initio and density functional theory (DFT) calculations. The same approach was used successfully in our previous workCitation32. The proposed inductive effect is considered by incorporating O, N and C atoms in the ring systematically. The electron-withdrawing ability of the X, Y and Z atoms in the model compounds follows the order O > N > C. The location of these atoms was varied with respect to the distance from the site of enzyme attachment, taking into account that the strength of inductive effect depends also on the distance. Adduct models (11), (13) and (14) mimic the hypothetical adducts of MAO-B inhibitors dihydrofuranone (8), pyrrolidinone (7) and oxazolidinone (6), respectively, for which experimental results are available. The remaining model molecules were designed for the purpose of comparison and deeper understanding of the inactivation hypothesis.
Figure 3. Reactions modeled to mimic the dissociation of the hypothetical enzyme-inhibitor covalent adducts. Both cis and the trans isomers were modeled for adducts 9–12.
Methods
Ab initio HF/6-31G* and density functional B3LYP/6-31G* theories were used throughout this study using Spartan 04 and Gaussian 03 softwaresCitation42,Citation43. For each reaction shown in , structures of the reactants and the transition states were fully optimized. Normal mode analysis was performed to characterize each stationary point. Reactant structures produce all real frequencies. Transition structures were characterized with one imaginary vibrational mode corresponding to the stretching motion of the C–S and C–N bonds of the methylthio amine fragment. For the purpose of an additional check, intrinsic reaction coordinate (IRC)Citation44 calculation was performed for the transition structure of the prototype molecule (14), which has proven that the transition state connects reactant and the product. Activation energy of each reaction was calculated by subtracting the energy of the reactant from that of the transition state. Thermal corrections to the zero-point energy, enthalpy and Gibbs free energy were calculated at 298.15 K and 1 atm. The effect of aqueous environment of the enzyme surface was also taken into account using SM5.4 solvation methodology developed by Chambers et al.Citation45. Calculated aqueous solvation energies were added to the gas phase energies, and listed in the tables.
Rate constants were calculated according to the well-known equationCitation46
where k is the rate constant, kb the Boltzmann constant, T the temperature (298.15 K), ΔG* the free energy of activation, h the Planck constant, c the concentration (taken as unity) and R the gas constant.
Prior to ab initio and DFT geometry optimizations, conformational analysis of each reactant molecule in was performed with semiempirical PM3 and SM5.4 methods using the conformer distribution facility in Spartan 04. The stable conformations were listed according to their energies in aqueous environment. Among them, the lowest energy conformation in which amine lone pair electron was antiperiplanar with respect to the C–S bond was selected, because such an orientation is known to facilitate the β-elimination reactions. The selected conformation was then used for the initial geometry in the optimizations of the reactants and the transition states using ab initio and DFT.
Results and discussion
During β-elimination step, C–S σ bond breaks and C–N π bond forms. These critical bond distances for reactants and transition states optimized with B3LYP are listed in . No remarkable inductive effect of the heteroatoms on these bond lengths was observed at X, Y and Z positions. The optimized geometries of the transition states and the reactants are demonstrated in Supplementary Figures S1 and S2. The optimization of the product complexes of these reactions is prohibitive due to the presence of charge separation in gas phase. Therefore, in order to ensure that the optimized transitions states connect the reactants to the products, IRC for the adduct (14) was performed as a representative of the adduct models 9–15, and is shown in . The critical bond distances (C–S and C–N) of the transition state lie between those of the reactant and the product complex.
Figure 4. IRC plot for the dissociation reaction of model adduct 14.
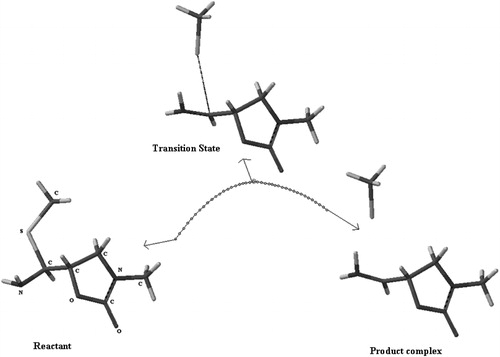
Table 1. The length (Å) of breaking and forming bonds for reactants and transition states optimized at B3LYP/6-31G* level and imaginary frequencies of the transition states.
According to Silverman, the reason of inactivation of enzyme is the stability of the intermediate because the more stable the adduct the slower the rate of return of enzyme. Thus, activation energy of the dissociation reaction (β-elimination) should exhibit an inverse relation with the elimination rate and a direct relation with the duration of elimination. In other words, as the stability of the intermediate and thus, the elimination time increase, activation energy increases and the reaction rate decreases. When MAO-B was inhibited with the heterocyclic inhibitors 6–8 and then the rate of return of enzyme activity was measured with dialysis, it was found that the stability order of the adducts was oxazolidinone (6) > trans-lactone (8-trans) > cis-lacton (8-cis) > lactam (7)Citation40. If the hypothesized inactivation mechanism is correct, then the activation energies for the elimination reactions of the corresponding adduct models should follow the order: 14 > 11-trans > 11-cis > 13. Therefore, computed activation energies and rate constants for the elimination reactions of the adduct models have been evaluated in the light of this experimentally known trend, and tabulated in and .
Table 2. Activation energies (kcal/mol) calculated at HF/6-31G* level in gas and aqueous phases, solvation energies (within parentheses) and calculated rate constants (s−1) in aqueous phase.
Table 3. Activation energies (kcal/mol) calculated at B3LYP/6-31G* level in gas and aqueous phases, solvation energies (within parentheses) and calculated rate constants (s−1) in aqueous phase.
When and are examined in terms of electronic and Gibbs free energies, both ab initio and DFT calculations predicted similar trends. From the activation energies computed with HF/6-31 G* in the gas phase, the relative order of adduct stability is found to be 12-trans > 15 > 12-cis ≈ 11-trans > 11-cis > 14 > 10-cis > 9-trans > 13 > 9-cis > 13′ > 10-trans. On the other hand, the relative order in aqueous phase is 15 > 12-trans ≈ 12-cis > 14 > 11-trans > 10-cis > 11-cis > 13 > 9-trans > 13′ > 9-cis ≈ 10-trans, which agrees well with experimental observation. The model intermediate 15 is found to be the most stable intermediate since it involves three electron-withdrawing atoms (O, N and O at X, Y and Z positions, respectively). Structure 12 that involves two oxygen atoms at X and Z positions is the second most stable intermediate. Structure 10 involving only one oxygen atom at position X and 9 are found to be the most unstable ones. These findings are consistent with the inactivation hypothesis proposed by Silverman et al.Citation40.
The relative orders obtained from energy computations using B3LYP/6-31 G* in gas and aqueous phases are found to be 12-trans > 15 ≈ 14 > 11-cis > 12-cis > 13 > 11-trans > 9-cis > 9-trans > 10-trans > 13′ and 15 > 12-trans > 12-cis > 14 > 11-trans > 10-trans > 13 > 11-cis > 13′ > 9-cis > 9-trans, respectively. Except structure 13, the order of adduct stability in aqueous phase is the same as the experimentally determined order, 14 > 11-trans > 11-cis > 13, shown in bold face in the former sentence. On the other hand, it is possible that adduct may have some flexibility to adopt several conformations inside the enzyme. Thus, calculations performed with another stable conformation (13′) of the structure 13 revealed that the kinetic stability of adduct model 13′ is less than 11-cis. The order of stability was then obtained as 14 > 11-trans > 11-cis > 13′ which is in very good agreement with the result of dialysis. This suggests that the conformation of an intermediate can be an important factor for the enzyme inactivation. Considering the inductive effects of O, N and C atoms at the positions X, Y and Z in the modeled structures, all model adducts exhibited the expected order of stability as 15 > 12 > 14 > 11-trans > 10 > 11-cis > 13′ > 9. In this sequence, adduct 12 takes place between 15 and 14. The reason why 12 is more stable than 14 is that it has two oxygen atoms (X and Z=O) which are close to α-carbon, whereas 14 has an oxygen atom at position Z and a nitrogen atom at the position Y which is less electronegative than oxygen and also further away from α-carbon atom. Adduct models 10-trans and 11-trans have very close energies to each other, which is reasonable since each adduct contains an oxygen atom at the same distance to α-carbon. The reason why the increased electron-withdrawing ability makes the enzyme adduct more stable is that it stabilizes the sp3 hybridization of the α-carbon atomCitation34 (). Besides, the electron-withdrawing effect destabilizes the sp2 hybridization of the immonium ion.
Structural features of MAO enzymesCitation25–28 emphasize the hydrophobic nature of the active site composed of aromatic moieties that include tyrosines in the aromatic cage and the FAD cofactor. Since the calculated stability trend of adduct models in the aqueous phase confirms experimental findings, rather than in the gas phase, it is reasonable to assume that the proposed adducts can form with residues that are close to the surface of MAOs, which are expected to be in more hydrophilic environment. According to Silverman & HiebertCitation23, cys365 in MAO-B and the corresponding amino acid cys374 in MAO-A are logical candidates which are on the surface of the protein. The results of our calculations in aqueous solution are consistent with this rationale. However, we propose that another cysteine, cys172 in the entrance cavity of the crystal structure of MAO-B plays a role in inhibition through hydrogen-bonding interactions which is responsible for the inhibitory behavior of p-nitrobenzylamine in MAO-BCitation13,Citation47. Cys172 is also a reasonable candidate to give a covalent adduct with oxazolidinones because, if this is the case, radical 3 () generated by deprotonation via FAD would travel a shorter distance (11 Å) from FAD to cys172 to undergo radical combination. On the other hand, the radical should travel a much longer distance (24 Å) to reach cys365.
Conclusion
Activation energies related to the β-elimination reactions of eight heterocyclic methylthioamine derivatives were calculated using ab initio and DFT methods to represent the covalent adducts in the proposed inactivation mechanism. The adduct models having stronger electron-withdrawing atoms nearer to α-carbon exhibited larger activation energies. Calculated activation energies were in good agreement with the relative stabilities of the hypothetical adducts predicted in the literature by enzyme inactivation measurements. These results provide direct evidence and support the proposed inactivation mechanism. They also suggest that it is possible to rationally design new and selective inactivators, simply by incorporation of electron-withdrawing moieties to the molecules capable of binding to the enzyme.
HF/6-31G* and B3LYP/6-31G* show similar trend in prediction of activation energy of elimination step. For both computational methods, the calculated elimination rates in aqueous phase are in good agreement with experimental findings of Silverman et al.Citation40 Among the molecules in the model set, structure 15 is the most stable intermediate and 9 is the least stable one in aqueous phase. From these observations, it can be extracted that results obtained are independent to computational method and solvent effect should be considered in calculating the elimination rate. Another key point is that different conformations of intermediate may play an important role in determination of stability and computation of elimination rate as observed in two different conformations 13 and 13′.
Supplementary material
Supplementary data are available consisting of 3-dimensional view of the optimized structures, total energies and Gibbs free energies in gas phase and in aqueous medium.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was supported by the Marmara University Scientific Research Projects Commission (BAPKO), project no.: BSE-087/051201.
Acknowledgements
The authors thank Prof. K. Yelekçi for critical reading of the manuscript.
References
- Lu X, Rodrigues M, Gu W, Silverman RB. Inactivation of mitochondrial monoamine oxidase B by methylthio-substituted benzylamines. Bioorg Med Chem 2003;11:4423–30
- Ives JL, Heym J. Antidepressant agents. Annu Rep Med Chem 1989;24:21–9
- Tetrud VW, Langston JW. The effect of deprenyl (selegiline) on the natural history of Parkinson’s disease. Science 1989;245:519–22
- Silverman RB. Radical ideas about monoamine-oxidase. Acc Chem Res 1995;28:335–42, and the references therein
- Haefely WE, Burkard WP, Cesura AM, et al. Biochemistry and pharmacology of moclobemide, a prototype RIMA. Psychopharmacology 1992;106:6–14
- Cesura AM, Fletcher A. The new generation of monoamine oxidase inhibitors. Prog Drug Res 1992;38:171–297
- Binda C, Newton-Vinson P, Hubalek F, et al. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 2002;9:22–6
- Scrutton NS. Chemical aspects of amine oxidation by flavoprotein enzymes. Nat Prod Rep 2004;21:722–30
- Edmondson DE, Mattevi A, Binda C, Hubalek F. Structure and mechanism of monoamine oxidase. Curr Med Chem 2004;11:1983–93
- Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch Biochem Biophys 2010;493:13–25
- Dunn RV, Munro AW, Turner NJ, et al. Tyrosyl radical formation and propagation in flavin dependent monoamine oxidases. Chem BioChem 2010;11:1228–31
- Erdem SS, Karahan Ö, Yıldız İ, Yelekçi K. A computational study on the amine-oxidation mechanism of monoamine oxidase: insight into the polar nucleophilic mechanism. Org Biomol Chem 2006;4:646–8
- Akyüz MA, Erdem SS, Edmondson DE. The aromatic cage in the active site of monoamine oxidase B: effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J Neural Trans 2007;114:693–8
- Erdem SS, Büyükmenekşe B. Computational investigation on the structure–activity relationship of the biradical mechanism for monoamine oxidase. J Neural Trans 2011;118:1021–9
- Miller JR, Edmondson DE. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 1999;38:13670–83
- Silverman RB, Cesarone JM, Lu X. Stereoselective ring-opening of 1-phenylcyclopropylamine catalyzed by monoamine oxidase-b. J Am Chem Soc 1993;115:4955–61
- Silverman RB, Zelechonok Y. Evidence for a hydrogen-atom transfer mechanism or a proton fast electron-transfer mechanism for monoamine-oxidase. J Org Chem 1992;57:6373–4
- Silverman RB, Lu X, Zhou JJP, Swihart A. Monoamine-oxidase b-catalyzed oxidation of cinnamylamine 2,3-oxide – further evidence against a nucleophilic mechanism. J Am Chem Soc 1994;116:11590–1
- Silverman RB, Ding CZ, Borillo JL, Chang JT. Mechanism-based enzyme inactivation via a deactivated cyclopropane intermediate. J Am Chem Soc 1993;115:2982–3
- Wang X, Silverman RB. 2-(Iodoethenyl)benzyl amines as potential probes for radical intermediates formed during monoamine oxidase-catalyzed oxidations. J Org Chem 1998;63:7357–63
- Silverman RB, Zhou JP, Eaton PE. Inactivation of monoamine-oxidase by (aminomethyl)cubane – 1st evidence for an alpha-amino radical during enzyme catalysis. J Am Chem Soc 1993;115:8841–2
- Silverman RB. Electron transfer chemistry of monoamine oxidase. In: Mariano PS, ed. Advances in electron transfer chemistry. Greenwich: JAI Press; 1992:177–213
- Silverman RB, Hiebert CK. Inactivation of monoamine oxidase-a by the monoamine oxidase-b inactivators 1-phenylcyclopropylamine, 1-benzylcyclopropylamine, and n-cyclopropyl-alpha-methylbenzylamine. Biochemistry 1988;27:8448–53
- Zhong BY, Silverman RB. Identification of the active site cysteine in bovine liver monoamine oxidase B. J Am Chem Soc 1997;119:6690–1
- Binda C, Hubalek F, Li M, et al. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. FEBS Lett 2004;564:225–8
- de Colibus L, Binda C, Lustig A, et al. Three-dimensional structure of human monoamine oxidase A (MAO-A): relation to the structures of rat MAO-A and human MAO-B. Proc Natl Acad Sci 2005;102:12684–9
- Son SY, Ma J, Kondou Y, et al. Structure of human monoamine oxidase A at 2.2-Å resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci 2008;105:5739–44
- Ma J, Yoshimura M, Yamashita E, et al. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J Mol Biol 2004;338:103–14
- Lu X, Rodriguez M, Ji H, et al. Irreversible inactivation of mitochondrial monoamine oxidases. In: Chapman SK, Perham RN, Scrutton NS, eds. Flavins and flavoproteins. Proceedings of the 14th International Symposium; 2002 Jul 14–18; Cambridge. Berlin: Rudolf Weber; 2002:817–30
- Vintém APB, Price NT, Silverman RB, Ramsay RR. Mutation of surface cysteine 374 to alanine in monoamine oxidase A alters substrate turnover and inactivation by cyclopropylamines. Bioorg Med Chem 2005;13:3487–95
- Lu X, Rodriguez M, Gu W, Silverman RB. Inactivation of mitochondrial monoamine oxidase B by methylthio-substituted benzylamines. Bioorg Med Chem 2003;11:4423–30
- Erdem SS, Yelekci K. Computer modeling of oxygen containing heptylamines as monoamine oxidase inactivators. J Mol Struct (Theochem) 2001;572:97–106
- Borstnar R, Repic M, Krzan M, et al. Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: a computational study. Eur J Org Chem 2011;2011:6419–33
- Silverman RB, Ding CZ. Chemical-model for a mechanism of inactivation of monoamine-oxidase by heterocyclic-compounds – electronic effects on acetal hydrolysis. J Am Chem Soc 1993;115:4571–6
- Ding CZ, Silverman RB. Transformation of heterocyclic reversible monoamine oxidase-b inactivators into irreversible inactivators by N-methylation. J Med Chem 1993;36:3606–10
- Ding CZ, Silverman RB. 4-(Aminomethyl)-1-aryl-2-pyrrolidinones, a new class of monoamine oxidase-b inactivators. J Enzyme Inhib 1992;6:223–31
- Ding CZ, Silverman RB. Selective inactivation of monoamine-oxidase-b by aminoethyl 3-chlorobenzyl ether. Bioorg Med Chem Lett 1993;3:2077–8
- Lu XL, Silverman RB. Monoamine oxidase B-catalyzed reactions of cis- and trans-5-aminomethyl-3-(4-methoxyphenyl)dihydrofuran-2(3H)-ones. Evidence for a reversible redox reaction. J Am Chem Soc 1998;120:10583–7
- Ding CZ, Silverman RB. 5-(Aminomethyl)-3-aryldihydrofuran-2(3H)-ones, a new class of monoamine oxidase-B inactivators. J Med Chem 1992;35:885–9
- Silverman RB, Ding CZ, Gates KS. Design and mechanism of monoamine oxidase inactivators from a chemical perspective. In: Testa B, Kyburz E, Fuhrer W, Giger R, eds. Perspectives in medicinal chemistry. Basel: Verlag Helvetica Chimica Acta; 1993:73
- Jones TZE, Fleming P, Eyermann CJ, et al. Orientation of oxazolidinones in the active site of monoamine oxidase. Biochem Pharmacol 2005;70:407–16
- Spartan 04, Wavefunction Inc., Irvine, CA, USA. Available from: http://www.wavefun.com
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 03 revision D.01. Wallingford, CT: Gaussian Inc.; 2004
- Gonzalez C, Schlegel HB. An improved algorithm for reaction-path following. J Chem Phys 1989;90:2154–62
- Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. A model for aqueous solvation based on class IV atomic charges and first-solvation-shell effects. J Phys Chem B 1996;100:16385–98
- Steinfield JI, Francisco JS, Hase WL. Chemical kinetics and dynamics. Upper Saddle River (NJ): Prentice Hall; 1989:321–3
- Li M, Binda C, Mattevi A, Edmondson DE. Functional role of the “Aromatic Cage” in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 2006;45:4775–84