
Abstract
In continuation of our research efforts toward the identification and optimization for novel inhibitors of interaction between human immunodeficiency virus type 1 integrase and cellular cofactor LEDGF/p75, we designed and synthesized a new series of 4-benzylindole derivatives. Most of the title compounds proved to be able to block this protein–protein interaction (PPI), with a percentage ranging from 30% to 90% at 100 µM. The most promising derivative was compound 10b showing IC50 value of 6.41 µM. The main structure–activity relationships (SAR) are discussed and rationalized by docking studies.
Introduction
In the last years, the global community has made great progress in responding to the acquired immunodeficiency syndrome (AIDS) epidemic but there are yet about 40 million individuals living with the human immunodeficiency virus type 1 (HIV-1) and every day thousands more people are newly infected with HIVCitation1. United Nations Member States set clear targets for significantly reducing HIV infection and AIDS death and for scaling up treatment by 2015.
The goal is to strengthen and sustain a valid response to AIDS including the prevention of transmission of HIV as well as providing support and care to those already living with the virus, to prevent the HIV/AIDS epidemic from becoming a severe pandemic. Particularly, it is necessary to ensure that 15 million people can be receiving the antiretroviral therapy, thus reducing the transmission of HIV at least among children.
The overall number of people living with HIV has increased as result of new infections and the beneficial effects of the more widely available highly active anti-retroviral therapy (HAART), which changed AIDS from a rapidly lethal disease into a chronic manageable condition; nevertheless, new efforts are needed.
The currently FDA approved anti-HIV drugs belong to several different groups such as nucleoside reverse transcriptase inhibitors (NRTIs), nucleotide reverse transcriptase inhibitors (NtRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors (FIs), co-receptor inhibitors (CRIs) and integrase inhibitors (INIs)Citation2.
Despite HAART remarkably decreases viral load and provides a significant improvement in the life expectancy of HIV/AIDS patients, several factors, including the emergence of drug-resistant viral strains, drug toxicity, the poor ability of patients to adhere to the prescribed therapy and costs, require to refine the current therapy and develop new therapeutic paradigms targeting other steps in the viral replication processCitation3,Citation4.
In this context, HIV-1 integrase (IN), the enzyme which catalyzes the integration of viral cDNA into the host genome, represents a validated target for the development of new drugs against HIV-1 infectionCitation5–9. Furthermore, discovery of the keto-enol acid class, often referred as diketoacid (DKA) class, and their analogues and bioisosters as IN inhibitors was an additional advance in the validation of this enzyme as a therapeutically viable antiretroviral drug target, representing major leads in the development of anti-HIV-1 IN drugs.
More recently, it was demonstrated that the multifunctional IN interacts with viral DNA and its key cellular cofactor lens epithelium-derived growth factor (LEDGF/p75) to effectively integrate the reverse transcript into a host cell chromosomeCitation10,Citation11.
LEDGF/p75 binds HIV-1 IN via a small IN-binding domain (LEDGFIBD, residues 347–429) within its C-terminal region. IBD is both necessary and sufficient for interaction with HIV-1 INCitation12.
Taking into account that the association between IN and the cellular cofactor LEDGF/p75 can be considered as one of the most promising IN interaction for the design of protein–protein disrupting therapeuticsCitation13, we have directed our research toward the identification of new small molecules that are able to inhibit the protein–protein interaction (PPI) between the enzyme and its cofactor.
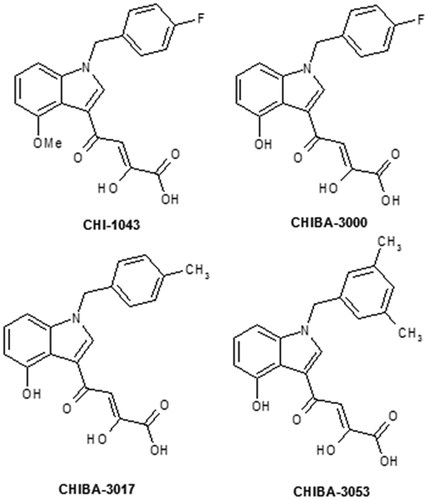
In previous research from our laboratory, it was found that N-benzyl-indoles, bearing diketo acid moiety, were able to exert a specific blockade of the IN/LEDGF interactionCitation14–Citation18 and among them we found CHI-1043, CHIBA-3000 and some methylbenzyl derivatives such as CHIBA-3017 and CHIBA-3053 () as interesting PPI inhibitorsCitation14,Citation19.
Figure 1. Chemical structure of CHI-1043, CHIBA-3000, CHIBA-3017 and CHIBA-3053.
Encouraged by these promising results, which could represent an interesting starting point for the development of novel dual drugs, and with the aim of achieving further information about the IN/LEDGF protein interaction, we focused our efforts to synthesize new indole analogues. Hence, several N-benzylsubstituents were inserted and structural modification of diketo acid portion was performed, replacing this critical moiety in a corresponding closed system which was able to maintain the important polar contacts with IBD. Moreover, docking simulations were carried out to investigate the possible binding mode of title compounds.
Material and methods
Computational studies
Docking studies were performed using GOLD software packageCitation20 version 4.1.1 (Gold, UK) and GoldScore scoring function as described in our previous paperCitation21.
The crystal structure of the dimeric INCCD complexed with the LEDGF/p75IBD was retrieved from the RCSB Protein Data Bank (entry code 2B4J)Citation22 and used for our docking simulations. The LEDGF structure was removed and hydrogen atoms were added to the IN protein in Discovery Studio 2.5.5Citation23. The standard default settings were used in all calculations. A 20.0 Å radius active site was drawn on the original position of the LEDGFIBD dipeptide Ile365-Asp366 and automated cavity detection was used. Results differing by less than 0.75 Å in ligand-all atom RMSD were clustered together. Two rotatable bonds of the diketo acid moiety of the ligands were kept fixed during docking calculation. The molecules containing the diketo acid moiety were simulated in the mono-ionized formCitation24.
Chemistry
All commercially available reagents and solvents were used without any further purification. The microwave-assisted reactions were carried out in a CEM Focused Microwave Synthesis System, Model Discover (CEM, Matthews, NC), working at the power necessary for refluxing under atmospheric conditions. Melting points were determined on a BUCHI Melting Point B-545 apparatus (Buchi, Milano, Italy) and are uncorrected. Elemental analyses (C, H, N) were carried out on a Carlo Erba Model 1106 Elemental Analyzer (Carlo Erba, Arese (MI), Italy) and the results are within ±0.4% of the theoretical values. Merck silica gel 60 F254 (Darmstadt, Germany) plates were used for analytical TLC; column chromatography was performed on Merck silica gel 60 (230–400 mesh) and Flash Chromatography (FC) on a Biotage SP1 EXP (Biotage, Uppsala, Sweden). 1H-NMR spectra were recorded in CDCl3 with TMS as internal standard or [D6]DMSO on a Varian Gemini-300 spectrometer (Varia, Gemini, USA). Chemical shifts were expressed in δ (ppm) and coupling constants (J) in hertz (Hz). All the exchangeable protons were confirmed by the addition of D2O.
Synthesis of 3-acetyl-4-methoxy-1H-indole (2a) and 3-acetyl-4-hydroxy-1H-indole (2b) was carried out following the previously reported procedureCitation14,Citation15,Citation25. Phosphoryl chloride (0.92 mL, 10 mmol) was added to ice cold dimethylacetamide (2.79 mL, 30 mmol) under stirring and cooling in ice. 4-Methoxy-1H-indole 1a (147.18 mg, 1 mmol) or 4-hydroxy-1H-indole 1b (133.15 mg, 1 mmol) was added and the reaction mixture was stirred at room temperature for 12 h, and then poured over ice and basified with a 4N aqueous sodium hydroxide solution. The mixture was extracted with ethyl acetate and dried over Na2SO4. After the removal of solvent under reduced pressure, the residue was powdered by treating with diethyl ether and recrystallized from dichloromethane.
For the obtained derivatives 2a and 2b, spectral data are in accordance with the literatureCitation14,Citation25.
General procedure for the synthesis of 3-acetyl-1-benzyl-1H-indoles (3a, 4–5a,b)
Using the synthetic procedure previously reported by usCitation14,Citation15,Citation25–27, 3-acetyl-4-methoxy-1H-indole (2a) (189 mg, 1 mmol) or 3-acetyl-4-hydroxy-1H-indole (2b) (159 mg, 0.001 mol) was dissolved in DMF (1 mL) at 0 °C and dry sodium hydride (120 mg, 5 mmol) or K2CO3 (138 mg, 1 mmol) was added. After stirring for 2 min, the suitable benzyl bromide (1.5 mmol) was added dropwise. The resulting solution was placed in a cylindrical quartz tube (diam. 2 cm), stirred and irradiated in a microwave oven at 100 W and at continuous temperature (50 °C) for 10 min. A saturated NaHCO3 solution was added. The reaction mixture was extracted with ethyl acetate (10 mL × 3) and dried over Na2SO4. After removal of the solvent under reduced pressure, the crude mixture was purified by flash chromatography using a mixture of cyclohexane:ethyl acetate (60:40) as eluent.
3-Acetyl-1-(4-methyl benzyl)-4-methoxy-1H-indole (3a). Spectral data are in accordance with the literatureCitation26.
3-Acetyl-1-(4-trifluoromethyl benzyl)-4-methoxy-1H-indole (4a). m.p. 152–154 °C, yield 85%; 1H-NMR (δ) 2.70 (s, 3H, CH3), 3.98 (s, 3H, CH3), 5.36 (s, 2H, CH2), 6.71 (d, J = 8.0, 1H, ArH), 6.84 (d, J = 8.0, 1H, ArH), 7.15–7.23 (m, 3H, ArH), 7.57 (d, J = 8.5, 2H, ArH), 7.73 (s, 1H, ArH). Anal. Calcd for C19H16F3NO2 – C: 65.70; H: 4.64; N: 4.03. Found – C: 65.87; H: 4.91; N: 4.33.
3-Acetyl-1-(4-trifluoromethyl benzyl)-4-hydroxy-1H-indole (4b). m.p. 190–192 °C, yield 51%; 1H-NMR (δ) 2.54 (s, 3H, CH3), 5.38 (s, 2H, CH2), 6.66–6.75 (m, 2H, ArH), 7.17 (t, J = 7.9, 1H, ArH), 7.25 (d, J = 7.1, 2H, ArH), 7.62 (d, J = 7.4, 2H, ArH), 7.70 (s, 1H, ArH). Anal. Calcd for C18H14F3NO2 – C: 64.86; H: 4.23; N: 4.20. Found – C: 64.99; H: 4.54; N: 4.35.
3-Acetyl-1-(4-tert-butyl benzyl)-4-methoxy-1H-indole (5a). m.p. 143–145 °C, yield 73%; 1H-NMR (δ) 1.29 (s, 9H, CH3), 2.68 (s, 3H, CH3), 3.97 (s, 3H, CH3), 5.25 (s, 2H, CH2), 6.69 (d, J = 7.9, 1H, ArH), 6.96 (d, J = 7.9, 1H, ArH), 7.09 (d, J = 8.5, 2H, ArH), 7.17 (d, J = 7.9, 1H, ArH), 7.34 (d, J = 8.5, 2H, ArH), 7.72 (s, 1H, ArH). Anal. Calcd for C22H25NO2–C: 78.77; H: 7.51; N: 4.18. Found – C: 78.95; H: 7.74; N: 4.51.
3-Acetyl-1-(4-tert-butyl benzyl)-4-hydroxy-1H-indole (5b). m.p. 184–186 °C, yield 75%; 1H-NMR (δ) 1.30 (s, 9H, CH3), 2.52 (s, 3H, CH3), 5.27 (s, 2H, CH2), 6.71–7.67 (m, 8H, ArH), 11.53 (s, 1H, OH). Anal. Calcd for C21H23NO2 – C: 78.47; H: 7.21; N: 4.36. Found – C: 78.28; H: 7.43; N: 4.21.
General procedure for the synthesis of ethyl 4-[1-benzyl-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoates (6a, 7–8a,b).
Adopting the synthetic procedure previously reported by usCitation14,Citation15,Citation19,Citation26,Citation27, a mixture of suitable 3-acetyl-1-benzyl-1H-indole (3a, 4–5a,b) (1 mmol), diethyl oxalate (219 mg, 1.5 mmol) and a catalytic amount of NaOCH3 was suspended in anhydrous THF (2 mL). The reaction mixture was placed in a cylindrical quartz tube (diam. 2 cm), stirred and irradiated at continuous temperature in a microwave oven for two successive time intervals under the same conditions (250 W, 2 min, 50 °C). The solvent was concentrated under reduced pressure and collected yellow solid was crystallized from ethanol and diethyl ether (1:4).
Ethyl 4-{1-(4-methyl-benzyl)-4-methoxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoate (6a). Spectral data are in accordance with the literatureCitation26.
Ethyl 4-{1-[4-(trifluoromethyl)benzyl]-4-methoxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoate (7a). m.p. 150 °C dec., yield 89%; 1H-NMR (δ) 1.25 (t, J = 7.2, 3H, CH3), 3.86 (s, 3H, CH3), 4.15 (d, J = 5.5, 2H, CH2), 5.55 (s, 2H, CH2), 6.64–7.70 (m, 9H, ArH and CH). Anal. Calcd for C23H20F3NO5 – C: 61.75; H: 4.51; N: 3.13. Found – C: 61.99; H: 4.40; N: 3.37.
Ethyl 4-{1-[4-(trifluoromethyl)benzyl]-4-hydroxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoate (7b). m.p. 263–265 °C, yield 96%; 1H-NMR (δ) 1.21 (t, J = 7.3, 3H, CH3), 4.09 (q, J = 7.3, 2H, CH2), 5.45 (s, 2H, CH2), 6.25 (s, 1H, OH), 6.64–8.20 (m, 9H, ArH and CH), 13.52 (s, 1H, OH). Anal. Calcd for C22H18F3NO5 – C: 60.97; H: 4.19; N: 3.23. Found – C: 61.06; H: 4.33; N: 3.40.
Ethyl 4-{1-[(4-tert-butyl)benzyl]-4-methoxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoate (8a). m.p. 253 °C dec., yield 96%; 1H-NMR (δ) 1.20 (s, 9H, CH3), 1.24 (t, J = 7.3, 3H, CH3), 3.81 (s, 3H, CH3), 4.08 (d, J = 7.3, 2H, CH2), 5.23 (s, 2H, CH2), 6.54–8.50 (m, 9H, ArH and CH). Anal. Calcd for C25H27NO5 – C: 71.70; H: 6.71; N: 3.22. Found – C: 71.95; H: 6.89; N: 3.41.
Ethyl 4-{1-[(4-tert-butyl)benzyl]-4-hydroxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoate (8b). m.p. 219–221 °C, yield 92%; 1H-NMR (δ) 1.20 (t, J = 7.3, 3H, CH3), 1.21 (s, 9H, CH3), 4.09 (q, J = 7.3, 2H, CH2), 5.27 (s, 2H, CH2), 5.36–8.13 (m, 10H, ArH, OH and CH), 13.71 (s, 1H, OH). Anal. calcd for C25H27NO5 – C: 71.24; H: 6.46; N: 3.32. Found – C: 71.33; H: 6.31; N: 3.53.
General procedure for the synthesis of 4-[1-benzyl-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acids (9–10a,b)
Following the synthetic procedure previously reported by usCitation14,Citation15,Citation19,Citation26,Citation27, the appropriate ethyl 4-[1-benzyl-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoate (7–8a,b) (1 mmol) was dissolved in methanol (5 mL) and treated with 2N NaOH (5 mL). The reaction mixture was stirred at room temperature for 1.5 h and then acidified with conc. HCl to give corresponding acids (9–10a,b). The desired products were crystallized from a mixture of ethanol and diethyl ether (1:4).
4-{1-[4-(Trifluoromethyl)benzyl]-4-methoxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoic acid (9a). m.p. 280 °C dec, yield 72%; 1H-NMR (δ) 3.90 (s, 3H, CH3), 5.64 (s, 2H, CH2), 6.79–7.71 (m, 8H, ArH and CH), 8.57 (s, 1H, ArH). Anal. Calcd for C21H16F3NO5 – C: 60.15; H: 3.85; N: 3.34. Found – C: 60.44; H: 4.05; N: 3.56.
4-{1-[4-(Trifluoromethyl)benzyl]-4-hydroxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoic acid (9b). m.p. 298–300 °C, yield 80%; 1H-NMR (δ) 5.50 (s, 2H, CH2), 6.27–8.02 (m, 9H, ArH and CH), 14.56 (s, 1H, OH). Anal. Calcd for C20H14F3NO5 – C: 60.44; H: 3.38; N: 3.36. Found – C: 60.61; H: 3.57; N: 3.49.
4-{1-[4-(tert-Butyl)benzyl]-4-methoxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoic acid (10a). m.p. 215–217 °C, yield 15%; 1H-NMR (δ) 1.19 (s, 9H, CH3), 3.75 (s, 3H, CH3), 5.28 (s, 2H, CH2), 6.51–9.06 (m, 9H, ArH and CH). Anal. Calcd for C24H25NO5 – C: 70.74; H: 6.18; N: 3.44. Found – C: 70.98; H: 6.31; N: 3.72.
4-{1-[4-(tert-Butyl)benzyl]-4-hydroxy-1H-indol-3-yl}-2-hydroxy-4-oxobut-2-enoic acid (10b). m.p. 196–198 °C, yield 74%; 1H-NMR (δ): 1.21 (s, 9H, CH3), 5.41 (s, 2H, CH2), 6.55–9.06 (m, 9H, ArH and CH), 11.33 (s, 1H, OH). Anal. Calcd for C23H23NO5 – C: 70.21; H: 5.89; N: 3.56. Found – C: 70.37; H: 5.61; N: 3.28.
General procedure for the synthesis of 4-[1-benzyl-1H-indol-3-oyl)-3-hydroxyfuran-2(5H)-ones (11a, 12–13a,b)Citation28
A solution of 40% aqueous formaldehyde in water (4 mL) was added to a mixture of appropriate diketoester derivative (6a, 7–8a,b) (1 mmol) in diethyl ether (5 mL). The stirring was then continued until clear layers were formed (usually within 1–2 h). Sometimes, an additional 4 mL of water was added if the reaction was especially thick or when the solid appeared to react slowly. The clear aqueous bottom layer was removed and the organic layer extracted twice with 5 mL of water. The combined aqueous extracts were cooled and acidified with 3 mL of concentrated hydrochloric acid. Then the saturated solution was cooled overnight thus ensuring complete product formation. The resulting solid was collected, dried and recrystallized from ethanol.
4-{1-[4-(Methyl)benzyl]-4-methoxy-1H-indol-3-oyl}-3-hydroxyfuran-2(5H)-one (11a). m.p. 120–122 °C, yield 71%; 1H-NMR (δ) 2.26 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 5.16 (s, 2H, CH2), 5.44 (s, 2H, CH2), 6.64–8.21 (m, 8H, ArH). Anal. Calcd for C23H21NO4 – C, 73.58; H, 5.64; N, 3.73. Found – C, 73.78; H, 5.42; N, 3.91.
4-{1-[4-(Trifluoromethyl)benzyl]-4-methoxy-1H-indol-3-oyl}-3-hydroxyfuran-2(5H)-one (12a). m.p. 170–172 °C, yield 63%; 1H-NMR (δ): 3.77 (s, 3H, CH3), 5.05 (s, 2H, CH2), 5.59 (s, 2H, CH2), 6.68–7.71 (m, 7H, ArH), 8.21 (s, 1H, ArH). Anal. Calcd for C22H18F3NO5 – C: 60.97; H: 4.19; N: 3.23. Found – C: 61.13; H, 4.36; N, 3.45.
4-{1-[4-(Trifluoromethyl)benzyl]-4-hydroxy-1H-indol-3-oyl}-3-hydroxyfuran-2(5H)-one (12b). m.p. 249–251 °C, yield 61%; 1H-NMR (δ) 5.00 (s, 2H, CH2), 5.58 (s, 2H, CH2), 6.44–7.71 (m, 7H, ArH), 9.77 (s, 1H, ArH), 12.79 (s, 1H, OH). Anal. Calcd for C21H14F3NO5 – C: 60.44; H: 3.38; N: 3.36. Found – C: 60.63; H, 3.56; N, 3.59.
4-{1-[4-(Tert-butyl)benzyl]-4-methoxy-1H-indol-3-oyl}-3-hydroxyfuran-2(5H)-one (13a). m.p. 179–181 °C, yield 19%; 1H-NMR (δ) 1.22 (s, 9H, CH3), 3.76 (s, 3H, CH3), 5.04 (s, 2H, CH2), 5.41 (s, 2H, CH2), 6.68–7.34 (m, 8H, ArH), 8.19 (s, 1H, OH). Anal. Calcd for C24H27NO5 – C: 71.24; H: 6.46; N: 3.32. Found – C: 71.37; H: 6.58; N: 3.55.
4-{1-[4-(Tert-butyl)benzyl]-4-hydroxy-1H-indol-3-oyl}-3-hydroxyfuran-2(5H)-one (13b). m.p. 212–214 °C, yield 71%; 1H-NMR (δ) 1.22 (s, 9H, CH3), 5.12 (s, 2H, CH2), 5.44 (s, 2H, CH2), 6.53–7.35 (m, 8H, ArH), 8.81 (s, 1H, OH). Anal. Calcd for C24H23NO5 – C: 71.10; H: 5.72; N: 3.45. Found – C: 71.32; H: 5.56; N: 3.64.
LEDGF/p75-HIV-1 integrase interaction screening
The AlphaScreen assay was performed as previously describedCitation29. Reactions were performed in 25 µL final volume in 384-well Optiwell™ microtiter plates (Perkin-Elmer, Benelux). The reaction buffer contained 25 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.01% (v/v) Tween-20 and 0.1% (w/v) bovine serum albumin. His6-tagged integrase (300 nM final concentration) was incubated with the compounds at 4 °C for 30 min. The compounds were added in varying concentrations from 1 up to 100 nM. Later, 100 nM of recombinant flag-LEDGF/p75 was added and incubation was extended by another hour at 4 °C. Subsequently, 5 µL of Ni-chelate-coated acceptor beads and 5 µL of anti-flag donor beads were added to a final concentration of 20 µg/mL of both beads. Proteins and beads were incubated at 30 °C for 1 h in order to allow association to occur. Exposure of the reaction to direct light was prevented as much as possible and the emission of light from the acceptor beads was measured in the EnVision plate reader (PerkinElmer, Benelux) and analyzed using the EnVision manager software (Envision, Wichita, KS).
Results and discussion
The most interesting previously reported compounds were benzyl-indole derivatives CHI-1043, CHIBA-3000, CHIBA-3017 and CHIBA-3053 () which proved to be active showing an IC50 value in the low-micromolar range (36.16, 76.44, 67.96 and 3.5 μM, respectively)Citation14,Citation15,Citation19,Citation25.
First, in this study we focused our attention on the benzyl moiety as a relevant lipophilic feature regulating the IN-LEDGF/p75 inhibitory process. Particularly, by means of DRY probe in GRID software, we previously disclosed the hydrophobic region in the LEDGFIBD binding site, corresponding to IN residue Trp131 (15). This approach is common to other studies and applied also for the analysis of several classical HIV targetCitation30,Citation31. Thus, the high inhibitory efficacy of the most efficacious inhibitors could be explained for their ability to map well the DRY area related to the crucial residue Trp131. On this basis, we have explored the effect of bulkier hydrophobic substituents on the benzyl group, such as tert-butyl and trifluoromethyl, that could increase the ability to cover the hydrophobic area on LEDGFIBD binding site as well as the lipophilicity of our compounds.
In addition, we incorporated the 1,3-diketoacid motif into a closed system characterized by the same chemical functionalities, in order to explore the influence of this chemical manipulation on binding inhibitory effect. Following an earlier reported method, the synthesis of the target compounds was accomplished using the synthetic pathways shown in Citation15,Citation27,Citation28.
Scheme 1. Reagents and conditions: (i) POCl3, CH3CON(CH3)2, RT, 12 h; (ii) benzyl bromide, K2CO3 or NaH, DMF, MW: 10 min at continuous temperature (50 °C), 100 W; (iii) diethyl oxalate, dry CH3ONa, THF, two separate steps under the same conditions MW: 2 min, at continuous temperature (50 °C), 250 W; (iv) 2 N NaOH, MeOH, RT, 1.5 h; (v) CH2O, Et2O/H2O, RT, 2 h.
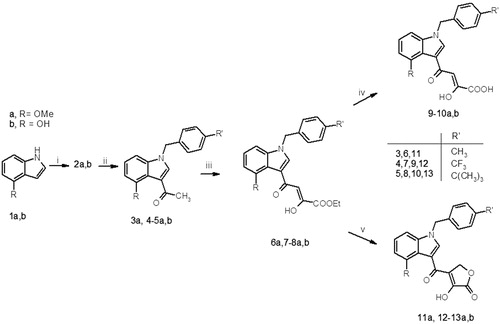
Suitable 4-substituted indoles (1a,b) were converted into the corresponding 3-acetyl derivatives (2a,b) under Vilsmeier–Haack conditions, in the presence of DMF, acid chloride, such as phosphoryl chloride, and an excess of N.N-dimethylacetamide. The obtained intermediates 2a,b were N-alkylated by reaction with the suitable benzyl bromide. This synthetic step was carried out by the addition of sodium hydride for the methoxy substituted derivatives (a), and potassium carbonate for the hydroxyl substituted derivatives (b). In both cases, the synthesis was performed under microwave (MW) irradiation conditions, thus reducing reaction time, usual thermal reagents degradation and increasing the yields. Reaction of benzyl derivatives (3a and 4–5a,b) with diethyl oxalate under the same conditions: 50 °C for 2 min under 250 W (MW) provided key diketoester intermediates (6a and 7–8a,b), which were converted into the corresponding diketoacids 9–10a,b, by hydrolysis in basic medium, or cyclized in the hydroxyl-furanone derivatives (11a and 12–13a,b), by treatment with formaldehyde aqueous solution. In order to profile inhibitory effects we tested all new synthesized compounds, CHI-1310 and CHI-1164 in AlphaScreen assay ().
Table 1. Inhibition of IN-LEDGF/p75 interaction of new synthesized compounds, CHI and CHIBA derivatives.
The obtained results showed that derivatives exhibited inhibitory effects at 100 µM concentration with a percentage ranging from 30% to 99%. Analysis of IC50 values pointed out the relevance of the introduction of hydrophobic substituents on benzyl group as previously predicted by our molecular modeling studiesCitation15.
In this series of molecules, both for the diketo acids derivatives and 3-hydroxyfuran-2(5H)-ones, generally the presence of hydroxyl group at C4 of benzene fused ring positively influences the inhibitory effects. Overall, the contemporary presence of the alkyl substitution on the benzyl moiety and the hydroxyl group on the indole system provided the most interesting results.
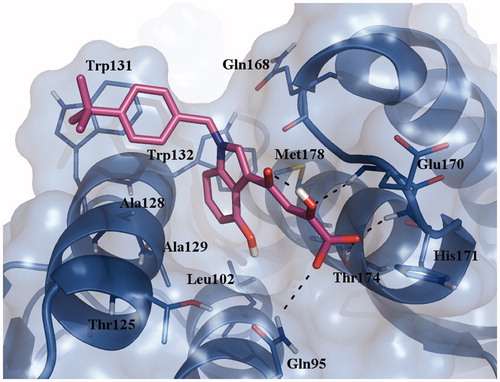
The best activity was displayed when the hydroxyl group of the indole nucleus was associated to the t-butyl substituent of the benzyl moiety (10b IC50 = 6.41 µM). Docking result for the most active compound of the series (10b) into the LEDGFIBD binding pocket on INCCD is reported in and shows nearly the same binding mode of previously reported diketo acid derivatives CHI-1043 and CHIBA-3053Citation21. In particular, the diketo acid moiety forms hydrogen bonds with the main chain nitrogens of residues Glu170 and His171 and the side chain of residues Thr174 and Gln95. The fused benzene ring of the indole nucleus projects into the IN hydrophobic pocket, and the N-benzyl substituent presents hydrophobic contacts for the crucial Trp131 residue of chain B. Overall, this new work supports our previous findings on the main structural requirements for IN-LEDGF/p75 inhibition.
Figure 2. Binding mode of compound 10b on INCCD. Hydrogen bonds are shown as dotted lines. This figure was produced with pymol32.
Declaration of interest
This work was supported by the European Commission (HEALTH-F3-2008-201032) (THINC project). The authors declare no conflict of interest.
References
- Global HIV/AIDS Response. Epidemic update and health sector progress towards universal access progress report; 2011
- Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem 2010;53:521–38
- Sechi M, Rizzi G, Bacchi A, et al. Design and synthesis of novel dihydroquinoline-3-carboxylic acids as HIV-1 integrase inhibitors. Bioorg Med Chem 2009;17:2925–35
- Katlama C, Murphy R. Emerging role of integrase inhibitors in the management of treatment-experienced patients with HIV infection. Ther Clin Risk Manag 2009;5:331–40
- Neamati N, Marchand C, Pommier Y. HIV-1 integrase inhibitors: past, present, and future. Adv Pharmacol 2000;49:147–65
- d’Angelo J, Mouscadet JF, Desmaele D, et al. HIV-1 integrase: the next target for AIDS therapy? Pathol Biol (Paris) 2001;49:237–46
- Neamati N. A small-molecule antagonist of virion assembly. Expert Opin Investig Drugs 2001;10:1767–70
- Anthony NJ. HIV-1 integrase: a target for new AIDS chemotherapeutics. Curr Top Med Chem 2004;4:979–90
- Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov 2005;4:236–48
- Busschots K, Voet A, De Maeyer M, et al. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J Mol Biol 2007;365:1480–92
- Greene WC, Debyser Z, Ikeda Y, et al. Novel targets for HIV therapy. Antiviral Res 2008;80:251–65
- Cherepanov P, Sun ZY, Rahman S, et al. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol 2005;12:526–32
- Al-Mawsawi LQ, Neamati N. Allosteric inhibitor development targeting HIV-1 integrase. ChemMedChem 2011;6:228–41
- De Luca L, Barreca ML, Ferro S, et al. Pharmacophore-based discovery of small-molecule inhibitors of protein–protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem 2009;4:1311–6
- De Luca L, Ferro S, Gitto R, et al. Small molecules targeting the interaction between HIV-1 integrase and LEDGF/p75 cofactor. Bioorg Med Chem 2010;18:7515–21
- De Luca L, Ferro S, Morreale F, Chimirri A. Inhibition of the interaction between HIV-1 integrase and its cofactor LEDGF/p75: a promising approach in anti-retroviral therapy. Mini Rev Med Chem 2011;11:714–27
- De Luca L, Ferro S, Morreale F, et al. Inhibitors of the interactions between HIV-1 IN and the cofactor LEDGF/p75. ChemMedChem 2011;6:1184–91
- De Luca L, Ferro S, Morreale F, et al. Fragment hopping approach directed at design of HIV IN-LEDGF/p75 interaction inhibitors. J Enzyme Inhib Med Chem 2012. [Epub ahead of print]. doi:10.3109/14756366.2012.703184
- De Luca L, Gitto R, Christ F, et al. 4-[1-(4-Fluorobenzyl)-4-hydroxy-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acid as a prototype to develop dual inhibitors of HIV-1 integration process. Antiviral Res 2011;92:102–7
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48
- De Luca L, Morreale F, Chimirri A. Insight into the fundamental interactions between LEDGF binding site inhibitors and integrase combining docking and molecular dynamics simulations. J Chem Inf Model 2012;52:3245--54
- Cherepanov P, Ambrosio AL, Rahman S, et al. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci USA 2005;102:17308–13
- Accelrys, Catalyst: http://www.accelrys.com
- Sechi M, Bacchi A, Carcelli M, et al. From ligand to complexes: inhibition of human immunodeficiency virus type 1 integrase by beta-diketo acid metal complexes. J Med Chem 2006;49:4248–60
- De Luca L, Barreca ML, Ferro S, et al. A refined pharmacophore model for HIV-1 integrase inhibitors: optimization of potency in the 1H-benzylindole series. Bioorg Med Chem Lett 2008;18:2891–5
- Ferro S, Grazia SD, De Luca L, et al. Microwave assisted organic synthesis (MAOS) of small molecules as potential HIV-1 integrase inhibitors. Molecules 2011;16:6858–70
- De Luca L, De Grazia S, Ferro S, et al. HIV-1 integrase strand-transfer inhibitors: design, synthesis and molecular modeling investigation. Eur J Med Chem 2011;46:756–64
- Ferro S, Barreca ML, De Luca L, et al. New 4-[(1-benzyl-1H-indol-3-yl)carbonyl]-3-hydroxyfuran-2(5H)-ones, beta-diketo acid analogs as HIV-1 integrase inhibitors. Arch Pharm (Weinheim) 2007;340:292–8
- Al-Mawsawi LQ, Christ F, Dayam R, et al. Inhibitory profile of a LEDGF/p75 peptide against HIV-1 integrase: insight into integrase-DNA complex formation and catalysis. FEBS Lett 2008;582:1425–30
- Ortuso F, Langer T, Alcaro S. GBPM: GRID-based pharmacophore model: concept and application studies to protein–protein recognition. Bioinformatics 2006;22:1449–55
- Alcaro S, Artese A, Ceccherini-Silberstein F, et al. Computational analysis of Human Immunodeficiency Virus (HIV) Type-1 reverse transcriptase crystallographic models based on significant conserved residues found in Highly Active Antiretroviral Therapy (HAART)-treated patients. Curr Med Chem 2010;17:290–308
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos (CA): DeLano Scientific LLC; 2008. Available from: http://www.pymol.org