
Abstract
Recently the literature described the binding of Haptoglobin (HP) with ecotin, a fold-specific serine-proteases inhibitor with an anticoagulant profile and produced by Escherichia coli. In this work, we used some in silico and in vitro techniques to evaluate HP 3D-fold and its interaction with wild-type ecotin and two variants. Our data showed HP models conserved trypsin fold, in agreement to the in vitro immunological recognition of HP by trypsin antibodies. The analysis of the three ecotin–HP complexes using the mutants RR and TSRR/R besides the wild type revealed several hydrogen bonds between HP and ecotin secondary site. These data are in agreement with the in vitro PAGE assays that showed the HP-RR complex in native gel conditions. Interestingly, the ternary complex interactions varied depending on the inhibitor structure and site-directed mutation. The interaction of HP with TSRR/R involved new residues compared to wild type, which infers a binding energy increase caused by the mutation.
Introduction
Haptoglobin (HP) is a hemoglobin (HB)-binding protein present in the plasma and synthesized by the liverCitation1. This α2-sialoglycoprotein is important in several human physiological processes by interacting with different proteins such as the plasmatic free HB. HP forms a stable complex with HB that together with the macrophage CD163 receptor promotes the rapid elimination of the HB-reactive ironCitation2–4. This mechanism prevents the oxidative effects caused by the iron permanence in the circulationCitation5,Citation6.
In fact, HP contributes to different biologic events that go beyond simple removal of the toxic HB from plasma. HP is also an acute-phase protein with immunomodulatory propertiesCitation7,Citation8. The increase of HP plasmatic concentration may act as a marker for monitoring infections responses, tumoral processes or general inflammation status in patientsCitation9,Citation10.
Humans present three HP phenotypes due to the presence of two HP gene alleles (HP1 and HP2) that arose by an intragenic duplication of the HP1 allele. In the encoded HP2 protein, the cysteine residue connecting the α-chains is duplicated. Thus HP2-1 and HP2-2 phenotypes display a spectrum of various HP (αβ)-multimers, which possibly modulate these biological profilesCitation6.
HP presents two chains – light (α1 – 9 Kda and α2 – 18 Kda) and heavy (β – 38 Kda) – that associate to form the so-called monomeric HP (HP1). HP dimer (haptoglobin-1-1 (HP-1-1)) is formed by these αβ-chains covalently linked by a disulfide bond between the Cys15 residue of each β-chainCitation11,Citation12. Interestingly, the HP light(α) chain is similar to the complement control proteins (CCP) whereas the heavy(β) chain presents a serine-protease-like sequence but with no complete catalytic site or enzymatic activityCitation13–15.
Until now, the interactions between HP and macromolecular targets of physiological relevance have been explored using molecular modeling approachesCitation11,Citation12,Citation16. However, Andersen et al.Citation4 reported a crystallographic structure of porcine HP–HB complex revealing the core residues to the HP–HB interaction and the protective role of HP in the hem-induced reactive oxygen species generation.
In recent studies developed by our group, we showed that HP interacts with ecotin, a periplasmic Escherichia coli-derived protein (16 kDa) and a “fold-specific” inhibitor. Ecotin has an unusually broad specificity to trypsin-like proteases, thus presenting different biological profiles (e.g. anticoagulant, antiplatelet and anticancer)Citation17,Citation18. This serine-protease inhibitor (142 amino acid residues) shows a singular binding mode using two distinct binding sites to recognize the enzyme targetCitation19. Thus, this inhibitor is able to form a tetrameric structure with trypsin-like serine proteasesCitation20.
Serine proteases with a trypsin fold are involved in many biological processes, including blood coagulationCitation21, metastasis of cancer cellsCitation22, fibrinolysisCitation23, mammary gland involutionCitation24 and envenomation processCitation25. Some of these enzymes are inhibited by ecotin such as trypsin, chymotrypsin, elastase, factor Xa, kallikrein and factor XIIaCitation20,Citation26. Interestingly, ecotin can be found in circulation until 12–14 h after intravenous administration in contrast to other proteins with similar molecular weight (∼30 min)Citation27. Apparently, this pharmacokinetic feature occurs due to ecotin binding to HP that would act as a reservoir or as a carrier protein for ecotin in blood, thus amplifying its pharmacodynamic profileCitation20.
To identify theoretically the binding mode of ecotin to a serine-protease-related protein and to reinforce HP as a target in mammals blood to this inhibitor, herein we used an in silico approach to evaluate the human HP fold. In addition, we also analyzed human HP similarity to serine-proteases and to the recent porcine crystallographic structure to further evaluate its interaction with ecotin and two variants M84R/M85R (RR), D70R, V81T, T83S, M84R and M85R (TSRR/R) designed against two different serine proteases (u-PA and thrombin, respectively)Citation27.
Materials and methods
Native page and western-blot analysis
Protein samples were electrophoresed on 10% SDS-PAGE or Native-PAGE according to laemmliCitation28. For immunoblot analysis, protein bands were transferred onto a nitrocellulose membrane and treated with tris-buffered saline with 0.1% (v/v) Triton X-100 pH 7.4 (TBST, SIGMA®, St. Louis, MO) containing 5% (w/v) non-fat dry milk. The membrane was probed with a 1:5000 dilution of the specific antibody in the same buffer for 1 h and washed thrice with TBST. A 1:5000 dilution of goat antirabbit horseradish peroxidase conjugated serum (Pierce) was applied in TBST-1% milk and incubated for 1 h with the membrane. The membrane was washed and antibody-bound protein bands were detected by enhanced chemiluminescenceCitation29.
For the Native gel, ecotin-RR (8 μg) was incubated with HP (8 μg) in 20 mM tris-HCl (pH 7.5) after testing different concentrations for selecting the best result. Following a 1 h at 37 °C, two aliquots were taken for analysis. One was subjected to electrophoresis on a nondenaturing gel (20% acrylamide) to observe the ecotin–HP complex formation, as described by Maruyama and coworkersCitation30. Urokinase-type plasminogen activator (u-PA)(1 IU) was added to the other aliquot, and the reaction was started after 2 min at 37 °C by the addition of SPECTROZYME®UK (chromogenic substrate for the amidolytic assay of urokinase -- American Diagnostica Inc., Stamford, CT) (0.2 mM, final concentration), as described by Castro and coworkersCitation20). Absorbance was followed at 450 nm.
Molecular modeling
Dimeric HP-1-1: construction and comparative evaluation
Haptoglobin 1 (HP1) sequence was obtained from the database of genes (GenBank) of the National Center for Biotechnology Information (NCBI) (accession number X00637) after decodification from the human DNA sequence. Protein similarity was determined for both heavy and light chains by multiple sequence alignments performed in Clustal W2 software (www.ebi.ac.uk/Tools/msa/clustalw2), using selected proteins previously describedCitation3,Citation12,Citation31.
Briefly, the construction of dimeric HP structure was performed in three steps including the construction of HP: (1) light(α) and heavy(β) subunits, (2) monomer and (3) dimerCitation32. On that purpose, we used the Swiss-Model and Deepview/Swiss-PDB Viewer Version 4.04 programs (http://swissmodel.expasy.org/; http://spdbv.vital-it.ch/)Citation33,Citation34 and zymogen and active catalytic domain of complement protease c1r (PDB ID:1gpza and 1md8a, respectively) as templatesCitation14,Citation35,Citation36. Initially the 18N-terminal amino acids of the HP1 sequences, which represent a signal peptide, were previously removed, once these are released during the proteolytic maturation of the polypeptide. Then, after constructing HP1 monomers as described by Abreu et al.Citation32, we structurally aligned them with the dimer protease C1r crystal structure (PDB ID: 1gpz) in an antiparallel position as proposed by Polticelli and coworkersCitation16,Citation35. To construct the HP dimeric structure, we used the report of the Ettrich et al.Citation12 electronic microscopy data to align the subunits and establish the disulfide bond between the cysteines 15(Cys15) of the light chains(α).
Construction of theoretical complexes of the HP-1-1 with the ecotin variants
The construction of ecotin–HP complexes was performed as described by Castro and collaborators in 2001Citation37. Initially the crystal structure of the mutant ecotin homodimeric (Y69F, D70P) bound to mutant anionic trypsin II (D102N) (PDB ID: 1ezu) was selected as templateCitation31,Citation38,Citation39. Then the ecotin was aligned with the wild type or mutant (RR – M84R,M85R and TSRR/R – D70R, V81T, T83S, M84R/M85R) ecotin whereas the trypsin was aligned with the crystal structure of u-PA (PDB ID: 1gja)Citation40. Then, for the construction of the ternary complex (enzyme--ecotin--HP), the HP1 dimer model were aligned with the ecotin–u-PA final complex using the program Deep View/Swiss-PDB Viewer 4.0Citation14.
Optimization, validation and interactions analyses
All models were submitted to successive geometry optimization steps using the Gromos96 force field, available in the program Deepview/Swiss-PDB Viewer Version 4.0Citation14, to minimize energy. This program also allowed the evaluation of other parameters such as electrostatic potential map, area and volume. The models were validated by the analysis of the Ramachandran plot. We also used the Procheck program in the PDBsum server to analyze the interactions (http://www.ebi.ac.uk/pdbsum)Citation41. Since the crystallographic structure of Porcine HP bound to HB (PDB ID: 4F4O) was recently releasedCitation4, we compared it with our theoretical model of the human HP using Swiss PDB Viewer to establish the overall root mean square deviation and to verify the structural feasibility of HP physiological protective role (binding to HB) while bound to ecotin.
Results and discussion
Recently we reported the interaction of HP with ecotin, a trypsin-fold enzyme inhibitor using affinity columnsCitation20. In this work we enlightened this binding using an in silico approach to analyze HP-1-1 three-dimensional structure, the HP–ecotin complex binding mode and the feasibility of a ternary HP–ecotin–serine-protease complex.
Constructing HP-1-1
HP-1 light(α) and heavy(β) chains
The multiple sequence alignment evaluation of the primary sequences of HP1 light(α) and heavy(β) chains showed that HP1 light chain(α) primary structure is similar to apolipoprotein – H (PDB = 1c1za), complement system proteases C1r (PDB ID: 1gpza) and C1s (PDB ID: 1elva) whereas HP1 heavy chain(β) conserved several aminoacids residues present in Trypsin superfamilyCitation35,Citation42,Citation43. The similarity reached from 21% to 33% with low number of identical residues when compared to C1r proteases, elastase, chymase, trypsin, chymotrypsin, thrombin and coagulation factor X ().
Figure 1. Primary and Secondary structure analyses of HP-1 light(α) and heavy(β) chains (HP1a and HP1b, respectively). The multiple sequence alignment show the identical (*), conserved (:) or semi conserved (.) aminoacid residues in the proteins analyzed by using the Clustal–Omega program (http://www.ebi.ac.uk/clustalo). HP was compared to similar proteins including the complement system proteases C1r (zymogen – 1gpza, and catalytic active domain – 1md8a) and C1s (1elva), apolipoprotein-H (1c1za), chymotrypsin (1ca0), elastase (1brup), chymase (1pjp), trypsin (1sgt), thrombin (1a2c) and coagulation factor X (1c5 m). The secondary structures were predicted by using the Jpred program (http://www.compbio.dundee.ac.uk/~www–jpred), and are shown in the protein sequence (β-strands in green, α-helices in red and extended regions in blue). Dashes represent gaps and the sequence C-terminal of complement system proteases C1r zymogen (1gpza) and C1s (1elva) and apolipoprotein-H (1c1za) was suppressed and represented by a gray block in HP1a sequence alignment. The binding sites for HB and the macrophage scavenger receptor CD163 (CD163) are marked in light pink and underlined, respectively. VPEKKT motif of the HP loop involved in the recognition of CD163 is marked in yellow. The position of the catalytic triad of the serine proteases are boxed and numbered according to the Trypsin superfamily. See the colored picture on the online version.

Despite the low number of identical residues, the theoretical prediction of the secondary structures of the HP1 light(α) and heavy(β) chains pointed to the conservation of the main structure for both chains as previously discussed by other authors. In the HP1b, the prediction for the C-terminal region (α-helix) and the core of the structure (β-sheets) are related to the β–β barrel formation expected for Trypsin superfamily members (). Interestingly, the HP-binding sites for both HB and macrophage CD163 receptor are not conserved regions for other trypsin family members. On the other hand, the usual serine proteases catalytic triad (His57, Asp102, Ser195) is not fully present in HP (Lys57, Asp102, Ala195), resulting in the lack of an enzymatic activity.
The theoretical model of HP1 light(α) chain using C1r complement protease (PDB ID: 1gpza) as template showed an ellipsoidal structure containing six β-sheets ()Citation35. The model was similar to that reported by Ettrich et al.Citation12 and to other complement system proteins crystal structure. Meanwhile, HP1 heavy(β) chain model was constructed based on the monomeric structure of the active catalytic domain of complement protease C1r (PDB ID: 1md8a) and using Polticelli et al. dataCitation16,Citation36. This model revealed a conserved serine proteases globular shape, with two groups of β-sheets and two α-helices as previously predicted (). As expected, the Western blotting analysis of HP1 using Trypsin and HP antibodies reinforced our theoretical data as the recognition by both Abs suggested similar structural regions (). The data are in accordance to the current literature that reports the purification of HP through an ecotin-RR affinity column, which implies the full recognitionCitation27.
Figure 2. Comparison of the theoretical models of HP-1 light(α) and heavy(β) chains (HP1a and HP1b, respectively) with homologous proteins. Each protein secondary structures (β-strands in green, α-helices in red and extended regions in blue) on the left, and the electrostatic potential map (positive and negative regions in blue and red respectively) on the right were predicted by using Deepview/Swiss-PDB Viewer. See the colored picture on the online version.
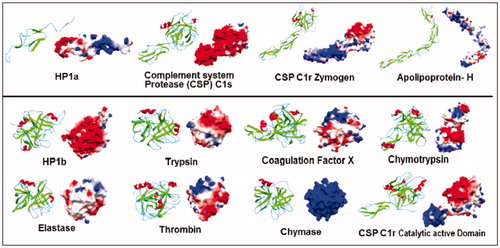
Figure 3. Comparison of the theoretical models of HP-1 monomeric (left) and dimeric (right) forms colored by the type of chain – light(α) and heavy(β) (yellow and purple, respectively), secondary structures (β-strands in green, α-helices in red and extended regions in blue) and electrostatic potential distribution (positive and negative regions in blue and red, respectively). Important structural regions are pointed including HB and macrophages CD163 receptor binding sites and the main disulfide bonds (arrows). See the colored picture on the online version.
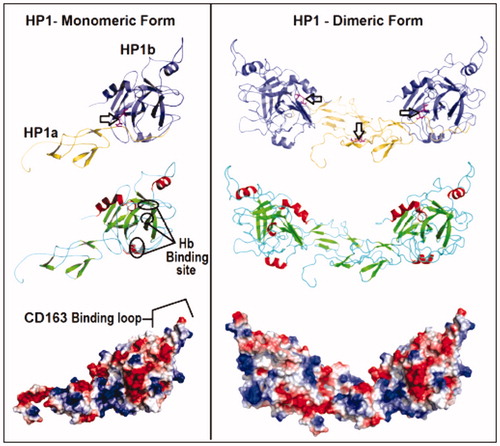
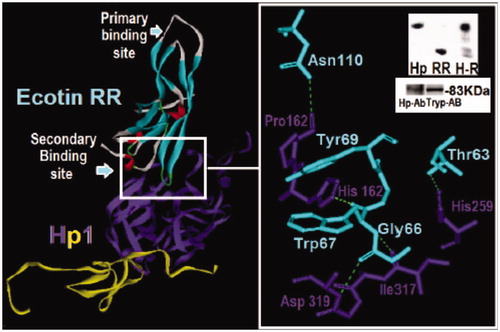
Figure 4. Structural analysis of the dimeric HP and ecotin-RR theoretical complex. Only one HP1 monomer (HP1a and HP1b) is depicted for clarity. HP1a is in yellow and HP1b in purple whereas ecotin β-strands are shown in light blue, α-helices in red and extended regions in light gray. The region of the interaction of ecotin-RR secondary binding site and HP1 residues are zoomed and show the hydrogen bonds (light green traces) formed on the right. Inset: Native PAGE of HP and ecotin-RR incubation leading to the complex H-R formation (up) and the Western-blotting assay using HP and Trypsin antibodies to recognize HP1 alone (down), reinforcing that HP is a Trypsin-related protein probably at both 3D-folding structure and immunogenic levels. See the colored picture on the online version.
Interestingly, despite the similarity to serine proteases, the analysis of the HP electrostatic potential map revealed a different charge distribution for each chain evaluated and for the enzymes evaluated. The negatively charged profile with few positive sites of HP1 heavy(α) chain in contrast to the light(β) chain probably fine-tune the HB binding followed by macrophage CD163 receptor recognition (). Thus, despite the conserved fold structure, apparently the electronic, electrostatic and conformational features still guarantee the specificity of these proteins, without affecting the immunogenic structure.
HP-1: monomer (HP1) and dimer (HP-1-1)
We used the complement protease C1r (PDB ID:1gpza) as template to align the HP1 light(α) and heavy(β) chains and generate the disulfide bonds between cysteines (Cys72-Cys189) for constructing the so-called HP monomer (HP1), as previously reportedCitation3,Citation16.
The final HP1 monomeric model showed an area of 16 Å2 and volume of 42.6 Å3. The parameters of the potential map were evaluated revealing a distributed electronegative profile (). Interestingly, the HP-binding region for macrophage CD163 receptor is externalized, which suggests the feasibility of interacting with HB and CD163 simultaneously ().
The Ramachandran plot evaluation of HP1 monomer was similar to that of the crystal structure of the complement protease C1r zymogen catalytic domain used as template (PDB ID: 1gpza). Thus 208 amino acids residues were in favored regions (73.2%), 74 in allowed areas (25.3%) and only 4 (Ala60, His131, Gln152 and Lys 214) in prohibited regions (1.4%) (not shown).
The theoretical model of the dimeric HP-1-1 was assembled by aligning two monomers. The structure was stable and similar to those described by other authors including Wejman et al.Citation16,Citation44 (). According to the potential electrostatic map analysis, HP-1-1 dimer presented an electronegative profile similar to the monomeric form (). The validation step using the procheck program showed a similar protease C1r (PDB ID: 1gpz) pattern with 72.7% of the amino acids residues in favored regions (413 amino acids), 25.9% in allowed areas (147 amino acids) and 1.4% (8 amino acids) in prohibited regions (not shown).
HP-1-1–ecotin theoretical complexes: evaluating a specific fold inhibitor
Currently, many researches aim to understand the relationship between the protein structure, the biological activity, binding interactions, the recognition and affinity. According to the literature, in order to understand protein selectivity, both sequences analysis and three-dimensional structures should be considered. Some of these studies evaluate the protein targets and their three-dimensional complexes to get into their biological functions and interactionsCitation32,Citation45,Citation46.
In this work, we used molecular modeling techniques to explore the binding mode of the ternary complex formed by HP-1-1 (dimer), ecotin (wild type or variants) and the urokinase-type plasminogen activator (u-PA – a trypsin-like serine-protease). Since HP does not affect ecotin ability to inhibit u-PA according to the literatureCitation20, the analysis of a feasible secondary binding mode proposed by the literature was verifiedCitation39. Thus, u-PA and HP were bound to ecotin primary and secondary sites, respectively, based on the Laboissiere et al. dataCitation47.
Our theoretical data showed that ecotin-RR is able to bind to HP through its secondary binding (HP-1-1 – Ecot, Asp320 – Gly66, His162 – Trp67 and Asn110 – Pro163) as suggested by the literatureCitation20 and reinforcing the trypsin-like structure of HP. According to the data reported in the literature about the HP purification from plasma through an ecotin-RR affinity columnCitation27, the in vitro Trypsin antibodies recognition assay and the ecotinRR-HP binding assay using native gel electrophoresis () also added more significance to the theoretical data.
In this work we constructed the ternary complex since it is theoretically feasible that HP-1-1 interacts with ecotin through its secondary binding site whereas simultaneously allows ecotin inhibition of proteases (e.g. u-PA)Citation20 ().
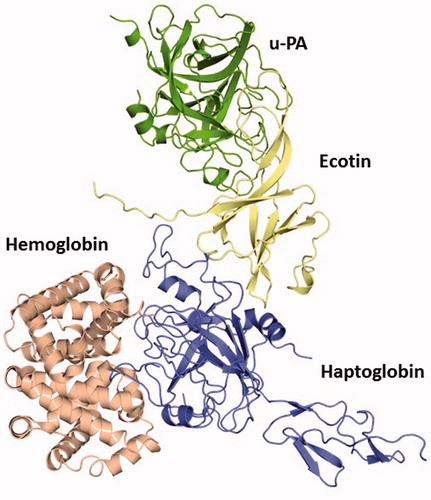
Figure 5. Comparison of ternary models formed by HP-1 dimer form (HP1) (blue), urokinase-type plasminogen activator (u-PA) (red) and wild-type ecotin (green) in (a) or with mutation in the primary binding site (M84R/M85R) (RR – dark yellow) in (b) or in both ecotin binding sites (D70R, V81T, T83S, M84R and M85R) (TSRR/R gray) in (c). The ecotin–aminoacid residues involved in HP and u-PA binding are in CPK. The insets report the number of aminoacid residues involved in hydrogen bonds that are higher for HP1 when a secondary binding mutation is added as in TSRR/R. The number (2) points the presence of two interactions simultaneously. See the colored picture on the online version.
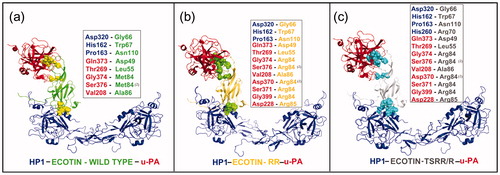
Interestingly, our results led to feasible ternary complexes, but with an interaction pattern slightly different (). Significantly, the interface region involving the u-PA catalytic site and ecotin primary site presented a higher number of interactions (e.g. hydrogen bonds and hydrophobic interactions) than that of ecotin secondary binding site and the dimeric HP (). According to the literature, mutations in primary and secondary sites of ecotin allow different interactions with u-PA (39). Wang et al.Citation48 showed that the wild-type ecotin has lower affinity to u-PA than its mutants Met84Arg and Met85Arg (RR), which are in accord to our molecular modeling results.
Our data reinforced that each mutation modulates ecotin interaction with this enzyme increasing the interactions network. Thus, probably due to an increase of positive charges in the binding region, the two ecotin mutant theoretical models revealed ecotin-RR (M84R and M85R) with more interactions with u-PA than wild-type ecotin (e.g. RR-u-PA hydrogen bonds: Arg85 – Asp228 and Arg84 – Asp370, Ser371, Gly399) ().
Apparently, only the mutation in the secondary binding site of ecotin TSRR/R led to an increase of feasible interactions with HP-1-1 due to the redistribution of hydrogen bonds in this region (e.g. Arg70 of ecotin TSRR/R and His260 of HP-1-1) ().
Compared to the wild-type ecotin, tThe interaction of HP with ecotin mutants (mainly TSRR/R) generates an improved connection pattern concerning the involvement of several new residues. This included the hydrogen bond between Arg70 of ecotin TSRR/R and His260 of HP-1-1, and a new set of bonds that increased the interaction between these molecules (). These data reinforced HP as a feasible reservoir or carrier for ecotin in plasma, which may be responsible for the increased ecotin circulation time in the blood (12–14 h)Citation27.
Apparently, chances are low to the formation of ecotin–HP complex through ecotin primary site since HP-1-1 lacks the serine-protease characteristic active site, leading to a structural rearrangement, steric impairment and ultimately avoidance of ecotin binding (not shown)Citation46.
Altogether, our experimental and theoretical data reinforced the hypothesis of HP-1-1 as a trypsin-related target for ecotin in mammals blood. Experiments incubating bovine serum with the ecotin-RR column confirmed HP as the major ecotin-binding protein in the plasma, also reinforcing this hypothesisCitation27.
Finally, according to our theoretical results, the HP-binding region for CD163 receptor (Val241, Glu243, Lys244 and Thr246)Citation3 and the 212–221 loop of HP heavy chain involved in binding to HBCitation37 were not affected by the ecotin interaction, inferring no interference to HP biological profile.
Porcine and human HPs: evaluating the HP molecular model
Finally, our human HP model was compared to the recent crystallographic structure of porcine HP, which revealed an overall root mean square deviation of 1.88 Å. This reinforced our model as a good theoretical prediction to the Human HP 3D structure, also suggesting that even when bond to other macromolecules, such as HB, conformational changes are unlikely to occur in HP. In addition, when compared to the porcine HP–HB complex, we observed the conservation of the 212–221 loop as a non-occupied position in our human HP model when complexed with ecotin. This suggests that human HP is able to establish feasible interactions with HB through this loop to protect it from oxidative modification even when bound to ecotin WT and engineered variants with maintenance of the HP physiological role. The complex of ecotin, the target enzyme (u-PA), the HP and HB is theoretically feasible since all the binding regions are free to hold this structure interacting together (). Thus, these results reinforced the recent experimental data published that suggested the ability of HP acting as reservoir of this inhibitor without losing its own physiologic activity or affecting ecotin biological profile.
Figure 6. The quaternary complex HB–human HP–ecotin--uPA constructed based on the structural alignment with the crystal structure of the complex porcine HP–HB (PDB ID: 4F4O). This theoretical data revealed that even bound to ecotin, HP is able to establish feasible interactions to protect key HB residues from oxidative modification after exposure to hem-induced ROS. Thus, this complex points to the maintenance of HP physiological role despite its binding to this inhibitor. HB (light red), HP (blue), ecotin (yellow) and u-PA (green). See the colored picture on the online version.
Conclusions
In this work we evaluated the binding mode of a ternary complex formed by HP-1-1 dimer, u-PA and anticoagulant ecotin variants to identify their interactions and the feasible role of HP as a carrier of ecotin in mammals blood. Each ternary complex with ecotin variants (wild type, RR and TSRR/R) displayed a particular interaction panel and pointed for the feasibility of the maintenance of each protein biological activity (HP and ecotin). All together, our experimental and theoretical data reinforced the potential of HP as a reservoir or a carrier protein in blood for ecotin.
Declaration of interest
The authors report no conflicts of interest.
We thank the support of Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal Docente (CAPES), and Programa de Pós-Graduação da Universidade Federal Fluminense (UFF) for the financial support and H.C.C. and P. S. fellowships. TT and CSC were supported by NIH grant CA072006.
References
- Levy AP, Asleh R, Blum S, et al. Haptoglobin: basic and clinical aspects. Antioxidants Redox Signal 2010;12:293–304
- Alayash AI. Haptoglobin: old protein with new functions. Clin Chim Acta 2011;412:493–8
- Nielsen MJ, Petersen SV, Jacobsen C, et al. A unique loop extension in the serine protease domain of haptoglobin is essential for CD163 recognition of the haptoglobin-hemoglobin complex. J Biol Chem 2007;28:1072–9
- Andersen CBF, Torvund-Jensen M, Nielsen MJ, et al. Structure of the haptoglobin-HB complex. Nature 2012;489:456–9
- Theilgaard-Mönch K, Jacobsen LC, Nielsen MJ, et al. Haptoglobin is synthesized during granulocyte differentiation, stored in specific granules, and released by neutrophils in response to activation. Blood 2006;108:353–61
- Van Vlierberghe H, Langlois M, Delanghe J. Haptoglobin polymorphisms and iron homeostasis in health and in disease. Clin Chim Acta 2004;345:35–42
- Wassell J. Haptoglobin: function and polymorphism. Clin Labo 2000;46:547–52
- Wicher KB, Fries E. Evolutionary aspects of hemoglobin scavengers. Antioxidants Redox Signal 2010;12:249–59
- Ye B, Cramer DW, Skates SJ, et al. Haptoglobin-alpha subunit as potential serum biomarker in ovarian cancer. Clin Cancer Res 2003;9:2904–11
- Zhang S, Shu H, Luo K, et al. N-linked glycan changes of serum haptoglobin beta chain in liver disease patients. Mol BioSyst 2011;7:1621–8
- Boonyapranai K, Tsai HY, Chen MC, et al. Glycoproteomic analysis and molecular modeling of haptoglobin multimers. Electrophoresis 2011;32:1422–32
- Ettrich R, Brandt W, Baumruk V, et al. Study of chaperone-like activity of human haptoglobin: conformational changes under heat shock conditions and localization of interaction sites. Biol Chem 2002;383:1667–76
- Di Cera E. Serine proteases. IUBMB Life 2009;61:510–15
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 1997;18:2714–23
- Varfolomeev SD, Gariev IA, Uporov IV. Catalytic sites of hydrolases: structures and catalytic cycles. Russian Chem Rev 2005;74:61
- Polticelli F, Bocedi A, Minervini G, Ascenzi P. Human haptoglobin structure and function -- a molecular modelling study. FEBS J 2008;275:5648–56
- Yang SQ, Craik CS. Engineering bidentate macromolecular inhibitors for trypsin and urokinase-type plasminogen activator1. J Mol Biol 1998;279:1001–11
- Zani ML, Moreau T. Phage display as a powerful tool to engineer protease inhibitors. Biochimie 2010;92:1689–704
- Bachovchin DA, Cravatt BF. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat Rev Drug Discov 2012;11:52–68
- Castro HC, Monteiro RQ, Assafim M, et al. Ecotin modulates thrombin activity through exosite-2 interactions. Int J Biochem Cell Biol 2006;38:1893–900
- Krarup A, Wallis R, Presanis JS, et al. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One 2007;2:e623
- Dass K, Ahmad A, Azmi AS, et al. Evolving role of uPA/uPAR system in human cancers. Cancer Treatment Rev 2008;34:122–36
- Parmar N, Albisetti M, Berry LR, Chan AKC. The fibrinolytic system in newborns and children. Clin Lab 2006;52:115–24
- Watson CJ. Post-lactational mammary gland regression: molecular basis and implications for breast cancer. Expert Rev Mol Med 2006;8:1–15
- De Lima DC, Alvarez Abreu P, De Freitas CC, et al. Snake venom: any clue for antibiotics and cam? Evid Based Complement Alternat Med 2005;2:39–47
- McGrath ME, Gillmor SA, Fletterick RJ. Ecotin: lessons on survival in a protease-filled world. Protein Sci 1995;4:141–8
- Sathler PC, Craik CS, Takeuchi T, et al. Engineering ecotin for identifying proteins with a trypsin fold. Appl Biochem Biotechnol 2010;160:2355–65
- Laemmli UK. Cleavage of structural proteins during the assembly of bacteriophage T4. Nature 1970;227:680–5
- McGrath ME, Hines WM, Sakanari JA, et al. The sequence and reactive site of ecotin. A general inhibitor of pancreatic serine proteases from Escherichia coli. J Biol Chem 1991;266:6620–5
- Maruyama M, Sugiki M, Yoshida E, et al. Broad substrate specificity of snake venom fibrinolytic enzymes: possible role in haemorrhage. Toxicon 1992;30:1387–97
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Molr Biol 1990;215:403–10
- Abreu PA, Albuquerque MG, Rodrigues CR, Castro HC. Structure–function inferences based on molecular modeling, sequence-based methods and biological data analysis of snake venom lectins. Toxicon 2006;48:690–701
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006;22:195–201
- Kiefer F, Arnold K, Künzli M, et al. The SWISS-MODEL repository and associated resources. Nucleic Acids Res 2009;37:D387–92
- Budayova-Spano M, Lacroix M, Thielens NM, et al. The crystal structure of the zymogen catalytic domain of complement protease C1r reveals that a disruptive mechanical stress is required to trigger activation of the C1 complex. EMBO J 2002;21:231–9
- Budayova-Spano M, Grabarse W, Thielens NM, et al. Monomeric structures of the zymogen and active catalytic domain of complement protease c1r: further insights into the c1 activation mechanism. Structure 2002;10:1509–19
- Castro HC, Silva DM, Craik C, Zingali RB. Structural features of a snake venom thrombin-like enzyme: thrombin and trypsin on a single catalytic platform? Biochim Biophys Acta – Protein Struct Mol Enzymol 2001;1547:183–95
- Bernstein FC, Koetzle TF, Williams GJ., et al. The protein data bank: a computer-based archival file for macromolecular structures. J Mol Biol 1977;112:535–42
- Gillmor SA, Takeuchi T, Yang SQ, et al. Compromise and accommodation in ecotin, a dimeric macromolecular inhibitor of serine proteases1. J Mol Biol 2000;299:993–1003
- Katz BA, Sprengeler PA, Luong C, et al. Engineering inhibitors highly selective for the S1 sites of Ser190 trypsin-like serine protease drug targets. Chem Biol 2001;8:1107–21
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993;26:283–91
- Schwarzenbacher R, Zeth K, Diederichs K, et al. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J 1999;18:6228–39
- Gaboriaud C, Rossi V, Bally I, et al. Crystal structure of the catalytic domain of human complement c1s: a serine protease with a handle. EMBO J 2000;19:1755–65
- Wejman JC, Hovsepian D, Wall JS, et al. Structure of haptoglobin and the haptoglobin-hemoglobin complex by electron microscopy. J Mol Biol 1984;174:319–41
- Tanramluk D, Schreyer A, Pitt WR, Blundell TL. On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine. Chem Biol Drug Des 2009;74:16–24
- Varshney A, Sen P, Ahmad E, et al. Ligand binding strategies of human serum albumin: how can the cargo be utilized? Chirality 2010;22:77–87
- Laboissiere MC, Young MM, Pinho RG, et al. Computer-assisted mutagenesis of ecotin to engineer its secondary binding site for urokinase inhibition. J Biol Chem 2002;277:26623–31
- Wang CI, Yang Q, Craik CS. Isolation of a high affinity inhibitor of urokinase-type plasminogen activator by phage display of ecotin. J Biol Chem 1995;270:12250–6