
Abstract
A new class of novel thiazole-(benz)azole derivatives was synthesized to investigate their anticancer activity. The structure of the compounds was confirmed by IR, 1H-NMR, and MS spectral data and elemental analyses. Anticancer effect of the compounds was evaluated against A549 and C6 tumor cell lines. MTT, analysis of DNA synthesis, acridine orange/ethidium bromide staining method and analysis of caspase-3 activation assays were performed for anticancer activity investigations. Compounds 6f and 6g, which carry 5-chloro and 5-methylbenzimidazole groups showed significant anticancer activity. Potential of these compounds to direct tumor cells to apoptotic pathway, which is a precondition of anticancer action, was also observed.
Introduction
Malignancy is caused by abnormalities in cells, which might be due to inherited genes or caused by outside exposure of the body to chemicals, radiation or even infectious agentsCitation1,Citation2. Chemotherapy is one of the techniques adopted for the treatment and eradication of cancerous cells. DNA-damaging agents displaying various mechanisms of action have achieved widespread use as chemotherapeutics and still constitute a basic tool set in the treatment of cancerCitation3,Citation4. On the other hand, significant side-effects, such as nausea, vomiting, diarrhea, hair loss, serious infections and growth of tumor-cell population are often encountered during chemotherapyCitation5. Thus, the limitations of current anticancer drugs highlight the need for the discovery of new anticancer agents.
Thiazoles represent a class of heterocyclic compounds of great importance in anticancer drug research. Interest in the antineoplastic activity of thiazoles has increased after discovery of the thiazole-based cytotoxic agents tiazofurinCitation6–8, distamycinCitation9, bleomycinCitation10,Citation11, netropsin and thia-netropsinCitation12. Several studies including the anticancer activity of certain thiazole analogs were reported by medicinal chemistsCitation13–20.
In addition to thiazoles, some other azole compounds have also attracted much attention due to their prominent utilization as antitumor agentsCitation21. For instance, derivatives of triazoleCitation22, imidazoleCitation23, benzimidazoleCitation24,Citation25, benzoxazoleCitation26–28 and benzothiazoleCitation29 have shown anticancer properties. Triazole derivatives anastrozole and letrozole are the aromatase inhibitors, currently finding widespread application in the clinicCitation30. Dacarbazine, an imidazole compound, inhibits DNA, RNA and protein synthesis by alkylation mechanismCitation31. Bisbenzimidazole compounds such as Hoechst 33342 and Hoechst 33258 have anticancer ability to inhibit DNA topoisomerase I and many other cellular processesCitation32. Natural bis(benzoxazole) product UK-11 shows cytotoxic action on cancer cells, selectivelyCitation33. Benzothiazole derivative phortress is another selective anticancer compound that shows anticancer activity by inducing protein expression and forming a DNA adductCitation34.
As described above, in general, azole compounds causes a DNA-damage and exhibit anticancer activity by varying mechanisms of action. Prompted from these observations, we synthesized a series of azole and benzazole compounds and investigated their anticancer activity profiles in a recent studyCitation35. In such study, N-phenyl-2-(benz)azolylthio acetamide substructure indicated essential impact on cytotoxic action. Hence, in the present study we synthesized novel compounds that bear thiazole and N-phenyl-2-(benz)azolylthio acetamide substructures and investigated their effect on DNA synthesis of carcinogenic cell lines and apoptosis.
Experimental section
Chemistry
All chemicals were purchased from Merck (Nottingham, UK) or Sigma-Aldrich Chemical (Poole, UK) companies. All melting points (m.p.) were determined by Electrothermal 9100 digital melting point apparatus (Electrothermal, Essex, UK) and were uncorrected. 1H-NMR data were recorded by Bruker 500 MHz spectrometer (Bruker Bioscience, Billerica, MA). M + 1 peaks were determined by AB Sciex-3200 Q-TRAP LC/MS/MS system (500 Old Connecticut Path, Framingham, MA). Elemental analyses were performed on a Perkin Elmer EAL 240 (Perkin Elmer, Norwalk, CT) elemental analyzer.
4′-Acetaminoacetophenone (1)
4′-Aminoacetophenone (0.05 mol, 6.75 g) and triethylamine (0.06 mol, 8.34 mL) were dissolved in THF (100 mL) with a constant stirring at 0–5 °C, then acetyl chloride (0.06 mol, 4.78 mL) was added dropwise to this solution. The reaction mixture was allowed to stirring for 1 h at room temperature. After evaporation of solvent, the obtained solid was washed with water, filtered, dried and recrystallized from ethanol. Yield: 82%; m.p. 168 °C (reference 168–170 °C)Citation36. IR: (KBr) νmax (cm−1): 3350 (amide N–H), 3068 (aromatic C–H), 1692 (ketone C=O), 1664 (amide C=O), 1369–1180 (C–N) and 841 (1,4-disubstituted benzene).
4-(2-Bromoacetyl)acetanilide (2)
Compound 1 (0.04 mol, 7.08 g) and HBr (0.5 mL) were dissolved in acetic acid (30 mL) and bromine (0.044 mol, 2.27 mL) was added dropwise at room temperature. After completion of the addition of bromine, reaction mixture was allowed to additional stirring for 1 h and then poured into ice-water (100 mL). Precipitated product was filtered, washed with water, dried and then recrystallized from ethanol. Yield: 86%; m.p. 188 °C (reference 185–187 °C)Citation37. IR: (KBr) νmax (cm−1): 3363 (amide N–H), 3062 (aromatic C–H), 1698 (ketone C=O), 1665 (amide C=O), 1378–1196 (C–N) and 848 (1,4-disubstituted benzene).
4-(2-Methyl-4-thiazolyl)acetanilide (3)
Method A: Compound 2 (0.03 mol, 7.68 g) and thioacetamide (0.03 mol, 2.25 g) in ethanol (100 mL) were stirred at room temperature for 48 h. The precipitated product was filtered, dried and recrystallized from ethanol. Yield: 78%; m.p. 144 °C (reference 141–142 °C)Citation38. IR: (KBr) νmax (cm−1): 3365 (amide N–H), 3062 (aromatic C–H), 1665 (amide C=O), 1367–1211 (C–N) and 843 (1,4-disubstituted benzene).
Method B: Compound 2 (0.003 mol, 0.77 g) and thioacetamide (0.03 mol, 2.25 g) were dissolved in ethanol (30 mL). This solution was transferred into a beaker (150 mL), a bigger beaker (400 mL) was closed onto the reaction mixture, and put into a domestic microwave oven. Reaction mixture was left under microwave irradiation (180 W) 2 min intervals and ethanol (10 mL) was added before each microwave irradiations. Reaction was routinely controlled by thin-layer chromatography. After third microwave irradiation, the residue was recrystallized from ethanol. Yield: 89%; m.p. 144 °C (reference 141–142 °C)Citation38.
4-(2-Methyl-4-thiazolyl)aniline (4)
Compound 3 (0.025 mol, 5.8 g) was refluxed in 10% HCl (100 mL) for 1 h. The mixture was cooled down, poured into iced water (100 mL) and made basic with 10% NaOH solution. The precipitated product was filtered, dried and recrystallized from ethanol. Yield: 92%; m.p. 136 °C (reference m.p. 133–135 °C)Citation38. IR: (KBr) νmax (cm−1): 3365 (amine N–H), 3361 (amine N–H), 3062 (aromatic C–H), 1367–1211 (C–N) and 843 (1,4-disubstituted benzene).
2-Chloro-N-[4-(2-methyl-4-thiazolyl)phenyl]acetamide (5)
Chloroacethyl chloride (0.02 mol, 1.6 mL) was added dropwise over 15 min to a magnetically stirred solution of compound 4 (0.02 mol, 3.8 g) and triethylamine (0.02 mol, 2.8 mL) in dry THF (15 mL). After completion of reaction, the solvent was evaporated under reduced pressure. Water was added to wash the resulting solid and the mixture was filtered, dried and recrystallized from ethanol to give compound 5. Yield: 83%; m.p. 156 °C. IR: (KBr) νmax (cm−1): 3367 (amide N–H), 3053 (aromatic C–H), 1676 (amide C=O), 1605–1403 (C=C, C=N), 1369–1214 (C–N) and 838 (1,4-disubstituted benzene). 1H-NMR (500 MHz, dimethyl sulphoxide, DMSO-d6) δ (ppm): 2.69 (3H, s, CH3), 4.32 (2H, s, CO–CH2), 7.76 (2H, d, J = 8.15 Hz, Ar-H), 7.81 (1H, s, thiazole C5-H), 7.87 (d, 2H, J = 8.10 Hz, Ar-H) and 10.50 (s, 1H, N–H). For C12H11ClN2OS calculated: 54.03% C, 4.16% H and 10.50% N; found: 54.65% C, 4.15% H and 10.47% N. MS [M + 1]+: m/z 267.5.
General procedure for synthesis of 2-(substituted-sulfanyl)-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide derivatives (6a–n)
A mixture of compound 5 (0.001 mol, 0.27 g), the appropriate mercapto-(benz)azole derivative (0.001 mol) and K2CO3 (0.001 mol, 0.14 g) in acetone was refluxed for 2 h. The cooled mixture was filtered, dried and recrystallized from ethanol to afford target compounds 6a–n.
2-[(Thiazoline-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)pheny])acetamide (6a)
Yield 81%; m.p. 105 °C. IR (KBr) νmax (cm−1): 3252 (amide N–H), 3064 (aromatic C–H), 2978 (aliphatic C–H), 1661 (amide C=O), 1608–1412 (C=C and C=N) and 1335–981 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 3.48 (t, 2H, J = 7.30 Hz, thiazoline C5–H), 4.10 (s, 2H, S–CH2), 4.14 (t, 2H, J = 7.30 Hz, thiazoline C4–H), 7.63 (d, 2H, J = 7.50 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.87 (d, 2H, J = 7.50 Hz, Ar-H) and 10.34 (s, 1H, N–H). For C15H15N3OS3 calculated: 51.55% C, 4.33% H and 12.02% N; found: 51.56% C, 4.35% H, and 12.01% N. MS [M + 1]+: m/z 350.
2-[(1-Methylimidazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6b)
Yield 85%; m.p. 193 °C. IR (KBr) νmax (cm−1): 3281 (amide N–H), 3024 (aromatic C–H), 2945 (aliphatic C–H), 1670 (amide C=O), 1597–1399 (C=C and C=N) and 1298–1021 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 3.61 (s, 3H, N–CH3), 3.89 (s, 2H, S–CH2), 6.98 (d, 1H, J = 7.17 Hz, imidazole C4–H), 7.26 (d, 1H, J = 7.14 Hz, imidazole C5–H), 7.61 (d, 2H, J = 8.30 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.87 (d, 2H, J = 8.10 Hz, Ar-H) and 10.47 (s, 1H, N–H). For C16H16N4OS2 calculated: 55.79% C, 4.68% H and 16.27% N; found: 55.74% C, 4.69% H and 16.25% N. MS [M + 1]+: m/z 345.
2-[(4-Methyl-4H-1,2,4-triazole-3-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6c)
Yield 82%; m.p. 187 °C. IR (KBr) νmax (cm−1): 3276 (amide N–H), 3028 (aromatic C–H), 2990 (aliphatic C–H), 1672 (amide C=O), 1569–1402 (C=C and C=N) and 1315–1007 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 3.61 (s, 3H, N–CH3), 4.07 (s, 2H, S–CH2), 7.61 (d, 2H, J = 8.50 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.88 (d, 2H, J = 8.20 Hz, Ar-H), 8.56 (s, 1H, triazole C5–H) and 10.47 (s, 1H, N–H). For C15H15N5OS2 calculated: 52.15% C, 4.38% H and 20.27% N; found: 52.16% C, 4.40% H and 20.29% N. MS [M + 1]+: m/z 346.
2-[(5-Methyl-1,3,4-thiadiazole-3-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6d)
Yield 91%; m.p. 186 °C. IR (KBr) νmax (cm−1): 3269 (amide N–H), 3029 (aromatic C–H), 2964 (aliphatic C–H), 1686 (amide C=O), 1566–1410 (C=C and C=N) and 1324–1012 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.68 (s, 3H, CH3), 2.71 (s, 3H, CH3), 4.29 (s, 2H, S–CH2), 7.64 (d, 2H, J = 8.60 Hz, Ar-H), 7.82 (s, 1H, thiazole C5–H), 7.89 (d, 2H, J = 8.30 Hz, Ar-H) and 10.47 (s, 1H, N–H). For C15H14N4OS3 calculated: 49.70% C, 3.89% H and 15.46% N; found: 49.71% C, 3.86% H and 14.48% N. MS [M + 1]+: m/z 363.
2-[(1H-Benzimidazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6e)
Yield 76%; m.p. 241 °C. IR (KBr) νmax (cm−1): 3274 (amide N–H), 3012 (aromatic C–H), 2973 (aliphatic C–H), 1681 (amide C=O), 1549–1399 (C=C and C=N) and 1268–984 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 4.30 (s, 2H, S–CH2), 7.46 (brs, 2H, Ar-H), 7.12–7.15 (m, 2H, Ar-H), 7.65 (d, 2H, J = 8.15 Hz, Ar-H), 7.81 (s, 1H, thiazole C5-H), 7.89 (d, 2H, J = 8.32 Hz, Ar-H), 10.62 (s, 1H, N–H) and 12.67 (s, 1H, benzimidazole N–H). For C19H16N4OS2 calculated: 59.98% C, 4.24% H and 14.73% N; found: 59.96% C, 4.23% H and 14.75% N. MS [M + 1]+: m/z 381.
2-[(5-Chloro-1H-benzimidazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6f)
Yield 86%; m.p. 233 °C. IR (KBr) νmax (cm−1): 3269 (amide N–H), 3053 (aromatic C–H), 2953 (aliphatic C–H), 1682 (amide C=O), 1576–1395 (C=C and C=N) and 1305–996 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.71 (s, 3H, CH3), 4.31 (s, 2H S-CH2), 7.16 (dd, 1H, J = 8.35 Hz and 7.92 Hz, Ar-H), 7.46 (d, 1H, J = 7.96 Hz, Ar-H), 7.52 (s, 1H, Ar-H), 7.65 (d, 2H, J = 8.20 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.89 (d, 2H, J = 8.35 Hz, Ar-H), 10.56 (s, 1H, N–H) and 12.86 (s, 1H, benzimidazole N–H). For C19H15ClN4OS2 calculated: 55.00% C, 3.64% H and 13.50% N; found: 55.04% C, 3.63% H and 13.51% N. MS [M + 1]+: m/z 415.5.
2-[(5-Methyl-1H-benzimidazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6g)
Yield 90%; m.p. 106 °C. IR (KBr) νmax (cm−1): 3268 (amide N–H), 3042 (aromatic C–H), 2969 (aliphatic C–H), 1686 (amide C=O), 1546–1419 (C=C and C=N) and 1279–1022 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.39 (s, 3H, CH3), 2.70 (s, 3H, CH3), 4.27 (s, 2H, S–CH2), 6.96 (d, 1H, J = 8.50 Hz, Ar-H), 7.25 (s, 1H, Ar-H), 7.35 (d, 1H, J = 8.15 Hz, Ar-H), 7.65 (d, 2H, J = 8.55 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.87 (d, 2H, J = 8.20 Hz, Ar-H), 10.59 (s, 1H, N–H) and 12.54 (s, 1H, benzimidazole N–H). For C20H18N4OS2 calculated: 60.89% C, 4.60% H and 14.20% N; found: 60.87% C, 4.63% H and 14.22% N. MS [M + 1]+: m/z 395.
2-[(5-Nitro-1H-benzimidazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6h)
Yield 78%; m.p. 207 °C (dekomp.). IR (KBr) νmax (cm−1): 3296 (amide N–H), 3042 (aromatic C–H), 2976 (aliphatic C–H), 1689 (amide C=O), 1604–1412 (C=C and C=N) and 1330–1064 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 4.38 (s, 2H, S–CH2), 7.62 (d, 1H, J = 7.96 Hz, Ar-H), 7.66 (d, 2H, J = 8.20 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.89 (d, 2H, J = 8.35 Hz, Ar-H), 8.07 (dd, 1H, J = 8.10 Hz and 7.97 Hz, Ar-H), 8.32 (s, 1H, Ar-H), 10.63 (s, 1H, N–H) and 12.63 (s, 1H, benzimidazole N–H). For C19H15N5O3S2 calculated: 53.63% C, 3.55% H and 16.46% N; found: 53.61% C, 3.57% H and 16.45% N. MS [M + 1]+: m/z 426.
2-[(Benzoxazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6i)
Yield 83%; m.p. 148 °C. IR (KBr) νmax (cm−1): 3269 (amide N–H), 3037 (aromatic C–H), 2989 (aliphatic C–H), 1667 (amide C=O), 1602–1450 (C=C and C=N) and 1301–1055 (C–N and C–O). 1H-NMR (500 MHz, DMSO-d6): 2.71 (s, 3H, CH3), 4.41 (s, 2H, S–CH2), 7.33–7.37 (m, 2H, Ar-H), 7.62–7.67 (m, 4H, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.90 (d, 2H, J = 8.35 Hz, Ar-H) and 10.56 (s, 1H, N–H). For C19H15N3O2S2 calculated: 59.82% C, 3.96% H and 11.02% N; found: 59.82% C, 3.98% H and 11.05% N. MS [M + 1]+: m/z 382.
2-[(5-Chlorobenzoxazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6j)
Yield 82%; m.p. 129 °C. IR (KBr) νmax (cm−1): 3276 (amide N–H), 3053 (aromatic C–H), 2949 (aliphatic C–H), 1676 (amide C=O), 1598–1422 (C=C and C=N) and 1310–1005 (C–N and C–O). 1H-NMR (500 MHz, DMSO-d6): 2.71 (s, 3H, CH3), 4.39 (s, 2H, S–CH2), 7.38 (d, 1H, J = 8.55 Hz, Ar-H), 7.65 (d, 2H, J = 8.80 Hz, Ar-H), 7.72–7.75 (m, 2H, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.89 (d, 2H, J = 8.40 Hz, Ar-H) and 10.59 (s, 1H, N–H). For C19H14ClN3O2S2 calculated: 54.87% C, 3.39% H and 10.10% N; found: 54.89% C, 3.40% H and 10.13% N. MS [M + 1]+: m/z 416.5.
2-[(5-Methylbenzoxazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6k)
Yield 79%; m.p. 158 °C. IR (KBr) νmax (cm−1): 3281 (amide N–H), 3024 (aromatic C–H), 2976 (aliphatic C–H), 1678 (amide C=O), 1598–1409 (C=C and C=N) and 1299–1006 (C–N and C–O). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.40 (s, 3H, CH3), 2.70 (s, 3H, CH3), 4.39 (s, 2H, S–CH2), 7.13 (d, 1H, J = 8.45 Hz, Ar-H), 7.43 (s, 1H, Ar-H), 7.52 (d, 1H, J = 8.50 Hz, Ar-H), 7.65 (d, 2H, J = 8.30 Hz, Ar-H), 7.81 (s, 1H, thiazole C5–H), 7.89 (d, 2H, J = 8.35 Hz, Ar-H) and 10.54 (1H, s, N–H). For C20H17N3O2S2 calculated: 60.74% C, 4.33% H and 10.62% N; found: 60.73% C, 4.36% H and 10.61% N. MS [M + 1]+: m/z 396.
2-[(5-Nitrobenzoxazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6l)
Yield 84%; m.p. 218 °C. IR (KBr) νmax (cm−1): 3268 (amide N–H), 3046 (aromatic C–H), 2994 (aliphatic C–H), 1669 (amide C=O), 1596–1400 (C=C and C=N) and 1302–1015 (C–N and C–O). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 4.48 (s, 2H, S–CH2), 7.65 (d, 2H, J = 8.35 Hz, Ar-H), 7.82 (s, 1H, thiazole C5–H), 7.89–7.93 (m, 3H, Ar-H), 8.26 (dd, 1H, J = 8.25 Hz and J = 4.15 Hz, Ar-H), 8.48 (d, 1H, J = 4.60 Hz, Ar-H) and 10.54 (s, 1H, N–H). For C19H14N4O4S2 calculated: 53.51% C, 3.31% H and 13.14% N; found: 53.54% C, 3.35% H and 13.17% N. MS [M + 1]+: m/z 427.
2-[(Benzothiazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6m)
Yield 87%; m.p. 130 °C. IR (KBr) νmax (cm−1): 3277 (amide N–H), 3039 (aromatic C–H), 2975 (aliphatic C–H), 1676 (amide C=O), 1569–1408 (C=C and C=N) and 1295–1011 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.70 (s, 3H, CH3), 4.42 (s, 2H, S–CH2), 7.37 (t, 1H, J = 7.56 Hz, Ar-H), 7.47 (t, 1H, J = 7.52 Hz, Ar-H), 7.66 (d, 2H, J = 7.82, Ar-H), 7.82 (s, 1H, thiazole C5–H), 7.84 (d, 1H, J = 8.50 Hz, Ar-H), 7.90 (d, 2H, J = 8.35 Hz, Ar-H), 8.03 (d, 1H, J = 8.32 Hz, Ar-H) and 10.54 (s, 1H, N–H). For C19H15N3OS3 calculated: 57.40% C, 3.80% H and 10.57% N; found: 57.42% C, 3.83% H and 10.54% N. MS [M + 1]+: m/z 427.
2-[(5-Chlorobenzothiazole-2-yl)sulfanyl]-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide (6n)
Yield 75%; m.p. 195 °C. IR (KBr) νmax (cm−1): 3294 (amide N–H), 3015 (aromatic C–H), 2986 (aliphatic C–H), 1659 (amide C=O), 1593–1402 (C=C and C=N) and 1296–1023 (C–N). 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 2.71 (s, 3H, CH3), 4.44 (s, 2H, S–CH2), 7.42 (dd, 1H, J = 8.30 Hz and 4.55 Hz, Ar-H), 7.67 (d, 2H, J = 8.40 Hz, Ar-H), 7.82 (s, 1H, thiazole C5–H), 7.90–7.91 (m, 3H, Ar-H), 8.07 (s, 1H, Ar-H) and 10.54 (s, 1H, N–H). For C19H14ClN3OS3 calculated: 52.83% C, 3.27% H and 9.73% N; found: 52.87% C, 3.29% H and 9.75% N. MS [M + 1]+: m/z 432.5.
Anticancer screening
Cell culture and drug treatment
C6 glioma cells were incubated in Dulbecco’s modified Eagle’s medium (Sigma, Deisenhofen, Germany) supplemented with 10% fetal calf serum (Gibco, Paisley, Scotland). A549 cells were incubated in 90% Roswell Park Memorial Institute medium(Roswell Park Memorial Institute, Buffalo, NY) supplemented with 10% fetal bovine serum (Gibco). All media were supplemented with 100 IU/mL penicillin–streptomycin (Gibco) and cells were incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Exponentially growing cells were plated at 2 × 104 cells/mL into 96-well microtiter tissue culture plates (Nunc, Roskilde, Denmark) and incubated for 24 h before the addition of the test compounds (the optimum cell number for cytotoxicity assays was determined in preliminary experiments). Stock solutions of the compounds were prepared in DMSO (Sigma-Aldrich) and further dilutions were made with a fresh culture medium (the concentration of DMSO in the final culture medium was <0.1% which had no effect on the cell viability).
MTT assay for cytotoxicity of compounds
The level of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) reduction was quantified as previously described in the literatureCitation39 with small modifications. After 24 h of preincubation, the tested compounds (6a–n) were added to give final concentration in the range of 6–200 µg/mL and the cells were incubated for 24 h. At the end of this period, MTT was added to final concentration of 0.5 mg/mL and the cells were incubated for 4 h at 37 °C. After removal of medium, the formazan crystals formed by MTT metabolism were solubilized by the addition of 200 µL DMSO to each well and absorbance was read at 540 nm with a microplate spectrophotometer (Bio-Tek, Winooski, VT). Every concentration was repeated in three wells and IC50 values (µM) were defined as the compound concentrations that reduced absorbance to 50% of control values.
Analysis of DNA synthesis
Analysis of DNA synthesis was measured by, BrdU (bromodeoxyuridine) cell proliferation colorimetric kit (Roche, Mannheim, Germany). First, cells were seeded into 96-well flat-bottomed microtiter plates at a density of 2 × 103. The tumor cells cultured in the presence of various concentrations of compounds 6b, 6c, 6e, 6f, 6g and mitoxantrone. Microtiter plates were incubated at 37 °C in a 5% CO2/95% air humidified atmosphere for 24 and 48 h. At the end of each day, the cells were labeled with 10 µL BrdU solution for 2 h and then fixed. Anti-BrdU-POD (100 µL) was added and incubated for 90 min. Finally, wells were washed with BPS and cells were incubated with substrate. Absorbance of the samples was measured with an ELX808-IU Bio-Tek apparatus at 492 nm. All experiments were repeated twice. For all compound doses, duplicate wells were used.
Spectrofluorometric analysis of caspase-3 activation
Caspase-3 activation was analyzed by Spectrofluorometric Caspase-3 Assay kit (BD Pharmingen, Franklin Lakes, NJ). Kit was designed to measure caspase-3 or DEVD-cleaving activity, an early marker of cells undergoing apoptosisCitation40. First, cells (1 × 106 cells/mL) were washed with phosphate-buffered saline (PBS), resuspended in cold cell lysis buffer and incubated for 30 min on ice. After 24 h incubation period with various concentrations of compounds 6f, 6g and mitoxantrone, cell lysates were prepared. For each reaction, 5 µL of reconstituted Ac-DEVD-AMC (synthetic tetrapeptide fluorogenic substrate for Caspase-3 activity) was added to a well containing 0.2 mL of 1 × HEPES buffer. Cell lysate (20 µL) was added to each well/reaction. Reaction mixtures were incubated for 1 h at 37 °C. The amount of AMC liberated from Ac-DEVD-AMC was measured using microplate reader (Perkin Elmer/Victor/X3) with an excitation wavelength of 380nm and an emission wavelength of 460 nm. Apoptotic cell lysates containing active Caspase-3 yielded a considerable emission as compared to controls. Also non-apoptotic control cell lysates AMC emission was accepted as 100% and other cell lysates emissions were measured according to control cells emissions. All experiments were repeated twice. For all doses, duplicate wells were used.
Acridine orange/ethidium bromide staining methods
C6 glioma cells were cultured at 1 × 105 cells/well onto six wells plate until 50% confluent. The medium was replaced with a fresh medium containing different concentrations of compounds 6f and 6g and mitoxantrone. The cells, without treatment, were used as negative controls. Cells were then incubated for 24 h in humidified atmosphere at 37 °C in CO2 5%. The medium was removed and cells were washed thrice with PBS. Cells were fixed with 70% ethanol and incubated for 5 min and then washed with PBS. A mixture of ethidium bromide:acridin orange (1:1) (100 µg/mL) was added and incubated for 5 min. Cells were washed with PBS, they were removed and covered on the object glass. Then assessment was carried out under fluorescence microscope (Olympus, Tokyo, Japan)Citation41. Acridine orange/ethidium bromide combination was used to visualize cells with aberrant chromatin organization. Acridine orange was used to visualize the number of cells which has undergone apoptosis.
Statistical analyses
SPSS (Chicago, IL) for Windows 15.0 was used for statistical analysis. Data were expressed as mean ± SD. Comparisons were performed by one-way analysis of variance test for normally distributed continuous variables and post hoc analyses of group differences were expressed by the Tukey test.
Results and discussions
Chemistry
Present study was undertaken to synthesize some novel thiazole–(benz)azole derivatives and investigate their probable anticancer effects. Synthetic way for compounds 6a–n was outlined in . Initially, 4′-aminoacetophenone was acetylated to prevent the occurrence of probable impurities, which may appear due to free amino group in further reaction steps, and 4′-acetaminoacetophenone (1) was obtained. Bromination of compound 1 in acetic acid gave 4-(2-bromoacetyl)acetanilide (2), which was then reacted with thioacetamide to afford 4-(2-methyl-4-thiazolyl)acetanilide (3) via the Hantzsch reaction. Hydrolysis of acetamide group of compound 3 in 10% HCl solution provided 4-(2-methyl-4-thiazolyl)aniline (4). Compound 4 in THF was acetylated with chloroacetyl chloride in the presence of triethylamine (5). Finally, compound 5 was reacted with corresponding (benz)azole-thiol derivatives under basic conditions to achieve the target compounds (6a–n). Hantzsch reaction was also carried out by microwaves irradiation (180 W, 2 min intervals; total 6 min) in a kitchen-type microwave oven. It was observed that yield (87%) of compound 3 was notably increased along with short reaction time.
Scheme 1. The synthetic protocol of the compounds (6a–n). Reagents: (i) acetyl chloride, TEA, THF, 0–5 °C; (ii) Br2, AcOH; (iii) thioacetamide, EtOH, room temperature or MWI; (iv) 10% HCl, EtOH, reflux; (v) chloroacetyl chloride, TEA, THF, room temperature; and (vi) appropriate thiol-benz(azole), K2CO3, acetone, reflux.
All the intermediates were analyzed in IR spectra to confirm each reaction step. During the analysis of compound 1, two significant carbonyl bands at 1692 and 1664 cm−1 were recorded for ketone and amide groups, respectively. Compound 2 gave same bands at 1698 and 1665 cm−1, whereas in the IR spectra of compound 3 disappearance of stretching band about 1700 cm−1 belonging to ketone carbonyl was an evidence for ring closure. Similarly, disappearance of amide carbonyl stretching bond at about 1660–1670 cm−1 showed that compound 4 was successfully obtained via deacetylation. On the other hand, for compound 5 such stretching bond was observed again at 1676 cm−1 due to the acetylation reaction of compound 4 with chloroacetyl chloride.
The structures of target compounds were assigned on the basis of spectroscopic and analytical data. Characteristic stretching absorptions for C=O and N–H bonds were observed at 1659–1689 and 3252–3296 cm−1, respectively. The stretching absorption at about 1608–1395 and 1335–981 cm−1 was recorded for C=C, C=N double bonds and C–O, C–N bonds, respectively. Disappearance of stretching absorption for the S–H bond at about 2550 cm−1 was an evidence for the occurrence of target compounds. In the 1H-NMR spectra, methyl substituent at the second position of thiazole ring appeared at 2.70–2.71 as a singlet. Aromatic proton (C5–H) of thiazole ring gave peak at 7.81–7.82 ppm as a singlet. Aromatic protons of phenyl moiety at 4th position of thiazole observed at about 7.61 and 7.87 were double doublets. Acetamide (NHCOCH2) peaks were observed at 3.89–4.48 ppm as a singlet and at 10.34–10.62 ppm as a broad singlet for methylene (CH2) and amide (NHCO) protons, respectively. The other peaks belonging to aromatic and aliphatic protons of variable side chains were recorded at estimated areas. In the mass spectra, M + 1 peaks agreed well with the calculated molecular weight of the target compounds. Elemental analysis results for C, H and N elements were satisfactory within calculated values of the compounds.
Biochemistry
The synthesized compounds 6a–n were tested against rat glioma (C6) and human lung (A549) cancer cell lines. Anticancer activity screening was carried out at four steps. Initially, cytotoxic potential of compounds on carcinogenic cells was tested by the MTT method. Mitoxantrone, a cytotoxic anticancer agent, was used as appositive control. Second, the compounds, which displayed comparable cytotoxicity to mitoxantrone were selected and tested for their inhibitory potency on DNA synthesis of carcinogenic cells. In the third step, Caspase-3 activation test was performed in order to observe carcinogenic cells undergoing apoptosis. Finally, acridine orange/ethidium bromide staining method was applied to visualize cells which have undergone apoptosis.
Cytotoxicity (MTT) method
MTT, based on the ability of metabolically active cells to convert the pale yellow MTT dye to a spectrophotometrically quantifiable blue formazan product, is one of the most preferred cytotoxicity tests applied to investigate potential of new compounds against cancer cell linesCitation29. In such test, C6 and A549 cell lines were incubated with various concentrations (6–200 µg/mL) of 6a–n that are maintained via serial dilutions. After completion of incubation period (24 h), cytotoxic activity of the compounds was examined and IC50 values were calculated (). Cytotoxic agent mitoxantrone was used as a positive control.
Table 1. IC50 (µM) values of the compounds 6a–n on A549 and C6 cells.
As seen in , on both cell type, the compounds 6b, 6c and 6e–g were more cytotoxic than the other derivatives due to their IC50 values about 122.6 µM and below. A549 cell line found to be more sensitive to the synthesized compounds than C6 cell line. The cytotoxic effect of the compounds 6b, 6c and 6e–g on A549 cells was comparable to that of mitoxantrone, whereas control agent showed higher cytotoxicity on C6 cells than the synthesized compounds.
MTT test results demonstrate that there are important differences in the cytotoxic activity of compounds 6a–n. Thus, it can be mentioned that varying azole or benzazole side groups on the chemical structure cause significant changes in cytotoxicity. According to cytotoxicity test results, for the azole side groups it can be declared that, 1-methylimidazol-2-yl and 1-methyl-1,2,4-triazol-2-yl enhance the cytotoxic activity more than thiazolin-2-yl and 5-methyl-1,3,4-thiadiazol-2-yl. In varying benzazole side groups, benzimidazol-2-yl has a greater effect than benzoxazol-2-yl and benzothiazol-2-yl. In addition, electronic characters of the substituents on fifth position of benzimidazole have an essential impact on cytotoxic effect. The cytotoxic effect of compounds 6f and 6g, which carry electron-donating chloro and methyl substituents on benzimidazole ring, was at the same level with that of nonsubstituted benzimidazole-bearing compound 6e. On the other hand, in compound 6h substitution of benzimidazole with electron withdrawing nitro group reduced the cytotoxicity. This result suggests that, substitution of benzimidazole ring with an electron-donating group improves the cytotoxicity.
Analysis of DNA synthesis
This immune staining procedure depends on measuring the incorporation of BrdU into nuclear DNA in place of thymidine during the S-phase of the cell cycle using specific anti-BrdU antibodiesCitation31. Thus, such method provides a colorimetric measurement for DNA synthesis inhibition ratio of the carcinogenic cells.
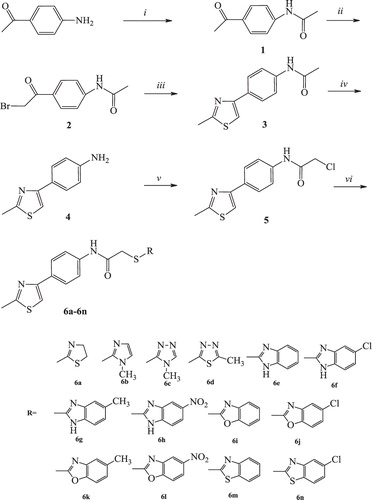
DNA synthesis of the carcinogenic cell lines was analyzed for compounds 6b, 6c and 6e–g, which indicated significant cytotoxic activity in the MTT test. Mitoxantrone was used as a positive control. For 24 and 48 h time periods, A549 and C6 cells were incubated with three different concentrations (IC50/2, IC50 and 2 × IC50) of the compounds. Tested compounds showed time- and dose-dependent inhibitory activity on the DNA synthesis of both cell lines ( and ). In all DNA synthesis inhibition results, C6 cells were the most resistant cells against all tested compounds. Compounds 6f and 6g had significant antiproliferative activity against both of cell lines.
Figure 1. DNA synthesis inhibitory activity of compounds 6b, 6c, 6e, 6f, 6g and mitoxantrone on A549 cells. Mean percent absorbance of untreated control cells was assumed 0% and three different concentrations (a = IC50/2, b = IC50, c = 2 × IC50) of test compounds and mitoxantrone were given. Data points represent means for two independent experiments ±SD of four independent wells. p < 0.05.
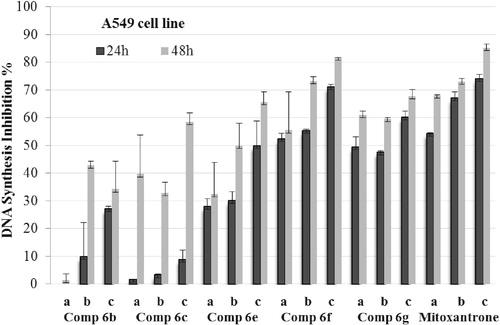
Figure 2. DNA synthesis inhibitory activity of compounds 6b, 6c, 6e, 6f, 6g and mitoxantrone on C6 cells. Mean percent absorbance of untreated control cells was assumed 0% and different concentrations (a = IC50/2, b = IC50, c = 2 × IC50) of test compounds and mitoxantrone were given. Data points represent means for two independent experiments ±SD of four independent wells. p < 0.05.
and demonstrate the effect of the selected compounds on DNA synthesis of A549 and C6 cells. As seen in the figures, 1-methylimidazole and 1-methyltriazole carrying compounds 6b and 6c have low inhibitory potency than benzimidazole carrying compounds 6e–g on the DNA synthesis of carcinogenic cells. It is clear that benzimidazole side provides more lipophilic character to compounds 6e–g, while imidazole and triazole sides in compounds 6b and 6c cannot facilitate similar lipophilicity. Thus, increasing lipophilic character in compounds 6e–g may enhance the DNA synthesis inhibition profile due to hydrophobic interaction with DNA. The inhibitory effect of the compounds 6f and 6g on DNA synthesis is higher than that of compound 6e. Such result refers that chloro or methyl substitution of benzimidazole side contributes the inhibition of DNA synthesis. Due to their comparable inhibitory potential to reference drug, compounds 6f and 6g were selected for further assays, which were performed to observe carcinogenic cells undergoing apoptosis.
Analysis of caspase-3 activation
The understanding of the cellular signaling processes leading to programmed cell death, called as apoptosis, is of utmost importance in cancer because damaged cells that do not enter the apoptotic pathway may proliferate unchecked and become a cancerous cell mass. Thus, letting tumor cells to apoptosis is a requirement for an anticancer agent. It is generally accepted that cytotoxic drugs eliminate malignant cells by inducing apoptosisCitation32,Citation33. The activation of caspases, a family of important signaling molecules with various tasks depending on subtype and organ, is a marker for apoptosisCitation34. The detection of activated caspase-3, the main effector caspase in the apoptotic enzyme cascade, by using fluorogenic substrates is a method commonly employed to observe the apoptosis levelCitation35.
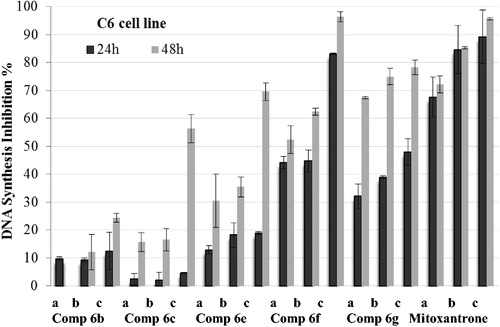
The compounds 6f and 6g, which displayed notable anticancer profile in both MTT and analysis of DNA synthesis assays, were tested for their caspase-3 activation potencies. Mitoxantrone was used as a control agent. A549 and C6 cells were maintained in cultures for 24 h and then exposed to Ac-DEVD-amc (1.0 mM) 30 min before exposure to three different concentrations (IC50/2 and IC50) of the compounds.
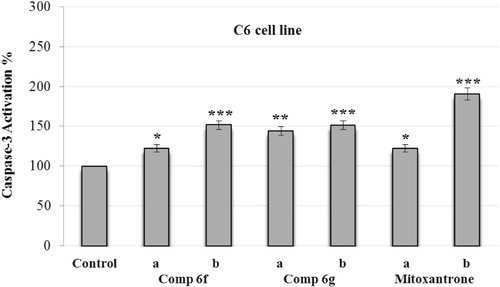
and present the caspase-3 activation percent of A549 and C6 cells that are induced by compounds 6f and 6g at different concentrations (IC50/2 and IC50). Synthesized compounds and control agent induced the caspase-3 activation in both cell lines. However, activation of caspase-3 seemed to be stronger on A549 cell line. When compared with control, compounds 6f and 6g and mitoxantrone caused 2.5-fold higher caspase-3 activity induction at IC50 concentrations. Besides, induction potent of compound 6g on caspase-3 activation was greater than both compound 6f and reference drug. These findings suggest that, as well as reference mitoxantrone, compounds 6f and 6g have potencies to direct tumor cells to apoptotic pathway, which is a precondition of anticancer action.
Figure 3. Effect of Ac-DEVD-amc on the activity of caspase-3 induced by compounds 6f, 6g and mitoxantrone in A549 cell line. A549 cells were maintained in cultures for 24 h and then exposed to Ac-DEVD-amc (1.0 mM) 30 min before exposure to two different concentrations (a = IC50/2 and b = IC50) of compounds 6f, 6g and mitoxantrone. Values represent mean ± SD from duplicate samples for each experiment. Significantly different from respective control cells: ***p < 0.001.
Figure 4. Effect of Ac-DEVD-amc on the activity of caspase-3 induced by compounds 6f, 6g and mitoxantrone in C6 cell line. C6 cells were maintained in cultures for 24 h and then exposed to Ac-DEVD-amc (1.0 mM) 30 min before exposure to two different concentrations (a = IC50/2 and b = IC50) of compounds 6f, 6g and mitoxantrone. Values represent mean ± SD from duplicate samples for each experiment. Significantly different from respective control cells: *p < 0.05, ** p < 0.01 and ***p < 0.001.
Acridine orange/ethidium bromide staining method
This method facilitates to visualize the changes in nuclear morphology induced by test compounds and thus observation of apoptotic cells becomes easier. For 24 h time period, A549 cells were treated with the compounds 6f and 6g, which indicated good results in prior assays. After washing with PBS, the cells were stained with a mixture of acridine orange–ethidium bromide solutions. The cells were viewed under an inverted fluorescent microscope and photographed.
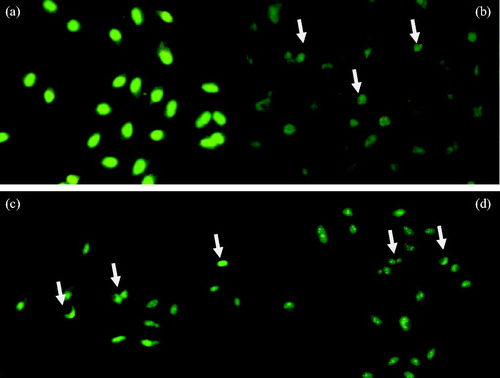
While the untreated A549 cells (control) were well spread with flattened morphology, apoptotic cells could be noticed in those treated with compounds 6f, 6g and mitoxantrone for 24 h (). Similarly, the nuclei were of the normal size (examined by staining the cells with ethidium bromide and acridine orange) in untreated cells, whereas treatment with IC50 of compounds 6f, 6g, and reference agent for 24 h resulted in condensation of nuclei (). Thus, the cancer cells treated with compounds 6f and 6g form apoptotic cells and exhibit nuclear condensation features that are characteristic of apoptosis. As a result, it can be declared that compounds 6f and 6g expedite the process of programmed cancer cell death.
Figure 5. Cellular and nuclear morphological changes of A549 cells following exposure to IC50 of compounds 6f, 6g and mitoxantrone for 24 h: (a) A549 control cells, (b) IC50 of mitoxantrone, (c) IC50 of compound 6f and (d) IC50 of compound 6g. Cells were distinguished according to the fluorescence emission and the morphological aspect of chromatin condensation in the stained nuclei. Viable cells have uniform bright green nuclei with organized structure. Early apoptotic cells have green nuclei, but perinuclear chromatin condensation is visible as bright green patches or fragments. White arrows indicate apoptotic cells. Colored version of the figure can be sought in the online version of this manuscript.
Conclusions
In the present study, novel N-[4-(2-methylthiazol-4-yl)phenyl]acetamide derivatives were synthesized and evaluated for their anticancer activity. Consecutive anticancer tests including cytotoxicity, analysis of DNA synthesis and induction of apoptosis revealed that compounds 6f and 6g, which carry benzimidazole and 5-chlorobenzimidazole side groups, were the most potent members in the series. In conclusion, result of this work has encouraged us to synthesize novel compounds that bear N-phenyl-2-(5-substitutedbenzimidazol-2-yl)thio acetamide substructure and investigate their anticancer profiles in further studies.
Declaration of interest
The authors report no conflicts of interest.
Related Research Data
References
- Liott LA, Steeg PS, Steller-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 1991;64:327–36
- Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993;73:161–5
- Dudkin VY, Wang, C, Arrington KL, et al. Pyridyl aminothiazoles as potent Chk1 inhibitors: optimization of cellular activity. Bioorg Med Chem 2012;12:2613–9
- Karikas GA, Schulpis KH, Reclos G, Kokotos G. Measurement of molecular interaction of aspartame and its metabolites with DNA. Clin Biochem 1998;31:405–7
- Filosa R, Pedut A, Caprariis P, et al. Synthesis and antiproliferative properties of N3/8-disubstituted 3,8-diazabicyclo[3.2.1]octane analogues of 3,8-bis[2-(3,4,5-trimethoxyphenyl)pyridin-4-yl]methyl-piperazine. Eur J Med Chem 2007;42:293–306
- Earle MF, Glazer RI. Activity and metabolism of 2-β-d-Ribofuranosylthiazole-4 carboxamide in human lymphoid tumor cells in culture. Cancer Res 1983;43:133–7
- Tricot GJ, Jayaram NH, Weber G, Hoffman RI. Tiazofurin: biological effects and clinical uses. Int J Cell Cloning 1990;8:161–70
- Tricot GJ, Jayaram NH, Lapis E, et al. Biochemically directed therapy of leukemia with tiazofurin, a selective blocker of inosine 5'-phosphate dehydrogenase activity. Cancer Res 1989;49:3696–701
- Zimmer C, Wahnert U. Nonintercalating DNA-binding ligands: specificity of the interaction and their use as tools in biophysical, biochemical and biological investigations of the genetic material. Prog Biophys Mol Biol 1986;47:31–112
- Umezawa H, Suhara Y, Taketa T, Maeda K. Purification of bleomycins. J Antibiot 1966;19:210–5
- Umezawa H, Takeuchi S, Hori T, et al. Studies on the mechanism of antitumor effect of bleomycin of squamous cell carcinoma. J Antibiot 1972;25:409–20
- Plouvier B, Houssin R, Baily C, Henichart J. Synthesis and DNA-binding study of a thiazole-containing analog of netropsin. J Heterocyclic Chem 1989;26:1643–7
- Lu Y, Li CM., Wang Z, et al. Discovery of 4-substitutedmethoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation and structure–activity relationships. J Med Chem 2009;52:1701–11
- Garuti L, Roberti M, Pession A, et al. Synthesis and antiproliferative activity of some thiazolylbenzimidazole-4,7-diones. Bioorg Med Chem Lett 2001;11:3147–9
- Bolos CA, Papazisi KT, Kortsaris AH, Voyatzi S. Antiproliferative activity of mixed-ligand dien-Cu(II) complexes with thiazole, thiazoline and imidazole derivatives. J Inorg Biochem 2002;88:25–36
- Yalowitz JA, Pankiewicz K, Patterson SE, Jayaram HN. Cytotoxicity and cellular differentiation activity of methylenebis(phosphonate) analogs of tiazofurin and mycophenolic acid adenine dinucleotide in human cancer cell lines. Cancer Lett 2002;181:31–8
- Li Z, Yang Q, Qian X. Novel thiazonaphthalimides as efficient antitumor and DNA photocleaving agents: effects of intercalation, side chains, and substituent groups. Bioorg Med Chem 2005;13:4864–70
- Cejudo R, Alzuet G, Gonzalez-Alvarez M, Garcia-Gimenez JL. DNA cleavage reaction induced by dimeric copper(II) complexes of N-substituted thiazole sulphonamides. J Inorg Biochem 2006;100:70–9
- Aliabadi A, Shamsa F, Ostad SN, et al. Synthesis and biological evaluation of 2-phenylthiazole-4-carboxamide derivatives as anticancer agents. Eur J Med Chem 2010;45:5384–9
- Fallah-Tafti A, Foroumadi A, Tiwari R, et al. Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities. Eur J Med Chem 2011;46:4853–8
- Gan L, Fang B, Zhou C. Synthesis of azole-containing piperazine derivatives and evaluation of their antibacterial, antifungal and cytotoxic activities. Bull Korean Chem Soc 2010;31:3684–92
- Rodriguez RJ, Acosta DJ. Comparison of ketoconazole- and fluconazole-induced hepatotoxicity in a primary culture system of rat hepatocytes. Toxicology 1995;96:83–92
- Boiani M, Gonzalez M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini-Rev Med Chem 2005;5:409–24
- Ramla MM, Omar MA, Tokuda H, El-Diwani H. Synthesis and inhibitory activity of new benzimidazole derivatives against Burkitt’s lymphoma promotion. Bioorg Med Chem 2007;15:6489–96
- Mann J, Baron A, Opoku-Boahen Y, et al. A new class of symmetric bis-benzimidazole-based DNA minor groove binding agents. J Med Chem 2001;44:138–44
- Ören İ, Temiz Ö, Yalcın İ, et al. Synthesis and antimicrobial activity of some novel 2,5- and/or 6-substituted benzoxazole and benzimidazole derivatives. Eur J Pharm Sci 1999;7:153–60
- Hisano T, Ichikawa M, Tsumoto K, Tasaki M. Synthesis of benzoxazoles, benzothiazoles and benzimidazoles and evaluation of their antifungal, insecticidal and herbicidal activities. Chem Pharm Bull 1982;30:2996–3004
- Nayyar A, Jain R. Recent advances in new structural classes of anti-tuberculosis agents. Curr Med Chem 2005;12:1873–86
- Jimonet P, Audiau F, Barreau M, et al. Riluzole series. Synthesis and in vivo “antiglutamate” activity of 6-substituted-2-benzothiazolamines and 3-substituted-2-imino-benzothiazolines. J Med Chem 1999;42:2828–43
- Wood PM, Lawrence LW, Thomas MP, et al. Aromatase and dual aromatase-steroid sulfatase inhibitors from the Letrozole and Vorozole templates. Chem Med Chem 2011;6:1423–38
- Trigg ME, Flanigan-Minnick A. Mechanisms of action of commonly used drugs to treat cancer. Commun Oncol 2011;8:358–69
- Ojha H, Murari BM, Anand S, et al. Interaction of DNA minor groove binder Hoechst 33258 with bovine serum albumin. Chem Pharm Bull 2009;57:481–6
- Kumar D, Jacob MR, Reynolds MB, Kerwina SM. Synthesis and evaluation of anticancer benzoxazoles and benzimidazoles related to UK-1. Bioorg Med Chem 2002;10:3997–4004
- Fichtner I, Monks A, Hose C, et al. The experimental antitumor agents Phortress and Doxorubicin are equiactive against human-derived breast carcinoma xenograft models. Breast Canc Res Treat 2004;87:97–107
- Özkay Y, Işıkdağ İ, İncesu Z, Akalın G. Synthesis of 2-substituted-N-[4-(1-methyl-4,5-diphenyl-1H-imidazole-2-yl)phenyl]acetamide derivatives and evaluation of their anticancer activity. Eur J Med Chem 2010;45:3320–8
- Peet NP, Sunder S, Barbuch RJ, et al. Reinvestigation of a 5H-dibenzo[d,h][1,3,6]triazonine synthesis. J Het Chem 1989;26:1611–7
- Xing R, Pan L, Wen X, et al. A practical and efficient procedure for the α-bromination of arylethanones. J Chinese Pharm Sci 2010;19:400–2
- Yurchenko RI, Malitskaya VP. Syntheses based on p-azido-ω-haloacetophenones. Z Organich Khim 1977;1:1980–7
- Mossmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Wang GW, Klein JB, Kang YJ. Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome C release and caspase-3 activation in cardiomyocytes. J Pharmacol Exp Ther 2001;298:461–8
- Cotter TG, Martin SJ. Techniques in apoptosis (a user’s guide). London: Portland Press; 1996:107–20