
Abstract
Herein, we designed and synthesized certain anilinoquinazoline derivatives bearing bulky arylpyridinyl, arylpropenoyl and arylpyrazolyl moieties at the 4′ position of the anilinoquinazoline, as potential dual HER2/EGFR kinase inhibitors. A detailed molecular modeling study was performed by docking the synthesized compounds in the active site of the epidermal growth factor receptor (EGFR). The synthesized compounds were further tested for their inhibitory activity on EGFR and HER2 tyrosine kinases. The aryl 2-imino-1,2-dihydropyridine derivatives 5d and 5e displayed the most potent inhibitory activity on EGFR with IC50 equal to 2.09 and 1.94 μM, respectively, and with IC50 equal to 3.98 and 1.04 μM on HER2, respectively. Furthermore, the anti-proliferative activity of these most active compounds on MDA-MB-231 breast cancer cell lines, known to overexpress EGFR, showed an IC50 range of 2.4 and 2.5 μM, respectively.
Introduction
The ErbB or epidermal growth factor (EGF) receptors are members of subclass 1 of the receptor tyrosine kinase (RTK) superfamily. There are four ErbB receptor family members: ErbB1 (EGFR, HER1), ErbB2 (HER2/neu), ErbB3 (HER3) and ErbB4 (HER4). The receptors are situated at the cell membrane and have an extracellular ligand-binding region, a transmembrane region and a cytoplasmic tyrosine-kinase domain. Ligand binding to the receptors results in receptor homo- and heterodimerization, activation of the intrinsic kinase domain and phosphorylation of specific tyrosine residues within the cytoplasmic tail. Proteins dock on these phosphorylated residues, leading to the activation of a variety of intracellular signaling pathways that promote cell growth, proliferation, differentiation and migrationCitation1. ErbB2 is the preferred dimerization partner for all the other ErbB receptors. ErbB receptors undergo various types of alteration in human tumors including gene amplification, receptor overexpression, activating mutations, overexpression of receptor ligands and/or loss of negative regulatory controls. The most robust example is that of amplification of HER2/neu in 25%–30% of breast cancers and is associated with a statistically significant shortening in disease-free and overall survivalCitation2.
Two predominant approaches have been developed for blocking the up-regulated HER signal pathway. One approach utilizes the anti-HER monoclonal antibody, which blocks the extracellular ligand-binding region of the receptor, thereby interfering with its activation and modulating the resultant intracellular signal cascade. The other approach for blocking the up-regulated HER signal pathway involves the use of orally active small molecule TKIs, these compounds act via competing with ATP for binding at the catalytic domain of tyrosineCitation3. In recent years, 4-anilinoquinazolines have emerged as a versatile template for inhibition of a diverse range of receptor tyrosine kinases. Among them, small molecule EGFR kinase inhibitors such as Gefitinib (Iressa)Citation4 and Erlotinib (Tarceva)Citation5 were introduced for cancer treatment recently. Subsequent research aimed at further exploration of the structure–activity relationship (SAR) of this novel template has led to the discovery of highly active compounds that target both EGFR and HER-2 kinases. for example. Lapatinib (Tykerb) which was approved in combination therapy for breast cancer patients already using CapecitabineCitation6.
In this study, we present a new sub-family of compounds containing 4-anilinoquinazoline scaffold as promising potent and selective EGFR, HER-2 inhibitors. Our strategy is directed toward designing a variety of ligands with diverse chemical properties hypothesizing that the potency of these molecules might be enhanced by adding alternative binding group such as 2-iminopyridine and 2H-pyrazole ring in the 4-position of aniline moiety (). This is a typical Structure Based Drug Design (SBDD) project. Starting from the “HITS” we have identified here, we are planning to investigate more substitution patterns utilizing the main pharmacophore as a core. We did not attempt to synthesize each and every derivative in the new series, but our main goal was to explore more substitution patterns not members. Such substitution pattern could target different regions of the ATP-binding site of the protein kinase domain to create differentially selective molecules. The design of our ligands was done based on previous SAR of 4-anilinoquinazolines and earlier work with quinazoline-based inhibitors of EGFR, which established that 6,7-dialkoxy substitution is compatible with good activity, and pivotal interactions between the receptor and the inhibitorsCitation7. In more recent approach, it was found that the dual inhibition of EGFR and ErbB-2 may offer increased activity over agents who target only one of these receptor kinases. After discovery of lapatinib, it was claimed that 4′ position of the aniline can tolerate a lot of bulky substituentsCitation8.
Figure 1. Structural modifications of the lead compounds (Gefitinib, Erlotinib and Lapatinib).
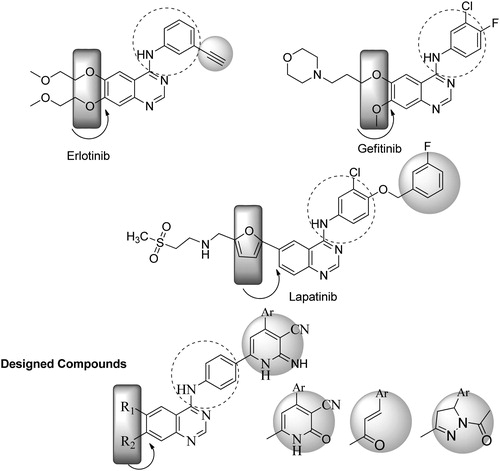
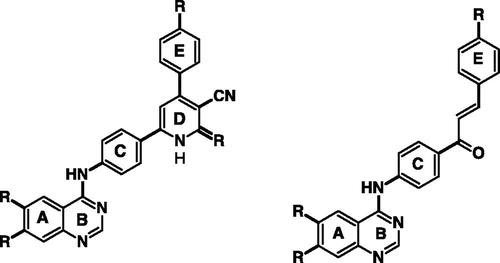
In this direction and in an approach to enhance the selectivity toward EGFR/ErbB-2, we introduced larger moieties at 4′ position of the aniline such as 4-aryl-2-imino pyridine moiety in a fashion similar to lapatinib, which binds in the ATP-binding cleft, so that the bulky group could be oriented deep in the back of the ATP binding site and makes predominantly hydrophobic interactions with the protein mimicking the 3-chloro-4-[(3-fluorobenzyl)oxy]aniline group of lapatinib (). The binding mode and docking energy of the designed compounds could be helpful tool for predicting their mechanism of antitumor activity. displays the two basic skeletons of the synthesized compounds.
Figure 2. The two basic skeletons of inhibitors in the new series.
Chemistry
Melting points were determined using a Stuart Scientific apparatus (Staffordshire, UK) and were uncorrected. IR spectra were recorded on a PerkinElmer FT-IR series (Waltham, MA) using KBr cell. 1H-NMR spectra were recorded on Brücker (Billerica, MA) 300 MHz in d scale. All the spectra were obtained on solutions in DMSO-d6 with TMS as internal standard; the values of the chemical shifts (d) are given in ppm, and coupling constants (J) are given in Hz. The electron impact (EI) mass spectra were recorded on Finnigan Mat SSQ 7000 (70 eV) mass spectrometer (Blue Lion Biotech, LLC, Snoqualmie, WA). Analytical thin layer chromatography (TLC) on silica gel plates containing UV indicator was employed routinely to follow the course of reactions and to check the purity of products. All reagents and solvents were purified and dried by standard techniques. Elemental microanalyses were performed at Microanalytical Center, Cairo University, and were within ±0.4% unless otherwise stated. The chemicals used in the synthesis are purchased from Aldrich (St. Louis, MO) and Merck (St. Louis, MO) and are of analytical grades. The preparation of compounds 2a,bCitation9, 3a,bCitation10 and 4a,bCitation9,Citation11 was performed according to reported procedures.
General procedures for the synthesis of 4-{4-[6-(4-Aryl-3-cyano-2-imino)-1,2-dihydropyridyl]phenylamino}quinazoline (5a–d) and 4-{4-[6-(4-Aryl-3-cyano-2-imino)-1,2-dihydropyridyl]phenylamino}-6,7-dimethoxy quinazolines (5e)
A mixture of the appropriate 4-anilinoquinazoline (4a–b) (0.01 mol), appropriate aldehyde (0.01 mol), malononitrile (0.01 mol) and ammonium acetate (0.08 mol) in absolute ethyl alcohol (30 ml) was heated under reflux, with stirring for 15–24 h. The reaction mixture was cooled and the formed precipitate was filtered, washed with cold ethyl alcohol, allowed to dry, and recrystallized from DMF/ethanol 1:2.
4-{4-[6-(4-(4-Bromophenyl)-3-cyano-2-imino)-1,2-dihydropyridyl]phenylamino}6,7-dimethoxyquinazoline (5e)
Dark green crystals, (yield 45%); m.p. 270 °C, IR (KBr, cm−1): 2214 (C≡N), 3321–3396–3479 (3NH), 2924–2846 (CH Aliphatic). 1H-NMR (300 MHz, DMSO): δ 8.40 (s, 1H, NH=CH–NH), 7.20 (s, 1H, CH=C–OCH3), 7.42 (s, 1H, CH=C–OCH3), 3.90 (s, 6H, OCH3–Ar), 9.10 (s, 1H, NH-Ar, D2O exchangeable), 6.85 (d, 2H, CH=CH–NH anilino), 8.05 (d, 2H, CH=CH–NH anilino), 10.00 (brs, 1H, NH, D2O exchangeable), 10.50 (brs, 1H, NH, D2O exchangeable), 7.90 (s, 1H, CH=C–Ar–H of pyridyl), 7.50 (d, 2H, CH=CH–Br), 7.60 (d, 2H, CH=CH–Br). MS (EI): 553 M+ 20%, 555.09 M+2 19.8%. Anal. for C28H21BrN6O2; C, 60.77%; H, 3.82%; N, 15.19%. Found: C, 61.03%; H, 3.97%; N, 15.41%.
General procedure for the preparation of (E)-3-Aryl-1-[4-(quinazolin-4-ylamino)phenyl]prop-2-en-1-one (7a–d) and (E)-3-Aryl-1-[4-(6,7-dimethoxyquinazolin-4-ylamino)phenyl]prop-2-en-1-one (7e)
A mixture of the appropriate 4-anilinoquinazoline (4a–b) (0.01 mol), appropriate aromatic aldehyde (0.01 mol) and 10% aqueous sodium hydroxide (10 ml) in ethanol (30 ml) was stirred at room temperature for about 3 h then kept at 0 °C overnight. The resulting solid was filtered off, rinsed with water, dried and recrystallized from ethanol.
(E)-3-(4-Chlorophenyl)-1-{4-[(6,7-dimethoxyquinazolin-4-yl)amino]phenyl}prop-2-en-1-one (7e)
Orange yellow crystals (yield 70%); m.p. above 300 °C. IR (KBr, cm−1): 1743 (C=O), 3337 (NH), 2924 (CH aliphatic), 3010 (CH aromatic). 1H-NMR (300 MHz, DMSO): δ 8.49 (s, 1H, NH=CH–NH), 7.24 (s, 1H, CH=C–OCH3), 7.40 (s, 1H, CH=C–OCH3), 3.9 (s, 6H, OCH3–Ar), 10.10 (s, 1H, NH–Ar, D2O exchangeable), 7.64 (d, 2H, CH=CH–NH), 7.71 (d, 2H, CH=CH–NH), 7.59–7.54 (d, J = 15 Hz, 1H, CH=CH–C=O), 8.06-8.01 (d, J = 15 Hz, 1H, CH=CH–C=O), 7.44 (d, 2H, CH=CH–Cl), 7.68 (d, 2H, CH=CH–Cl). MS (EI): 445 M+ 85.9%, 447 M+2 30%. Anal. for C25H20N3O3Cl; C, 67.34%; H, 4.52%; N, 9.42%. Found: C, 67.47%; H, 4.69%; N, 9.64%.
Computational methods and details
Protein preparation and receptor grid generation
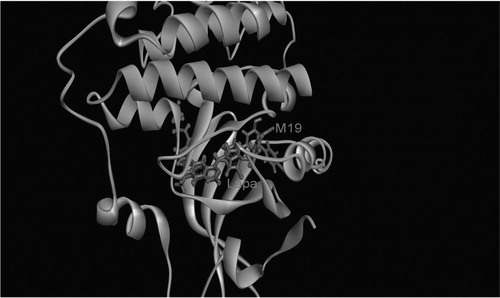
Crystal structure of the kinase domain of human EGFR co-crystallized with lapatinib was taken from the PDB (PDB entry: 1xkk)Citation12. Previous studies have shown that in certain cases, minor side-chain rearrangement may be crucial for allowing ligand–receptor interactionCitation13. In the current study, the most active ligand in vitro (5e) is superimposed on the original inhibitor (). This is followed by minimization and short molecular dynamic (MD) simulation for 1 ns in the NPT ensemble with AMBER (University of California, San Francisco, CA) to allow complex relaxation. The relaxed complex is used for the subsequent calculations. Complex is prepared using the protein preparation wizard using Maestro 9.2 (Schrödinger, LLC, New York, NY)Citation14. The structure is saturated by hydrogen atoms, all water molecules within 5 Å of the ligand but for a conserved water molecule (HOH) which is observed in the ATP hinge region of several kinasesCitation15. This bridge water molecule connects the N(3) of the quinazoline ring of the inhibitor to the backbone carbonyl oxygen of Gln791. Missing residues are added and refined using Prime 3.0Citation16 (Schrödinger, LLC, New York, NY). An ACE (N-acetyl) and NMA (N-methyl amide) groups are added to cap the uncapped N and C termini.
Figure 3. Superposition of the most active ligand, 5e on the original inhibitor, lapatinib.
Interactive hydrogen bond (H-bond) network optimization is carried out assuming a neutral pH. The protonation states of titratable amino acids are assigned at the same pH. Water orientation is modified to give the best interaction with the ligand and the receptor, an all atom impref minimization step is carried out to remove unfavorable steric clashes till it reaches convergence or a maximum RMSD of 0.3 Å from the original conformation. No steric clashes were reported after the final minimization step.
Based on the prepared protein structure in the previous step, receptor grid is prepared with the receptor grid generation module in Glide 5.7 (Schrödinger, LLC, New York, NY)Citation17 and sets up to the default values for vdW radii of 1 (no scaling). The binding site is determined as a box around the ligand that is centered inside the box. The ligand-Met793 H-bond was constrained.
Ligands preparation
The sequence of ligand preparation is crucial to obtain valid docking results. In a recent study, Butler et al. have shown that the active (bound) conformations of small molecular ligands lie in the vicinity of a near-local minima. It has been demonstrated that inadequate sampling of ligand conformational space can lead to a deleterious effect of the results of virtual screening experiments. That is, the scoring functions can bias toward the input geometry of the ligand. These findings clearly indicate that it is important to start the initial configuration of the ligands close to a minimum. For small scale situations in this study, it is possible for the 21 ligands to be studied using ab initio methods, but, in order to mimic more closely a real life situation, where ligands are prepared by a simple force field, or in the best case scenario, using semiempirical methods such as AM1, PM3, PM6Citation18; ligands are optimized at the PM6 semiempirical level of theory using the Gaussian 09 computational chemistry program (Gaussian, Inc., Wallingford, CT)Citation19. Electrostatic potential charges (ESP) are calculated at the B3LYP/cc-pVTZ level of theory using Jaguar (Schrödinger, LLC, New York, NY)Citation20.
Docking and scoring
The flexible docking module of Glide extra precision (Glide XP)Citation17,Citation21 is utilized. In order to increase the sampling space, a maximum of 50 000 initial ligand poses are kept in the initial phase of docking. A scoring window of poses within 1000 kcal mol−1 from the best scoring pose are retained, from which a maximum of 800 poses per ligand are subjected to 200 steps of energy minimization. To reduce the number of false positives in the docking experiment, the ligand pose is only considered when the predetermined constraint is satisfied. The output usually needs visual inspections to ensure appropriate positioning of the quinazoline ring in the hinge region. The effective region of docking is within a 4 Å boundary from the quinazoline core. When rescoring the docked poses, residues in the region of 6 Å are allowed to move flexibly, while retaining the predefined charges of the ligands previously calculated using the B3LYP/cc-pVTZ model. Ligand pose rescoring is done using the Prime/MM-GBSA module utility in Schrödinger, LLC, New York, NYCitation16,Citation21,Citation22.
Results and discussion
Synthesis
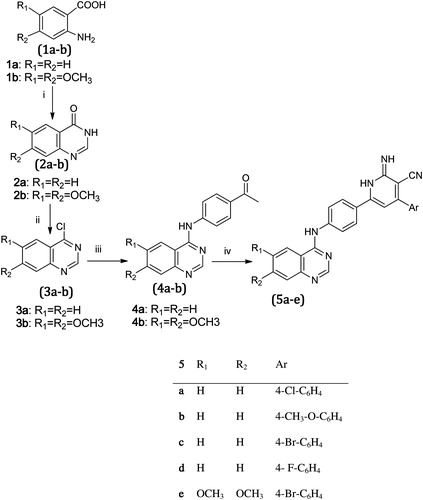
outlines the synthetic pathway used to obtain compounds 5a–e. Compounds (2a–b, 3a–b, 4a–b) were synthesized according to the previous reported methods. Accordingly, the 4-aryl-2-imino-pyridyl- quinazoline derivativesCitation9,Citation11 (5a–e) were obtained through a one-pot reaction of 4-chloroquinazolines (3a--b) with malononitrile, ammonium acetate and appropriate aldehyde in the presence of absolute ethanol as a solvent at reflux temperature. IR showed the appearance of cyano group between 2200 and 2218 cm−1, the 1H-NMR revealed the appearance of the NH groups between 9.9 and 10.3 ppm as singlets or broad peaks.
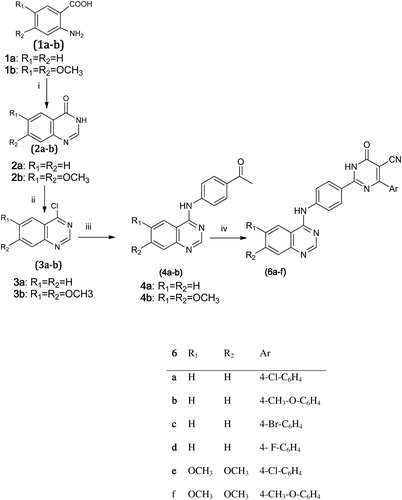
On the other hand, described the synthesis of the 4-aryl-2-oxo-pyridylquinazoline (6a–f). Compounds 4a–b were prepared as previously explained in followed by a one-pot reaction of these compounds with ethyl cyanoacetate, ammonium acetate, the appropriate aldehyde and ethanol as the reaction solvent at reflux temperature for 24 h. IR showed the appearance of cyano group between 2200 and 2218 cm−1 and C=O group at 1651 cm−1, the 1H-NMR revealed the appearance of a second NH group at 12.8 ppm which is also a strong olefinic proton appeared at 6.8 ppm.
Scheme 1. Synthesis of 2-iminopyridine derivatives (5a–e) Reagents and conditions: i = HCONH2/160 °C/4 h, ii = POCl3/TEA/90 °C/2 h, iii = p-aminoacetophenone/iPrOH/Reflux 2 h, iv = Malononitrile/NH4COOCH3/aldehyde ethanol/Reflux 18 h.
Scheme 2. Synthesis of 2-oxopyridine derivatives (6a–f) Reagents and conditions: iv = Ethyl cyanoacetate/NH4COOCH3/aldehyde/ethanol/Reflux 6 h.
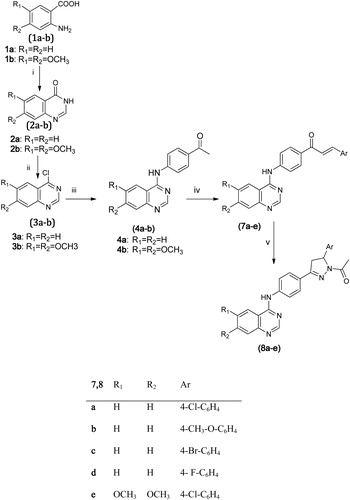
The targeted compounds (7a–e, 8a–e) were synthesized as shown in . Compounds 7a–e were prepared according to the general procedures by Kohler and ChadwellCitation23 applying Claisen Schmidt condensation of compounds 4a–b with substituted benzaldehydes using NaOH and water. 1H-NMR spectrum revealed the two characteristic doublets for the vinyl protons at δ 7.65–7.59 ppm and δ 8.06–8.01 or δ 8.18–8.13 ppm with a coupling constant J = 16 or 18 MHz confirming the trans-configuration. The latter compounds (7a–e) were treated with hydrazine hydrate in presence of acetic acid to afford compounds 8a–e showing distinctive doublet of doublets’ signals of the pyrazoline ring in the 1H-NMR spectrum.
Scheme 3. Synthesis of chalocne and pyrazoline derivatives (7a–e, 8a–e). Reagents and conditions: iv = aldehyde/ethanol/20%NaOH/0 °C overnight, v = NH2NH2.H2O/Glacial CH3COOH/reflux 5 h.
Biological evaluation (in vitro tyrosine kinase enzyme assay)
The in vitro kinase inhibition assay was determined for the synthesized compounds by KINEXUS Corporation, Vancouver, British Columbia, Canada.
The various protein kinase targets (EGFR and HER2) employed in the compound-profiling process were cloned, expressed and purified using proprietary methods. Quality control testing is routinely performed on each of the targets to ensure compliance to acceptable standards. 33P-ATP was purchased from PerkinElmer (Waltham, MA) and ADP-GloTM was purchased from Promega (Fitchburg, WI). All other materials were of standard laboratory grade.
The assay conditions for the various protein kinase targets were optimized to yield acceptable enzymatic activity. In addition, the assays were optimized to give high signal-to-noise ratio. Protein Kinase Assay – Radioactive Format Kinexus uses a radioactive assay format for profiling the evaluation of protein kinase targets and all assays are performed in a designated radioactive working area. Protein kinase assays were performed at ambient temperature for 20–40 min (depending on the target) in a final volume of 25 μl according to the following assay reaction recipe: 5 μl of diluted active protein kinase target (∼10–50 nM final protein concentration in the assay), 2.5 μl of peptide substrate, 3. μl of kinase assay buffer, 5 μl of compound or 10% DMSO, and 5 μl of 33P-ATP (250 μM stock solution, 0.8 μCi).
The assay was initiated by the addition of 33P-ATP and the reaction mixture was incubated at ambient temperature for 20–40 min, depending on the protein kinase target. After the incubation period, the assay was terminated by spotting 10 μl of the reaction mixture onto Multiscreen phosphocellulose P81 plate. The Multiscreen phosphocellulose P81 plate was washed three times for approximately 15 min each in a 1% phosphoric acid solution. The radioactivity on the P81 plate (EMD Millipore Corporation, Billerica, MA) was counted in the presence of scintillation fluid in a Trilux scintillation counter (GMI Inc., Ramsey, MN).
Blank control was set up for each protein kinase target which included all the assay components except the addition of appropriate substrate (replace with equal volume of kinase assay buffer). The corrected activity for each protein kinase target was determined by removing the blank control value.
The profiling data for various compounds against EGFR at 5 μM concentration compared to the positive control compound Staurosporine (Multikinase inhibitor) showed weak-to-good inhibition of EGFR activity (). The compound 5e showed the highest inhibition and the EGFR activity was inhibited by 74% by this compound compared to control. Compounds 5d and 7e also showed good inhibition and the EGFR activity was inhibited by 55% and 66%, respectively, by these compounds compared to control. Compounds 6c, 6a, 5b, 6e, 6f and 8e showed moderate inhibition and the EGFR activity was inhibited in the range of 35%–49% by these compounds compared to control. The rest of the compounds showed either weak inhibition (<30% inhibition compared to control) or no significant effects.
Table 1. Percent activity of EGFR/HER2 in the presence of various compounds using radiometric assay.
While the profiling data for various compounds against HER2 at 5 μM concentration showed weak-to-good inhibition of HER2 activity (), Compound 5e showed the highest inhibition and the HER2 activity was inhibited by 95% by this compound compared to control. Compound 5d also showed good inhibition and the HER2 activity was inhibited by 65% by this compound compared to control. Compounds 6b, 6c and 6e showed moderate inhibition and the HER2 activity was inhibited in the range of 35%–43% by these compounds compared to control. The rest of the compounds showed either weak inhibition (<30% inhibition compared to control) or no significant effects.
Based on the in vitro enzyme inhibition at 5 μM, the most potent compounds 5d, 5e and 7e were subjected to a tyrosine kinase enzyme assay at different concentration to evaluate the phosphotransferase activity of EGFR and/or HER2 protein kinase targets and determine the IC50 concentration of each. From the observed data (, Supplemental information ), the aryl 2-imino-1,2-dihydropyridine derivatives, 5d and 5e, displayed the most potent inhibitory activity on EGFR with IC50 equal to 2.09 and 1.94 μM, respectively, and for HER2 with IC50 equal to 3.981 and 1.036 μM, respectively; while 7e showed potent inhibitory activity on EGFR with an IC50 equal to 2.582 μM. Furthermore, the cytotoxicity assessment against MDA-MB breast cancer known to overexpress EGFR was performed for the selected three compounds and showed IC50 in the range of 2.4–2.5 μM.
Molecular modeling study
Docking study was carried out for the target compounds into the EGFR crystal structure complexed with Lapatinib (PDB code: Ixkk) using Glide extra precision (Glide XP) (Schrödinger)Citation17,Citation21. (Supplemental information ), the experimental percentages of inhibitions (%inhibition) are plotted against (a) XP G-score, (b) XP_LipophilicEvdW, (c) Prime/MM-GBSA DG and (d) Prime/MM-GBSA DG bind vdW scoring functions. This can be considered as a simple consensus scoring in which several scoring functions are used to rank the ligands. As can be seen from the Pearson’s correlation coefficient (r) and the coefficient of determination (r2), a moderate-to-good correlation is achieved. For example, the correlation of (a)–(d) is given by 0.42, 0.78, 0.30 and 0.42, respectively. The most correlated case is 4(b), the XP_LipophilicEvdW (r = 0.78 and r2 = 0.61) scoring function. It was shown previously that the highest correlation with experimental binding affinity in inhibitor-EGFR interaction stems from the lipophilic vdW interactionCitation15 (Supplemental information ). This is also verified in the current study where the highest correlation occurs with the vdW forces. Cho et al. also showed that for such receptors where binding is mostly driven by lipophilic interactions with several anchor points such as H-bonds, QM/MM docking is the best to pursue in that case by taking into account the protein polarizing effect on the ligand chargesCitation24, research in that direction is currently in progress.
The most important binding anchor is this inhibitor-Met793 H-bond (Supplemental information ). This H-bond guarantees the interaction between the quinazoline ring and the receptor and most probably ligands lacking this interaction are inactive. Another important binding motif is the two methoxy tails at the 6 and 7 positions of the quinazoline rings, accordingly, these tails seems to serve two functions: first, pushing the quinazoline ring into the hinge region thus cause tight packing and filling of the hinge region cavity; second, forms a strong lipophilic interaction and H-bonding with Leu718. This effect may account for the relatively high in vitro activity of 5e compared to 5c which has the same chemical structure of 5e but lacks these two methoxy tails, so, it lacks the lipophilic interaction with Leu718 and also it is less tightly packed into the hinge region, and having a slightly longer H-bonds interactions with Met793 (5e: 1.947 Å, 5c: 2.132 Å).
In addition to these hinge-specific interactions, non-specific contributions from individual amino acid residues may also exist. For example, the flexible part of the molecules (4-anilino moiety) is able to bind the selectivity pocket noticed in the DFG-out (inactive) conformation of the EGFR. This pocket is formed by amino acids Thr790, Met766, Leu777, Thr854 and Cys775.
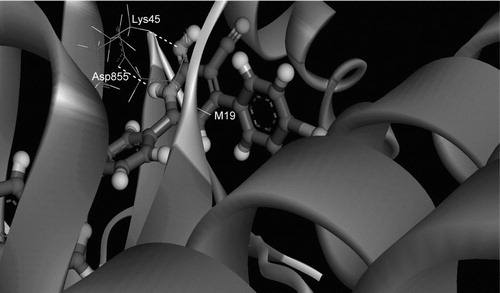
One may also expect that the substitution of the terminal flourobenzyl group by other bulky groups of different nature may give raise to different H-bonding interaction with those residues present in the selectivity pockets, and this has been already verified. Owing to the rigid receptor docking nature of Glide-XP, such H-bonding interaction cannot be identified easily and usually molecular dynamic simulations are of absolute necessityCitation25. It also needs certain degree of receptor flexibility of kinases which are known to be highly flexible. Indeed, running MD simulations that are long enough (60–80 ns) has verified that in addition to the hinge region H-bonds interactions, certain members of the new series are capable of forming other “extra-hinge” H-bonding interactions with EGFR. Particularly, those inhibitors whose ring “D” () is an iminopyridine ring, for example, 5a, 5b, 5d and 5e, are capable of forming two H-bonds with Asp855 and Lys745. displays the interaction of ring “D” of the most active inhibitor “5e” with these two amino acid residues, Asp855 and Lys745 after an 80 ns MD simulation in AMBER. This may also explain why these members may have higher %inhibition activity than those analogues that have a 2-pyridone ring instead, such as 6a, 6b, 6d, 6e and 6f. Research in this direction is currently in progress.
Figure 4. 3D inhibitor–protein interactions of the most active inhibitor (5e) with EGFR after 70 ns MD simulation.
Conclusion
Some compounds belonging to a class of anilinoquinazoline bearing bulky arylpyridinyl 5, 6, arylpropenoyl 7 and arylpyrazolyl 8 moieties at the 4′ position of the anilinoquinazoline scaffold were prepared. Compounds (5d, 5e and 7e) exhibited highly potent EGFR and HER2 inhibitory activity at 5 μM. IC50 was determined for these most active compounds on EGFR tyrosine kinase and displayed IC50 range equal to 2.09 and 1.94 μM for 5d and 5e, respectively, and 3.981 and 1.036 μM, respectively, on HER2. Moreover, the anti-proliferative activity of the same compounds against MDA-MB-231 breast cancer cell lines showed an IC50 of 2.4 and 2.5 μM, respectively. Molecular docking study further supported the strong inhibitory activity of 5d and 5e, which helps understanding the various interactions between the ligands and enzyme active sites in detail and thereby could enable us to design more selective inhibitors.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgements
MA acknowledges the Swinburne University Postgraduate Research Award (SUPRA). National Computational Infrastructure (NCI) at the Australian National University for the award under the Merit Allocation Scheme, Victorian Partnership for Advanced Computing (VPAC) and Swinburne University Supercomputing Facilities are also acknowledged.
Related Research Data
References
- Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol 2005;23:2445–59
- Olayioye MA. Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res 2001;3:385–9
- Levitzki A, Mishani E. Tyrphostins and other tyrosine kinase inhibitors. Annu Rev Biochem 2006;75:93–109
- Barlesi F, Tchouhadjian C, Doddoli C, et al. Gefitinib (ZD1839, Iressa) in non-small-cell lung cancer: a review of clinical trials from a daily practice perspective. Fund Clin Pharmacol 2005;19:385–93
- Ganjoo KN, Wakelee H. Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biologics 2007;1:335–46
- Ryan Q, Ibrahim A, Cohen MH, et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008;13:1114–19
- Rewcastle GW, Denny WA, Bridges AJ, et al. Tyrosine kinase inhibitors. 5. Synthesis and structure-activity relationships for 4-[(phenylmethyl)amino]- and 4-(phenylamino)quinazolines as potent adenosine 5′-triphosphate binding site inhibitors of the tyrosine kinase domain of the epidermal growth factor receptor. J Med Chem 1995;38:3482–7
- Abouzid K, Shouman S. Design, synthesis and in vitro antitumor activity of 4-aminoquinoline and 4-aminoquinazoline derivatives targeting EGFR tyrosine kinase. Bioorg Med Chem 2008;16:7543–51
- Rocco SA, Barbarini JE, Rittner R. Syntheses of some 4-anilinoquinazoline derivatives. ChemInform 2004;3:429–35
- Abouzid K, Youssef KM, Amine FM, et al. Synthesis of 1,4-disubstituted piperazines as potential antihypertensive agents. Egypt J Pharma Sci 1989;30:429–36
- Sharma VM. Inventor novel anticancer agents process for their preparation and pharmaceutical compositions containing them. Indian patent WO 2004069145; 2004
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72
- Zavodszky MI, Kuhn LA. Side-chain flexibility in protein–ligand binding: the minimal rotation hypothesis. Protein Sci 2005;14:1104–14
- Schrödinger Suite 2011 Protein Preparation Wizard; Epik version 2.2 S, LLC, New York, NY, 2011; Impact version 5.7, Schrödinger, LLC, New York, NY, 2011; Prime version 3.0, Schrödinger, LLC, New York, NY, 2011. Schrödinger Suite 2011 Protein Preparation Wizard; Epik version 2.2, Schrödinger, LLC, New York, NY, 2011; Impact version 5.7, Schrödinger, LLC, New York, NY, 2011; Prime version 3.0, Schrödinger, LLC, New York, NY, 2011
- Balius TE, Rizzo RC. Quantitative prediction of fold resistance for inhibitors of EGFR. Biochemistry 2009;48:8435–48
- Jacobson MP, Pincus DL, Rapp CS, et al. A hierarchical approach to all-atom protein loop prediction. Proteins Struct Funct Bioinf 2004;55:351–67
- Glide v, Schrödinger, LLC, New York, NY, 2011; Glide, version 5.7, Schrödinger, LLC, New York, NY, 2011
- Stewart J. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 2007;13:1173–213
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09 RA. Wallingford (CT): Gaussian, Inc.; 2009
- Jaguar v, Schrödinger, LLC, New York, NY, 2011; Jaguar, version 7.8, Schrödinger, LLC, New York, NY, 2011
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem 2006;49:6177–96
- Du J, Sun H, Xi L, et al. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM–GBSA calculation. J Comput Chem 2011;32:2800–9
- Kohler E, Chadwell H. Benzalacetophenone. Org Synth 1922;2:1–3
- Cho AE, Guallar V, Berne BJ, Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J Comput Chem 2005;26:915–31
- Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol 2011;9:71–9