
Abstract
A series of new 5-piperidinyl and 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones have been synthesized and evaluated for dual D2 and 5-HT1A receptor binding affinities. The synthesized ligands are structurally related to bifeprunox, a potential atypical antipsychotic, having potent D2 receptor antagonist and 5-HT1A receptor agonist properties. The Suzuki–Miyaura reaction of cyclic vinyl boronate with appropriate aryl halide yielded arylpiperidine, which was eventually transformed to piperidinyl-1H-benzo[d]imidazol-2(3H)-one. The reductive amination of the latter with appropriate biarylaldehdyes rendered the synthesis of 5-piperidinyl-1H-benzo[d]imidazol-2(3H)-ones. Likewise, the Buchwald–Hartwig coupling reactions of 1-boc-piperazine with appropriate aryl halide and subsequent removal of the boc group rendered arylpiperazine. The reductive amination of the latter with appropriate biarylaldehdyes accomplished the synthesis of 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones. The structure–activity relationship studies showed that cyclopentenylpyridine and cyclopentenylbenzyl groups contribute significantly to the dual D2 and 5-HT1A receptor binding affinities of these compounds.
Introduction
Schizophrenia is a severe psychiatric illness afflicting 1% of the population worldwide. Diagnosis is based on diverse and variably expressed symptoms which can be grouped as positive and negative. The positive symptoms include disorganized thought, delusions and auditory hallucinations, whereas the most characteristic negative symptoms are emotional flattening, poverty of speech and motivational deficitsCitation1. Although various molecular mechanisms have been proposed to provide antipsychotic activityCitation2, antagonism of the dopamine D2 receptor subtype remains the cornerstone of antipsychotic activityCitation3,Citation4. Chlorpromazine (1) and haloperidol (2a), the first-generation antipsychotics or typical antipsychotics, are dopamine antagonists and exhibit robust control of positive symptoms of schizophrenia, for instance hallucinations, agitation and delusions but fail to control the negative symptoms such as blunted affect, emotional withdrawal and cognitive deficits. In addition, motor retardation remains uncontrolled with these therapeutics for most schizophrenic patientsCitation5. Moreover, selective D2 receptor antagonists block nigrostriatal dopaminergic activity, which leads to extrapyramidal symptoms (EPS), such as dystonia and dyskinesia, and also result in the blockade of pituitary-located D2 receptors that control prolactin release, leading to hyperprolactinemiaCitation6a. The “second-generation” or atypical antipsychotics, such as clozapine (2b), combine D2 receptor antagonism with activity at other receptors, on the premise that a suitable balance of pharmacological activity should broaden the spectrum of therapeutic efficacy and reduce EPS. Clozapine exhibits partial agonist efficacy for 5-HT1A receptor-mediated stimulation of G-protein activationCitation6b, which accounts for part of the activity of clozapine in a model of anxiolytic-like activity: clozapine inhibited stress-induced ultrasonic vocalization in rats, an effect attenuated by selective 5-HT1A antagonist WAY-100635Citation6c. With respect to classical neuroleptics, clozapine shows significantly greater efficacy, including an improved effect on negative symptoms, and causes a marked increase in dopamine output in the prefrontal cortexCitation7. Clozapine, however, is associated with its own set of serious side effects, including weight gain, diabetes and an increased risk of seizures and agranulocytosisCitation8.
Although the utility of 5-HT1A receptor agonism in the treatment of schizophrenia is clearly evident, the optimal level of activation of this target is debatable. To achieve improved overall therapeutic benefit, combining D2 receptor blockade with 5-HT1A receptor activation rather than antagonism has been a subject of recent research attentionCitation9,Citation10. However, robust protection from EPS elicited by potent D2 receptor blockade likely requires high-efficacy agonism at 5-HT1A receptorsCitation11a; whereas lower efficacy at 5-HT1A receptors should be sufficient when combined with D2 receptor partial agonism. Several preclinical observations suggest that combining 5-HT1A and D2 receptor properties may provide a mutually complementary balance of pharmacological activity with reduced undesirable responsesCitation11b. Indeed, numerous mechanistic considerationsCitation12–14 and preclinical evidencesCitation15–17 support the potential of such a combination. Consequently, adoprazine (3) (SLV-313) and bifeprunox (4), bearing potent D2 receptor antagonist and 5-HT1A receptor agonist properties, were developedCitation18 (). Although compound 4 completed phase III clinical trials, its antipsychotic efficacy was determined to be inferior to that of risperidone and olanzapineCitation9 and, despite a satisfactory tolerance profile, the US Food and Drug Administration (FDA) did not grant marketing approval. The lack of sufficient antipsychotic efficacy of 4 likely reflects its marked agonism at D2 receptors. Thus, 4 suppresses basal firing rates of dopaminergic neurons in the ventral tegmental area and elicits circling behavior in rats unilaterally lesioned with 6-OH-DACitation9. In addition, the failure of 3 and 4 to oppose phencyclidine-induced social interaction deficits suggested that an appropriate “balance” of activity at these sites is necessary for activity in this modelCitation19. Thus, the need to discover compounds having varying ratios of D2 and 5-HT1A activities continuedCitation20.
Figure 1. Chlorpromazine, haloperidol, clozapine, adoprazine and bifeprunox.
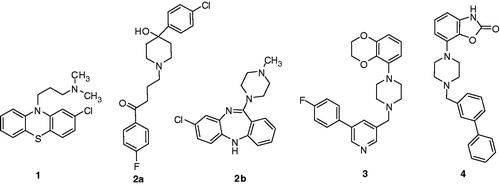
In an ongoing effort to develop new antipsychoticsCitation21,Citation22, we have synthesized a series of 5-piperidinyl-1H-benzo[d]imidazol-2(3H)-ones (5a–f) and 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones (6a–f), which are structural analogs of bifeprunox (4) (). Herein, we wish to disclose the synthesis, dual D2 and 5-HT1A receptor binding affinities and structure–activity relationship (SAR) of these compounds.
Figure 2. 5-piperidinyl-1H-benzo[d]imidazol-2(3H)-ones (5a–f) and 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones (6a–f).
![Figure 2. 5-piperidinyl-1H-benzo[d]imidazol-2(3H)-ones (5a–f) and 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones (6a–f).](/cms/asset/6371567f-c33b-4f1b-bb9a-4c5ae930c90d/ienz_a_776556_f0002_b.jpg)
Materials and methods
Chemistry
Instrumentation: Melting points were determined on a Büchi apparatus (Büchi Labortechnik AG, Flawil, Switzerland) and are uncorrected. Elemental analysis was carried out on a Perkin Elmer Elemental Analyzer Series 11 Model 2400 (PerkinElmer Inc., Waltham, MA). IR spectra were recorded on a Perkin Elmer 16F PC FTIR spectrophotometer (PerkinElmer Inc., Waltham, MA). 1H and 13C NMR spectra were measured in CDCl3 and d6-DMSO using TMS as internal standard on a JEOL JNM-LA 500 MHz spectrometer (JEOL USA Inc., Peabody, MA). Mass spectra were recorded on Agilent Technologies 6890N GC-MS system (Agilent Technologies Inc., Santa Clara, CA). Analytical TLC was carried out on silica gel 60 F254 plates (E. Merck, Darmstadt, Germany); column chromatography was carried out on Merck silica gel (200–400 mesh, E. Merck).
di-tert-Butyl 5-bromo-2-oxo-1H-benzo[d]imidazole-1,3(2H)-dicarboxylate (11): To a solution of compound 10 (0.4 g, 1.88 mmol) in anhydrous THF (15 mL) di-tert-butyl dicarbonate was added (1.64 g, 7.52 mmol) followed by the addition of 4-dimethylaminopyridine (0.23 g, 1.88 mmol) and the mixture was stirred overnight at room temperature. The mixture was diluted with ethyl acetate (25 mL) and washed successively with water (15 mL), brine (15 mL), dried over Na2SO4 and evaporated. Column chromatography on silica gel, eluting with ethyl acetate:hexanes (10:90) and then the ratio was changed to 20:80 to obtain the title compound 11. Yield: 94%, colorless amorphous solid, m.p. 109 °C–110 °C. IR (KBr, cm−1) 3041 (Ar-H), 2978 (Alph-H), 1691 (C=O), 1612, 1442 (C=C), 1222 (C-N), 1175 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.63 (s, 18H, OC(CH3)3), 7.33 (dd, J = 2.3, 8.4 Hz, 1H, H-6), 7.74 (d, J = 8.4 Hz, 1H, H-7), 8.08 (d, J = 2.4 Hz, 1H, H-4). 13C NMR (125.7 MHz, CDCl3) δ 27.76 (OC(CH3)3), 85.66 (OC(CH3)3), 115.26 (C-4), 117.26 (C-7), 125.15 (C-5), 127.08 (C-6), 146.73 (C-8), 148.17 (C-3), 154.68 (CO), 155. 46 (CO). Anal. Calcd for C17H21BrN2O5 (%): C, 49.41; H, 5.12; N, 6.78. Found (%): C, 49.35; H, 5.17; N, 6.68.
tert-Butyl 4-(4-amino-3-nitrophenyl)-5,6-dihydropyridine-1(2H)-carboxylate (14): To a nitrogen flushed flask containing the boronate (12) (1.39 g, 4.5 mmol), K2CO3 (1.86 g, 13.5 mmol) and PdCl2dppf (0.23 g, 0.28 mmol) was added a solution of the bromide (8) (1.03 g, 4.74 mmol) in DMF (30 mL). The mixture was heated to 80 °C and stirred under N2 overnight when TLC indicated completion of the reaction. The reaction was cooled to room temperature and filtered through a pad of celite. The filtrate was added to ethyl acetate (50 mL) and washed successively with water (20 mL), brine (3 × 15 mL), dried over Na2SO4 and evaporated. Column chromatography of the brown oily material on silica gel, eluting with ethyl acetate:hexanes (30:70) and then the ratio was changed to 50:50 to obtain compound 14. Yield: 52%, light yellow amorphous solid, m.p. 111 °C–112 °C. IR (KBr, cm−1) 3420, 3310 (NH2), 3030 (Ar-H), 2979 (Alph-H), 1690 (CO), 1612, 1515, 1414 (C=C), 1254 (C-N), 1159 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.45 (s, 9H, OC(CH3)3), 2.44 (br s, 2H, CH2), 3.59 (m, 2H, CH2), 4.08 (br s, 2H, CH2), 5.95 (br s, 1H, CH), 6.20 (br s, 2H, NH2), 6.76 (d, J = 8.8 Hz, 1H, H-5), 7.41 (dd, J = 2.8, 8.8 Hz, 1H, H-6), 8.02 (br s, 1H, H-2). 13C NMR (125.7 MHz, CDCl3) δ 26.88 (CH2), 28.39 (OC(CH3)3), 29.36 (CH2), 42.86 (CH2), 79.99 (OC(CH3)3), 118.87 (CH), 119.14 (C-5), 121.47 (C-2), 129.49 (C-1), 131.65 (C-3), 132.36 (C-6), 143.89 (C-4), 154.73 (CO). Anal. Calcd for C16H21N3O4 (%): C, 60.17; H, 6.63; N, 13.16. Found (%): C, 60.10; H, 6.68; N, 13.08.
tert-Butyl 4-(3,4-diacetamidophenyl)piperidine-1-carboxylate (15): Ra-Ni (20% w/w) was added to a solution of compound 14 (1.4 g, 4.39 mmol), acetic anyhydride (2.48 mL, 26.28 mmol) and sodium acetate (1.44 g, 17.52 mmol) in a mixture of THF (10 mL) and EtOH (20 mL) and the mixture was subjected to hydrogenation in a Parr apparatus at 60 psi for 8 h. After filtering over the pad of celite, the solution was concentrated to get a brown oily material, which was resolved over silica column eluting with ethyl acetate:hexanes (30:70) and then the ratio was changed to 60:40 to get 15. Yield: 82%, light brown gum. IR (KBr, cm−1) 3420, 3338 (NH2), 3033 (Ar-H), 2978 (Alph-H), 1691 (CO), 1620, 1524, 1408 (C=C), 1221 (C-N), 1154 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.47 (s, 9H, OC(CH3)3), 1.71 (m, 2H, CH2), 2.03 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.57 (m, 1H, CH), 2.75 (br s, 2H, CH2), 3.41 (br s, 2H, CH2), 4.19 (br s, 2H, CH2), 6.96 (dd, J = 2.4, 8.2 Hz, 1H, H-6), 7.18 (d, J = 2.3 Hz, 1H, H-2), 7.29 (d, J = 8.2 Hz, 1H, H-5), 8.80 (s, 1H, NH), 8.86 (s, 1H, NH). 13C NMR (125.7 MHz, CDCl3) δ 23.81 (COCH3), 24.83 (COCH3), 24.93 (CH2), 28.51 (OC(CH3)3), 33.04 (CH), 42.06 (CH2), 79.77 (OC(CH3)3), 123.51 (C-2/C-5), 124.49 (C-2/C-5), 125.60 (C-6), 128.70 (C-4), 130.66 (C-3), 143.82 (C-1), 154.91 (CO), 170.08 (CO). Anal. Calcd for C20H29N3O4 (%): C, 63.98; H, 7.79; N, 11.19. Found (%): C, 63.90; H, 7.85; N, 11.10.
tert-Butyl 4-(3,4-diaminophenyl)piperidine-1-carboxylate (16): To a solution of compound 15 (0.75 g, 2 mmol) in ethanol (20 mL) 4 M solution of KOH in H2O was added (4 mL, 16 mmol) and the mixture was stirred at 90 °C for 12 h. The mixture was concentrated under reduced pressure to get a brown oily material, which was resolved over silica column eluting with ethyl acetate:hexanes (50:50) and then the ratio was changed to 90:10 to get compound 16. Yield: 83%, dark brown gum. IR (KBr, cm−1) 3420, 3338 (NH2), 3033 (Ar-H), 2978 (Alph-H), 1691 (CO), 1620, 1524, 1408 (C=C), 1221 (C-N), 1154 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.49 (s, 9H, OC(CH3)3), 1.81 (m, 2H, CH2), 2.07 (m, 2H, CH2), 2.72–2.82 (m, 5H, CH, CH2), 4.24 (br s, 4H, 2NH2), 7.04 (dd, J = 2.5, 8.5 Hz, 1H, H-6), 7.32 (d, J = 2.4 Hz, 1H, H-2), 7.45 (d, J = 8.5 Hz, 1H, H-5). 13C NMR (125.7 MHz, CDCl3) δ 28.40 (OC(CH3)3), (CH), 29.95 (CH2), 33.72 (CH), 42.78 (CH2), 79.52 (OC(CH3)3), 123.50 (C-2/C-5), 123.66 (C-2/C-5), 126.60 (C-6), 127.43 (C-4), 140.29 (C-3), 154.96 (CO). Anal. Calcd for C16H25N3O2 (%): C, 65.95; H, 8.65; N, 14.42. Found (%): C, 65.87; H, 8.70; N, 14.33.
tert-Butyl 4-(3,4-diaminophenyl)-5,6-dihydropyridine-1(2H)-carboxylate (18): To a solution of compound 14 (2.1 g, 6.58 mmol) in a mixture of THF (15 mL) and EtOH (30 mL) Pd-C was added (0.21 g, 10% wet basis) and the mixture was subjected to hydrogenation in the Parr apparatus at 10 psi for 3 h. After filtering over the pad of celite, the solution was concentrated and loaded over silica column, eluting with ethyl acetate:hexanes (50:50) and then the ratio was changed to 90:10 to get compound 18. Yield 96%, light brown gum. IR KBr, cm−1) 3429, 3330 (NH2), 3036 (Ar-H), 2983 (Alph-H), 1691 (CO), 1628, 1520, 1415 (C=C), 1229 (C-N), 1160 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.46 (s, 9H, OC(CH3)3), 2.40 (br s, 2H, CH2), 3.43 (m, 2H, CH2), 4.02 (br s, 2H, CH2), 4.38 (br s, 4H, 2NH2), 5.93 (br s, 1H, CH), 7.06 (dd, J = 2.6, 8.2 Hz, 1H, H-6), 7.30 (d, J = 2.5 Hz, 1H, H-2), 7.46 (d, J = 8.2 Hz, 1H, H-5). 13C NMR (125.7 MHz, CDCl3) δ 26.89 (CH2), 28.33 (OC(CH3)3), 29.39 (CH2), 42.84 (CH2), 79.99 (OC(CH3)3), 118.77 (CH), 119.19 (C-5), 120.37 (C-2), 129.19 (C-1), 130.65 (C-3), 131.16 (C-6), 141.82 (C-4), 154.63 (CO). Anal. Calcd for C16H23N3O2 (%): C, 66.41; H, 8.01; N, 14.52. Found (%): C, 66.33; H, 8.07; N, 14.43.
tert-Butyl 4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)-5,6-dihydropyridine-1(2H)-carboxylate (13): A mixture of compound 18 (0.58 g, 2 mmol) and carbonyldiimidazole (CDI) (0.34 g, 2.1 mmol) in THF (10 mL) was stirred at 60 °C for 6 h. The solvent was removed in vacuo and the residue was triturated with diethyl ether and the resultant off-white solid was filtered, washed with diethyl ether to obtain compound 13. Yield: 68%, off-white solid, m.p. 244 °C–245 °C. IR (KBr, cm−1) 3433, 3191 (CONH), 3037 (Ar-H), 2978 (Alph-H), 1713, 1689 (C=O), 1604, 1433 (C=C), 1244 (C-N), 1178 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.39 (s, 9H, OC(CH3)3), 2.48 (br s, 2H, CH2), 3.54 (m, 2H, CH2), 3.93 (br s, 2H, CH2), 5.97 (br s, 1H, CH), 6.86 (d, J = 8.2 Hz, 1H, H-7), 6.95 (d, J = 2.4 Hz, 1H, H-4), 6.99 (dd, J = 2.4, 8.2 Hz, 1H, H-6), 10.60 (br s, 2H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 26.84 (CH2), 28.28 (OC(CH3)3), 32.33 (CH), 42.80 (CH2), 79.08 (OC(CH3)3), 105.16 (CH), 108.52 (C-4), 117.67 (C-7), 129.20 (C-6), 130.04 (C-8), 133.28 (C-5/C-9), 134.81 (C-5/C-9), 154.73 (CO), 155.67 (CO). Anal. Calcd for C17H21N3O3 (%): C, 64.74; H, 6.71; N, 13.32. Found (%): C, 64.68; H, 6.76; N, 13.24.
tert-Butyl 4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)piperidine-1-carboxylate (17): (1) Synthesis from 16. Following the same procedure adopted for the synthesis of 13, the title compound 17 was obtained from the reaction of compound 16 with CDI. Yield: 62%, off-white solid. (2) Synthesis from 13. To a solution of compound 13 (0.94 g, 3 mmol) in CH3OH (30 mL) Ra-Ni was added (0.20 g, 10% wet basis) and the mixture was subjected to hydrogenation in the Parr apparatus at 60 psi for 12 h. After filtering over the pad of celite, the solution was concentrated and recrystallized from diethyl ether to get compound 17. Yield: 90%, off-white solid, m.p. > 250 °C. IR (KBr, cm−1) 3290, 3175 (CONH), 3030 (Ar-H), 2989 (Alph-H), 1691 (CO), 1613, 1515, 1416 (C=C), 1220 (C-N), 1167 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.38 (s, 9H, OC(CH3)3), 1.86 (m, 2H, CH2), 2.06 (m, 2H, CH2), 2.40–2.48 (m, 3H, CH, CH2), 3.44 (obscured by H2O, 2H, CH2), 6.70–6.80 (m, 3H, H-4, H-6, H-7), 10.49 (br s, 1H, NH), 10.53 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 28.15 (OC(CH3)3), 29.25 (CH2), 33.24 (CH2), 41.72 (CH2), 78.72 (OC(CH3)3), 106.93 (C-4), 108.48 (C-7), 119.0 (C-6), 128.09 (C-8), 129.89 (C-9), 138.49 (C-5), 154.05 (CO), 155.61 (CO). Anal. Calcd for C17H23N3O3 (%): C, 64.33; H, 7.30; N, 13.24. Found (%): C, 64.25; H, 7.35; N, 13.15.
5-(Piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5): To a solution of compound 17 (0.95 g, 3 mmol) in a mixture of CH2Cl2 (15 mL) and CH3OH (20 mL) trifluoroacetic acid was added (3 mL) at 0 °C and the mixture was stirred for 6 h at room temperature. Solvents were evaporated under reduced pressure and triturating with diethyl ether gave the title compound 5. Yield: 90%, light yellow solid, m.p. 214 °C–215 °C. IR (KBr, cm−1) 3238 (CONH), 3043 (Ar-H), 2956 (Alph-H), 1692 (CO), 1612, 1446 (C=C), 1180 (C-N), 1126 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.73–188 (m, 3H, CH, CH2), 2.77 (m, 2H, CH2), 2.97 (m, 2H, CH2), 3.44 (obscured by H2O, 2H, CH2), 4.46 (m, 1H, CH), 6.74–6.84 (m, 3H, H-4, H-6, H-7), 10.50 (br s, 1H, NH), 10.54 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 30.04 (CH2), 33.37 (CH), 43.86 (CH2), 106.66 (C-4), 118.43 (C-7), 118.94 (C-6), 128.37 (C-8), 129.87 (C-9), 137.24 (C-5), 155.56 (CO). Anal. Calcd for C14H16F3N3O3 (%): C, 50.76; H, 4.87; N, 12.68. Found (%): C, 50.70; H, 4.92; N, 12.60.
tert-Butyl 4-(4-acetamido-3-nitrophenyl)piperazine-1-carboxylate (21): To an oven-dried flask, 1-boc-piperazine (3.19 g, 17.1 mmol), Cs2CO3 (5.82 g, 17.86 mmol), Pd2(dba)3 (1.44 g, 1.57 mmol), rac-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (0.89 g, 1.43 mmol), toluene (8 mL) and compound 7 (3.68 g, 14.26 mmol) were added. While stirring the reaction mixture at room temperature, the air in the flask was removed and replaced by N2. This process was repeated three times. The reaction temperature was brought to 110 °C and stirred for 8 h. Ethyl acetate (40 mL) was added to the mixture at room temperature, washed with H2O (15 mL), brine (10 mL), dried over Na2SO4 and evaporated. The brown oily material was chromatographed on a silica column, eluting with hexanes:ethyl acetate (3:7) and then the ratio was changed to 1:1 to obtain compound 18. Yield: 37%, light red solid. IR (KBr, cm−1) 3310, 3168 (NH), 3176 (Ar-H), 1709, 1678 (CO), 1519, 1416 (C=C), 1210 (C-N), 1151 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.49 (s, 9H, OC(CH3)3), 2.17 (s, 3H, COCH3), 3.15 (m, 4H, 2CH2), 3.60 (m, 4H, 2CH2), 7.22 (dd, J = 2.5, 8.5 Hz, 1H, H-6), 7.62 (d, J = 2.5 Hz, 1H, H-2), 7.57 (d, J = 8.5 Hz, 1H, H-5), 9.98 (br s, 1H, NH). 13C NMR (125.7 MHz, CDCl3) δ 25.59 (COCH3), 28.40 (OC(CH3)3), 48.92 (CH2), 52.31 (CH2), 79.94 (OC(CH3)3), 111.29 (C-2), 123.52 (C-5/C-6), 124.31 (C-5/C-6), 127.45 (C-4), 137.45 (C-3), 147.11 (C-1), 154.63 (CO), 168.78 (CO). Anal. Calcd for C17H24N4O5 (%): C, 56.03; H, 6.64; N, 15.38. Found (%): C, 55.96; H, 6.69; N, 15.30.
tert-Butyl 4-(4-amino-3-nitrophenyl)piperazine-1-carboxylate (22): Following the same procedure adopted for the synthesis of 16, the title compound 22 was obtained from the basic hydrolysis of compound 21. Yield: 90%, blood red solid, m.p. 128 °C–129 °C. IR (KBr, cm−1) 3477, 3325 (Ar-NH2), 3073 (Ar-H), 1673 (CO), 1516, 1412 (C=C), 1211 (C-N), 1161 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.47 (s, 9H, OC(CH3)3), 3.00 (m, 4H, 2CH2), 3.57 (m, 4H, 2CH2), 5.92 (br s, 2H, NH2), 6.79 (d, J = 8.1 Hz, 1H, H-5), 7.15 (m, 1H, H-6), 7.56 (d, J = 2.1 Hz, 1H, H-2). 13C NMR (125.7 MHz, CDCl3) δ 28.42 (OC(CH3)3), 43.21 (CH2), 50.49 (CH2), 79.97 (OC(CH3)3), 111.63 (C-2), 119.85 (C-5), 128.88 (C-6), 131.89 (C-4), 140.00 (C-3), 142.69 (C-1), 154.61 (CO). Anal. Calcd for C15H22N4O4 (%): C, 55.89; H, 6.88; N, 17.38. Found (%): C, 55.81; H, 6.94; N, 17.30.
tert-Butyl 4-(3,4-diaminophenyl)piperazine-1-carboxylate (23): Following the same procedure adopted for the synthesis of 18, the title compound 23 was obtained from compound 22. Yield: 71%, light brown solid, m.p. 118 °C–119 °C. IR (KBr, cm−1) 3427, 3336 (NH2), 3030 (Ar-H), 2973 (Alph-H), 1693 (CO), 1623, 1522, 1405 (C=C), 1249 (C-N), 1162 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.37 (s, 9H, OC(CH3)3), 2.77 (br s, 4H, 2CH2), 3.37 (br s, 4H, 2CH2), 3.99 (br s, 2H, NH2), 4.34 (br s, 2H, NH2), 6.02 (dd, J = 2.5, 8.6 Hz, 1H, H-6), 6.21 (d, J = 2.6 Hz, 1H, H-2), 6.38 (d, J = 8.6 Hz, 1H, H-5). 13C NMR (125.7 MHz, DMSO d6) δ 28.26 (OC(CH3)3), 39.99 (CH2), 50.82 (CH2), 79.18 (OC(CH3)3), 105.38 (C-2), 106.78 (C-6), 115.65 (C-5), 129.12 (C-4), 136.05 (C-3), 144.06 (C-1), 154.19 (CO). Anal. Calcd for C15H24N4O2 (%): C, 61.62; H, 8.27; N, 19.16. Found (%): C, 61.55; H, 8.31; N, 19.08.
4-Bromo-1,2-dinitrobenzene (24): To a cold solution of trifluoroacetic acid (25 mL) at 0 °C compound 8 was added (4.96 g, 22.96 mmol). After being stirred for 10 min, a solution of 33% hydrogen peroxide (13.75 mL, 133 mmol) was added and the mixture was stirred at room temperature for 0.5 h followed by stirring at 50 °C for 2 h. The mixture was poured into ice water and the solid obtained was filtered and washed with cold water to get the titled compound 24. Yield: 76%, yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.85–7.92 (m, 2H, H-5, H-6), 8.04 (br s, 1H, H-3). 13C NMR (125.7 MHz, CDCl3) δ 126.39 (C-2), 127.91 (C-3), 128.09 (C-4), 136.29 (C-5), 141.26 (C-1), 143.42 (C-2). Anal. Calcd for C6H3BrN2O4 (%): C, 29.18; H, 1.22; N, 11.34. Found (%): C, 29.12; H, 1.26; N, 11.26.
tert-Butyl 4-(3,4-dinitrophenyl)piperazine-1-carboxylate (25): To an oven-dried flask, 1-boc-piperazine (1.59 g, 8.5 mmol), Cs2CO3 (2.91 g, 8.93 mmol), Pd2(dba)3 (0.72 g, 0.78 mmol), rac-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (0.44 g, 0.71 mmol), toluene (6 mL) and compound 24 (1.76 g, 7.14 mmol) were added. While stirring the reaction mixture at room temperature, the air in the flask was removed and replaced by N2. This process was repeated three times. The reaction mixture was further stirred at room temperature for 0.5 h. Ethyl acetate (50 mL) was added to the mixture at room temperature, washed with H2O (15 mL), brine (10 mL), dried over Na2SO4 and evaporated. The brown oily material was chromatographed on a silica column, eluting with hexanes:ethyl acetate (30:70) and then the ratio was changed to 40:60 yield compound 25. Yield: 78%, yellow solid. IR (KBr, cm−1) 3073 (Ar-H), 1683 (CO), 1528, 1432 (C=C), 1226 (C-N), 1151 (C-O); 1H NMR (500 MHz, CDCl3) δ 1.48 (9H, s, OC(CH3)3), 3.02 (m, 4H, 2CH2), 3.58 (m, 4H, 2CH2), 7.17 (dd, J = 2.6, 8.8 Hz, 1H, H-6), 7.24 (d, J = 2.5 Hz, 1H, H-2), 7.71 (d, J = 8.5 Hz, 1H, H-4). 13C NMR (125.7 MHz, CDCl3) δ 28.30 (OC(CH3)3), 48.24 (CH2), 51.39 (CH2), 80.12 (OC(CH3)3), 124.23 (C-2), 124.92 (C-6), 127.49 (C-5), 128.17 (C-4), 141.62 (C-3), 146.94 (C-1), 154.60 (CO). Anal. Calcd for C15H20N4O6 (%): C, 51.13; H, 5.72; N, 15.90. Found (%): C, 51.07; H, 5.78; N, 15.82.
tert-Butyl 4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)piperazine-1-carboxylate (20): Following the same procedure adopted for the synthesis of 13, the title compound was obtained by the reaction of compound 23 with CDI. Yield: 66%, colorless solid, m.p. 250 °C–251 °C. IR (KBr, cm−1) 3292, 3165 (CONH), 3031 (Ar-H), 2979 (Alph-H), 1692 (CO), 1610, 1509, 1411 (C=C), 1222 (C-N), 1177 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.38 (9H, s, OC(CH3)3), 2.91 (m, 4H, 2CH2), 3.42 (m, 4H, 2CH2),), 6.52 (m, 2H, H-4, H-6), 6.78 (d, J = 8.6 Hz, 1H, H-7), 10.42 (br s, 1H, NH), 10.50 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 28.37 (OC(CH3)3), 40.01 (CH2), 50.66 (CH2), 79.30 (OC(CH3)3), 99.01 (C-4), 109.03 (C-6), 110.55 (C-7), 123.93 (C-8), 130.52 (C-3), 146.58 (C-5), 154.76 (CO), 155.89 (CO). Anal. Calcd for C16H22N4O3 (%): C, 60.36; H, 6.97; N, 17.60. Found (%): C, 60.28; H, 7.03; N, 17.51.
5-(piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6): Following the same procedure adopted for the synthesis of 5, the title compound was obtained from the treatment of compound 20 with trifluoroacetic acid. Yield: 93%, light purple solid, m.p. 110 °C–111 °C. IR (KBr, cm−1) 3220 (CONH), 3053 (Ar-H), 2936 (Alph-H), 1695 (CO), 1609, 1444 (C=C), 1186 (C-N), 1120 (C-O); 1H NMR (500 MHz, DMSO d6) δ 3.19 (m, 4H, 2CH2), 3.22 (m, 4H, 2CH2),), 6.59 (m, 2H, H-4, H-6), 6.80 (d, J = 8.8 Hz, 1H, H-7), 8.86 (br s, 2H, NH), 10.40 (br s, 1H, NH), 10.53 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 43.10 (CH2), 47.73 (CH2), 98.97 (C-4), 108.84 (C-6), 110.25 (C-7), 124.54 (C-8), 130.57 (C-3), 145.19 (C-5), 155.76 (CO). Anal. Calcd for C13H15F3N4O3 (%): C, 46.99; H, 4.55; N, 16.86. Found (%): C, 46.93; H, 4.60; N, 16.76.
5-(1-(biphenyl-4-ylmethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5a).
Representative procedure: To a solution of compound 5 (0.15 g, 0.45 mmol) and biphenyl-4-carbaldehyde (a) (0.1 g, 0.55 mmol) in DMSO (2 mL) at 0 °C Et3N was added (0.13 mL, 0.97 mmol). After being stirred for 0.5 h at room temperature, NaBH(OAc)3 (0.11 g, 0.53 mmol) was added and the mixture was stirred for 6 h. The reaction mixture was added to a saturated NaHCO3 solution (10 mL) and stirred for 15 min, followed by the addition of ethyl acetate (30 mL). The organic layer was separated and washed with saturated NaHCO3, brine, and dried over Na2SO4 and evaporated. The light yellow solid was crystallized from diethyl ether to obtain the title compound 5a. Yield: 51%, off-white solid, m.p. > 260 °C. IR (KBr, cm−1) 3429 (CONH), 3055 (Ar-H), 2966 (Alph-H), 1703 (CO), 1620, 1518, 1422 (C=C), 1226 (C-N), 1145 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.62–1.71 (m, 4H, 2CH2), 2.04 (m, 2H, CH2), 2.48 (m, 1H, CH), 2.92 (m, 2H, CH2), 3.51 (s, 2H, NCH2), 6.77–6.81 (m, 3H, 3Ar-H), 7.32 (m, 1H, Ar-H), 7.40–7.45 (m, 4H, 4Ar-H), 7.59–7.64 (m, 4H, 4Ar-H), 10.43 (br s, 1H, NH), 10.48 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 33.64 (CH2), 41.86 (CH), 53.66 (CH2), 59.45 (NCH2), 106.91 (Ar-C), 108.42 (Ar-C), 118.99 (Ar-C), 126.56 (Ar-C), 126.66 (Ar-C), 127.42 (Ar-C), 127.98 (Ar-C), 129.03 (Ar-C), 129.06 (Ar-C), 129.63 (Ar-C), 138.92 (Ar-C), 140.11 (Ar-C), 155.59 (C=O). Anal. Calcd for C25H25N3O (%): C, 78.30; H, 6.57; N, 10.96. Found (%): C, 78.22; H, 6.63; N, 10.86.
5-(1-((4′-Fluorobiphenyl-4-yl)methyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5b): Following the same procedure adopted for the synthesis of 5a, the title compound 5b was obtained from the reductive amination of compound 5 and aldehyde (b). Yield: 54%, off-white solid, m.p. > 260 °C. IR (KBr, cm−1) 3419 (CONH), 3045 (Ar-H), 2976 (Alph-H), 1702 (CO), 1622, 1511, 1428 (C=C), 1236 (C-N), 1170 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.66–1.76 (m, 4H, 2CH2), 2.16 (br s, 2H, CH2), 2.95 (m, 2H, CH2), 3.49 (s, 2H, NCH2), 6.77–6.82 (3H, m, 3Ar-H), 7.25 (m, 2H, 2Ar-H), 7.41 (d, J = 7.6 Hz, 2H, 2Ar-H), 7.58 (m, 2H, 2Ar-H), 7.66 (m, 2H, 2Ar-H), 10.44 (br s, 1H, NH), 10.49 (br. s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 33.22 (CH2), 41.51 (CH), 53.55 (CH2), 61.67 (NCH2), 106.88 (Ar-C), 108.42 (Ar-C), 115.64 (Ar-C), 115.87 (Ar-C), 118.98 (Ar-C), 126.48 (Ar-C), 126.57 (Ar-C), 128.01 (Ar-C), 128.62 (Ar-C), 128.69 (Ar-C), 129.86 (Ar-C), 136.49 (Ar-C), 138.11 (Ar-C), 138.65 (Ar-C), 155.56 (C=O), 160.94 (Ar-C), 162.87 (Ar-C). Anal. Calcd for C25H24FN3O (%): C, 74.79; H, 6.03; N, 10.47. Found (%): C, C, 74.70; H, 6.07; N, 10.38.
5-(1-((5-phenylpyridin-3-yl)methyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5c): Following the same procedure adopted for the synthesis of 5a, the title compound 5c was obtained from the reductive amination of compound 5 and aldehyde (c). Yield: 46%, off-white solid, m.p. 236 °C–237 °C. IR (KBr, cm−1) 3430, 3192 (CONH), 3048 (Ar-H), 2946 (Alph-H), 1693 (CO), 1629, 1516, 1423 (C=C), 1197 (C-N), 1115 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.62–1.70 (m, 4H, 2CH2), 2.08 (m, 2H, CH2), 2.92 (m, 2H, CH2), 3.60 (s, 2H, NCH2), 6.77–6.81 (m, 3H, 3Ar-H), 7.42 (m, 1H, Ar-H), 7.50 (m, 2H, 2Ar-H), 7.71 (m, 2H, 2Ar-H), 7.96 (s, 1H, H-4′), 8.50 (s, 1H, H-2′), 8.76 (s, 1H, H-6′), 10.44 (br s, 1H, NH), 10.48 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 33.59 (CH2), 41.78 (CH), 53.66 (CH2), 59.45 (NCH2), 106.91 (Ar-C), 108.37 (Ar-C), 118.98 (Ar-C), 126.95 (Ar-C), 127.02 (Ar-C), 127.96 (Ar-C), 128.26 (Ar-C), 129.22 (Ar-C), 129.26 (Ar-C), 134.02 (Ar-C), 134.66 (Ar-C), 138.86 (Ar-C), 146.35 (Ar-C), 149.07 (Ar-C), 155.56 (C=O). Anal. Calcd for C24H24N4O (%): C, 74.97; H, 6.19; N, 14.57. Found (%): C, 74.89; H, 6.25; N, 14.48.
5-(1-((5-(4-Fluorophenyl)pyridin-3-yl)methyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5d): Following the same procedure adopted for the synthesis of 5a, the title compound 5d was obtained from the reductive amination of compound 5 and aldehyde (d). Yield: 62%, light yellow solid, m.p. 241 °C–242 °C. IR (KBr, cm−1) 3410, 3191 (CONH), 3042 (Ar-H), 2956 (Alph-H), 1688 (CO), 1628, 1512, 1430 (C=C), 1170 (C-N), 1115 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.59–1.70 (m, 4H, 2CH2), 2.05 (m, 3H, CH, CH2), 2.91 (m, 2H, CH2), 3.58 (s, 2H, NCH2), 6.76–6.81 (m, 3H, 3Ar-H), 7.31 (m, 2H, 2Ar-H), 7.76 (m, 2H, 2Ar-H), 7.77 (s, 1H, H-4′), 8.48 (s, 1H, H-2′), 8.73 (s, 1H, H-6′), 10.43 (br s, 1H, NH), 10.47 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 33.59 (CH2), 41.79 (CH), 53.68 (CH2), 59.45 (NCH2), 106.91 (Ar-C), 108.38 (Ar-C), 116.02 (Ar-C), 116.21 (Ar-C), 119.0 (Ar-C), 127.98 (Ar-C), 129.07 (Ar-C), 129.13 (Ar-C), 129.20 (Ar-C), 129.84 (Ar-C), 133.57 (Ar-C), 134.02 (Ar-C), 134.33 (Ar-C), 134.64 (Ar-C), 138.87 (Ar-C), 146.28 (Ar-C), 149.07 (Ar-C), 155.59 (C=O). Anal. Calcd for C24H23FN4O (%): C, 71.62; H, 5.76; N, 13.92. Found (%): C, 71.51; H, 5.83; N, 13.82.
5-(1-(3-cyclopentenylbenzyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5e): Following the same procedure adopted for the synthesis of 5a, the title compound 5e was obtained from the reductive amination of compound 5 and aldehyde (e). Yield: 66%, light yellow solid, m.p. 238 °C–239 °C. IR (KBr, cm−1) 3322 (CONH), 3051 (Ar-H), 2966 (Alph-H), 1690 (CO), 1636, 1512, 1428 (C=C), 1223 (C-N), 1172 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.59–1.69 (m, 5H, CH, CH2), 1.93 (m, 2H, CH, CH2), 2.04 (m, 2H, CH, CH2), 2.62 (m, 2H, CH, CH2), 2.87 (m, 2H, CH2), 6.24 (s, 1H, CH), 6.76–6.82 (m, 3H, 3Ar-H), 7.16 (m, 1H, Ar-H), 7.24 (m, 1H, Ar-H), 7.30 (m, 1H, Ar-H), 7.38 (m, 1H, Ar-H) 10.44 (br s, 1H, NH), 10.48 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 22.89 (CH2), 32.83 (CH2), 32.96 (CH2), 33.49 (CH2), 39.00 (CH2), 41.79 (CH), 53.68 (CH2), 62.49 (NCH2), 106.90 (Ar-C), 108.38 (Ar-C), 118.96 (Ar-C), 124.26 (Ar-C), 126.10 (Ar-C), 127.81 (Ar-C), 128.29 (Ar-C), 129.84 (Ar-C), 136.08 (Ar-C), 139.05 (Ar-C), 142.00 (Ar-C), 155.66 (C=O). Anal. Calcd for C24H27N3O (%): C, 77.18; H, 7.29; N, 11.25. Found (%): C, 77.10; H, 7.36; N, 11.18.
5-(1-((5-cyclopentenylpyridin-3-yl)methyl)piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one (5f): Following the same procedure adopted for the synthesis of 5a, the title compound 5f was obtained from the reductive amination of compound 5 and aldehyde (f). Yield: 42%, light yellow solid, m.p. 249 °C–250 °C. IR (KBr, cm−1) 3342 (CONH), 3040 (Ar-H), 2956 (Alph-H), 1691 (CO), 1634, 1508, 1440 (C=C), 1221 (C-N), 1177 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.60–1.69 (m, 4H, CH, CH2), 1.95–2.05 (m, 6H, CH, CH2), 2.66 (m, 2H, CH2), 2.85 (m, 2H, CH2), 3.50 (s, 2H, NCH2), 6.41 (s, 1H, CH), 6.76–6.80 (m, 3H, 3Ar-H), 7.73 (s, 1H, H-4′), 8.34 (s, 1H, H-2′), 8.54 (s, 1H, H-6′), 10.43 (br s, 1H, NH), 10.47 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO-d6) δ 22.77 (CH2), 32.48 (CH2), 33.08 (CH2), 33.55 (CH2), 40.40 (CH2), 41.77 (CH), 53.65 (CH2), 59.51 (NCH2), 106.90 (Ar-C), 108.38 (Ar-C), 118.98 (Ar-C), 127.96 (Ar-C), 128.39 (Ar-C), 129.82 (Ar-C), 131.30 (Ar-C), 133.11 (Ar-C), 133.51 (Ar-C), 138.86 (Ar-C), 139.14 (Ar-C), 145.46 (Ar-C), 148.52 (Ar-C), 155.58 (C=O). Anal. Calcd for C23H26N4O (%): C, 73.77; H, 7.00; N, 14.96. Found (%): C, 73.69; H, 7.06; N, 14.88.
Table 1. Structures and affinities (Ki, nM) of compounds 5a–5f and 6a–6f on D2 and 5-HT1A receptors. Binding affinity values are expressed as means ± SEM of separate experiments, each performed in duplicate.
5-(4-(biphenyl-4-ylmethyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6a): Following the same procedure adopted for the synthesis of 5a, the title compound 6a was obtained from the reductive amination of compound 6 and aldehyde (a). Yield: 46%, off-white solid, m.p. > 260 °C. IR (KBr, cm−1) 3430 (CONH), 3060 (Ar-H), 2966 (Alph-H), 1705 (CO), 1622, 1511, 1438 (C=C), 1223 (C-N), 1169 (C-O); 1H NMR (500 MHz, DMSO d6) δ 2.51 (br s, 4H, 2CH2), 2.99 (br s, 4H, 2CH2), 3.53 (s, 2H, NCH2), 6.49 (m, 2H, 2Ar-H), 6.74 (m, 1H, Ar-H), 7.37–7.44 (m, 5H, 5Ar-H), 7.57–7.63 (m, 4H, 4Ar-H), 10.26 (br s, 1H, NH), 10.38 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 50.13 (CH2), 52.80 (CH2), 61.68 (NCH2), 97.83 (Ar-C), 108.56 (Ar-C), 109.39 (Ar-C), 123.93 (Ar-C), 124.49 (Ar-C), 127.28 (Ar-C), 128.90 (Ar-C), 129.49 (Ar-C), 130.38 (Ar-C), 136.72 (Ar-C), 137.86 (Ar-C), 139.94 (Ar-C), 146.42 (Ar-C), 155.56 (C=O). Anal. Calcd for C24H24N4O (%): C, 74.97; H, 6.29; N, 14.57. Found (%): C, 74.90; H, 6.35; N, 14.48.
5-(4-((4′-fluorobiphenyl-4-yl)methyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6b): Following the same procedure adopted for the synthesis of 5a, the title compound 6b was obtained from the reductive amination of compound 6 and aldehyde (b). Yield: 55%, off-white solid, m.p. > 260 °C. IR (KBr, cm−1) 3439 (CONH), 3065 (Ar-H), 2936 (Alph-H), 1707 (CO), 1632, 1501, 1448 (C=C), 1226 (C-N), 1179 (C-O); 1H NMR (500 MHz, DMSO d6) δ 2.52 (br s, 4H, 2CH2), 2.99 (br s, 4H, 2CH2), 3.53 (s, 2H, NCH2), 6.49 (m, 2H, 2Ar-H), 6.74 (d, J = 3.2 Hz, 1H, Ar-H), 7.26 (m, 2H, 2Ar-H), 7.39 (m, 2H, 2Ar-H), 7.58 (m, 2H, 2Ar-H), 7.67 (m, 2H, 2Ar-H), 10.26 (br s, 1H, NH), 10.37 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 50.13 (CH2), 52.79 (CH2), 61.65 (NCH2), 97.86 (Ar-C), 108.60 (Ar-C), 109.41 (Ar-C), 115.59 (Ar-C), 123.25 (Ar-C), 126.39 (Ar-C), 128.49 (Ar-C), 129.53 (Ar-C), 130.40 (Ar-C), 136.46 (Ar-C), 137.38 (Ar-C), 137.83 (Ar-C), 146.41 (Ar-C), 155.59 (C=O), 160.90 (Ar-C), 162.17 (Ar-C). Anal. Calcd for C24H23FN4O (%): C, 71.62; H, 5.76; N, 13.92. Found (%): C, 71.55; H, 5.83; N, 13.86.
5-(4-((5-phenylpyridin-3-yl)methyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6c): Following the same procedure adopted for the synthesis of 5a, the title compound 6c was obtained from the reductive amination of compound 6 and aldehyde (c). Yield: 44%, off-white solid, m.p. 241 °C–242 °C. IR (KBr, cm−1) 3433, 3195 (CONH), 3038 (Ar-H), 2936 (Alph-H), 1683 (CO), 1639, 1506, 1443 (C=C), 1177 (C-N), 1119 (C-O); 1H NMR (500 MHz, DMSO d6) δ 2.51 (br s, 4H, 2CH2), 2.99 (br s, 4H, 2CH2), 3.62 (s, 2H, NCH2), 6.50 (m, 2H, 2Ar-H), 6.73 (d, J = 3.2 Hz, 1H, Ar-H), 7.41 (m, 1H, Ar-H), 7.47 (m, 2H, 2Ar-H), 7.70 (m, 1H, Ar-H), 7.96 (s, 1H, H-4′), 8.51 (s, 1H, H-2′), 8.77 (s, 1H, H-6′), 10.26 (br. s, 1H, NH), 10.38 (br. s, 1H, NH). 13C NMR (125.7 MHz, DMSO-d6) δ 50.11 (CH2), 52.67 (CH2), 59.01 (NCH2), 97.88 (Ar-C), 108.58 (Ar-C), 109.44 (Ar-C), 123.29 (Ar-C), 126.93 (Ar-C), 127.09 (Ar-C), 128.16 (Ar-C), 129.15 (Ar-C), 130.40 (Ar-C), 133.57 (Ar-C), 134.56 (Ar-C), 135.19 (Ar-C), 136.99 (Ar-C), 146.37 (Ar-C), 149.04 (Ar-C), 155.58 (C=O). Anal. Calcd for C23H23N5O (%): C, 71.67; H, 6.01; N, 18.17. Found (%): C, 71.60; H, 6.06; N, 18.08.
5-(4-((5-(4-fluorophenyl)pyridin-3-yl)methyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6d): Following the same procedure adopted for the synthesis of 5a, the title compound 6d was obtained from the reductive amination of compound 6 and aldehyde (d). Yield: 42%, off-white solid, m.p. 246 °C–247 °C. IR (KBr, cm−1) 3413, 3190 (CONH), 3048 (Ar-H), 2946 (Alph-H), 1686 (CO), 1629, 1506, 1433 (C=C), 1176 (C-N), 1113 (C-O); 1H NMR (500 MHz, DMSO d6) δ 2.52 (br s, 4H, 2CH2), 2.99 (br s, 4H, 2CH2), 3.61 (s, 2H, NCH2), 6.50 (m, 2H, 2Ar-H), 6.73 (d, J = 3.2 Hz, 1H, Ar-H), 7.31 (m, 2H, 2Ar-H), 7.76 (m, 2H, 2Ar-H), 7.96 (s, 1H, H-4′), 8.50 (s, 1H, H-2′), 8.76 (s, 1H, H-6′), 10.26 (br s, 1H, NH), 10.38 (br s, 1H, NH). 13C NMR (125.7 MHz, DMSO d6) δ 50.08 (CH2), 52.64 (CH2), 58.98 (NCH2), 97.90 (Ar-C), 108.58 (Ar-C), 109.44 (Ar-C), 115.91 (Ar-C), 116.07 (Ar-C), 123.65 (Ar-C), 129.01 (Ar-C), 129.08 (Ar-C), 130.21 (Ar-C), 133.38 (Ar-C), 134.31 (Ar-C), 134.89 (Ar-C), 146.35 (Ar-C), 149.04 (Ar-C), 155.42 (C=O), 161.15 (Ar-C). Anal. Calcd for C23H22FN5O (%): C, 68.47; H, 5.50; N, 17.36. Found (%): C, 68.46; H, 5.54; N, 17.28.
5-(4-(3-cyclopentenylbenzyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6e): Following the same procedure adopted for the synthesis of 5a, the title compound 6e was obtained from the reductive amination of compound 6 and aldehyde (e). Yield: 62%, off-white solid, m.p. 244 °C–245 °C. IR (KBr, cm−1) 3410, 3183 (CONH), 3050 (Ar-H), 2948 (Alph-H), 1689 (CO), 1625, 1516, 1430 (C=C), 1178 (C-N), 1115 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.93 (m, 2H, CH2), 2.49 (br s, 2H, CH2), 2.63 (br s, 2H, CH2), 2.98 (br s, 4H, 2CH2), 3.49 (s, 2H, NCH2), 6.24 (br. s, 1H, CH), 6.49 (m, 2H, 2CH), 6.74 (d, J = 3.1 Hz, 1H, Ar-H), 7.18 (m, 1H, Ar-H), 7.28 (m, 1H, Ar-H), 7.32 (m, 1H, Ar-H), 7.38 (m, 1H, Ar-H), 10.27 (br s, 1H, NH), 10.39 (br s, 1H, NH). Citation13C NMR (125.7 MHz, DMSO d6) δ 22.86 (CH2), 32.78 (CH2), 32.94 (CH2), 50.11 (CH2), 52.77 (CH2), 62.08 (NCH2), 97.95 (Ar-C), 108.70 (Ar-C), 109.49 (Ar-C), 124.28 (Ar-C), 126.08 (Ar-C), 127.75 (Ar-C), 128.29 (Ar-C), 130.44 (Ar-C), 136.07 (Ar-C), 141.94 (Ar-C), 146.42 (Ar-C), 155.66 (C=O). Anal. Calcd for C23H26N4O (%): C, 73.77; H, 7.00; N, 14.96. Found (%): C, 73.68; H, 7.05; N, 14.86.
5-(4-((5-cyclopentenylpyridin-3-yl)methyl)piperazin-1-yl)-1H-benzo[d]imidazol-2(3H)-one (6f): Following the same procedure adopted for the synthesis of 5a, the title compound 6f was obtained from the reductive amination of compound 6 and aldehyde (f). Yield: 39%, off-white solid, m.p. 255 °C–256 °C. IR (KBr, cm−1) 3332 (CONH), 3030 (Ar-H), 2951 (Alph-H), 1689 (CO), 1644, 1506, 1450 (C=C), 1222 (C-N), 1176 (C-O); 1H NMR (500 MHz, DMSO d6) δ 1.95 (m, 2H, CH2), 2.51 (br s, 2H, CH2), 2.66 (m, 2H, CH2), 2.98 (br s, 4H, 2CH2), 3.54 (m, 2H, NCH2), 6.42 (br s, 1H, CH), 6.50 (m, 2H, 2Ar-H), 6.74 (m, 1H, Ar-H), 7.74 (s, 1H, H-4′), 8.36 (s, 1H, H-2′), 8.57 (s, 1H, H-6′), 10.26 (br s, 1H, NH), 10.40 (br s, 1H, NH). Citation13C NMR (125.7 MHz, DMSO d6) δ 22.69 (CH2), 32.40 (CH2), 33.01 (CH2), 50.06 (CH2), 52.66 (CH2), 59.06 (NCH2), 97.92 (Ar-C), 108.61 (Ar-C), 109.46 (Ar-C), 123.31 (Ar-C), 128.31 (Ar-C), 130.40 (Ar-C), 131.21 (Ar-C), 133.04 (Ar-C), 139.03 (Ar-C), 145.56 (Ar-C), 146.37 (Ar-C), 148.52 (Ar-C), 155.61 (C=O). Anal. Calcd for C22H25N5O (%): C, 70.38; H, 6.71; N, 18.65. Found (%): C, 70.30; H, 6.75; N, 18.55.
Pharmacology
Rat-cloned D2L dopaminergic receptors
Human-cloned dopamine D2L receptors stably expressed in C6 rat glioma cells (kindly donated by Professor Roberto Maggio, Università di L'Aquila, Italy) were radiolabeled with [3H]spiroperidol according to Scarselli et al. with minor modificationsCitation23. The incubation buffer (120 mM NaCl, 5.0 mM KCl, 5.0 mM MgCl2, 1 mM EDTA, 50 mM Tris-HCl, pH 7.4) contained 100 µg of dopamine D2L receptor membranes, 0.30–0.50 nM [3H]spiroperidol (Kd = 0.093 nM) and six to nine concentrations of drug solution in a final volume of 500 µL. The samples were incubated for 120 min at 25 °C, then the incubation was stopped by rapid filtration through Whatman GF/C glass fiber filters (presoaked in 0.5% polyethylenimine for 60 min). The filters were washed three times in 1 mL of ice-cold 50 mM Tris, 0.9% NaCl, pH 7.4. Nonspecific binding was determined in the presence of 10 µM haloperidol. The radioactivity bound to the filters was measured by liquid scintillation using a LS6500 Multi-Purpose scintillation Counter, Beckman.
Human-cloned 5-HT1A receptors
Human-cloned 5-HT1A serotonin receptors stably expressed in HEK293-EBNA cells (Perkin-Elmer, Waltham, MA) were radiolabeled with 1.0 nM [3H]-8-OH-DPATCitation24. Samples containing 32 μg of membrane protein, different concentrations of each compound ranging from 0.1 nM to 10 μM were incubated in a final volume of 500 μL of 50 mM Tris-HCl pH 7.4, 5 mM MgSO4 for 120 min at 37 °C. After this incubation time, samples were filtered through Whatman GF/C glass microfiber filters pre-soaked in polyethylenimine 0.5% for at least 30 min prior to use. The filters were washed twice with 1 mL of ice-cold buffer (50 mM Tris-HCl, pH 7.4). Nonspecific binding was determined in the presence of 10 μM 5-HT. The radioactivity bound to the filters was measured by liquid scintillation using the LS6500 Multi-Purpose scintillation Counter, Beckman.
Determination of Ki values
Ki values of the compounds were calculated by the Cheng–Prusoff equationCitation25. Binding affinity values are expressed as means ±SEM of separate experiments each performed in duplicate.
Results and discussion
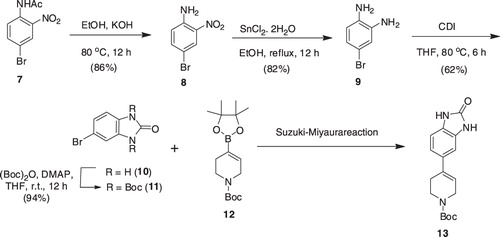
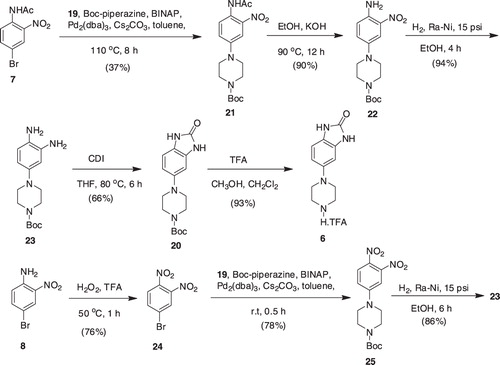
The synthesis of compounds 5a–f required the synthesis of intermediate 5, which was to be synthesized from 13. The synthesis of the latter was envisioned by the Suzuki–Miyaura reaction of bromo-1H-benzo[d]imidazol-2(3H)-one (10) with boronate (12) [22]. Preparation of 10 was commenced by the basic hydrolysis of acetamide (7) to produce nitroaniline (8), which in turn was transformed to benzenediamine (9) by the action of stannous chlorideCitation26. Reaction of 9 with CDI finally produced 10 in 62% yield. The Suzuki–Miyaura reaction of cyclic vinyl boronate (12)Citation22, derived from the vinyl triflates of N-protected tetrahydropyridines, with 10 under a variety of conditions (such as PdCl2dppf, KOAc, DMF, 80 °C; Pd(PPh3)4, toluene, ethanol, Na2CO3, reflux) were failed; the desired 13 could not be obtained rather the starting 10 was recovered (). The poor solubility of 10 in a variety of solvents is considered to be one of the factors for its lack of reactivity. Thus, intermediate 10 was protected with di-tert-butyl dicarbonate to furnish 11, which in turn was employed in the Suzuki–Miyaura reaction with boronate (12), under different conditions as described in the case of 10; however the desired product 13 was not detected.
Scheme 1. Synthesis of intermediate 13.
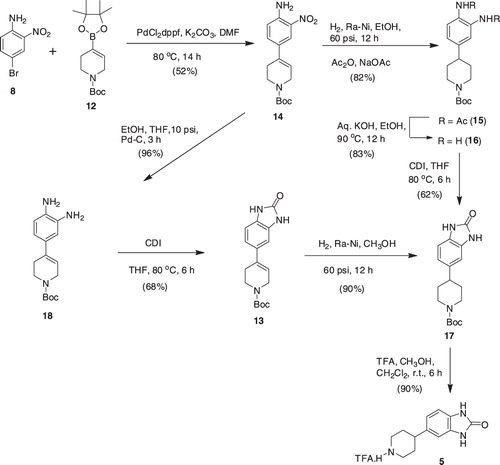
In an alternate approach, boronate (12) was reacted with nitroamine (8) under the Suzuki–Miyaura conditions to produce intermediate 14, which in turn was subjected to hydrogenation in the Parr apparatus at 60 psi. The hydrogenation required extended period of time (20 h) and was unclean, yielding the desired diamine (15) in a very low yield (20%) after column purifications. Thus, hydrogenation of compound 14 was performed in ethanol in the presence of acetic anhydride (six equivalents) and sodium acetate (four equivalents), which rendered reduction of both nitro group and double bond with concomitant acetylation of the amino groups, yielding intermediate 15 in 82% yield. Basic hydrolysis of the latter with aqueous KOH in ethanol ultimately generated diamine (16) in a good yield (83%). Reaction of 16 with CDI in THF, heating the mixture at 80 °C for 6 h, finally gave access to the key intermediate 17 in an overall yield of 42% from 14. Alternatively, intermediate 17 was also synthesized from 14 by hydrogenation of 14 over Pd-C at 10 psi to produce diamine (18), which was treated with CDI in THF to generate intermediate 13. Hydrogenation of the latter over Ra-Ni in methanol furnished intermediate 17 in an overall higher yield of 59% from 14. This approach did not require column purifications of intermediates (13 and 17) as was the case with intermediates 15 and 16 (). Exposure of intermediate 17 to trifluoroacetic acid in a mixture of methanol and dichloromethane finally furnished the desired key intermediate 5.
Scheme 2. Synthesis of intermediate 13, an alternative route.
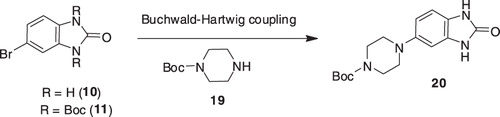
The synthesis of compounds 6a–f required the synthesis of intermediate 6, which was to be accomplished from intermediate 20. The Buchwald–Hartwig coupling of bromides 10 or 11 with tert-butyl piperazine-1-carboxylate 19, under different reaction conditions (PdCl2dppf, KOAc, DMF, 80 °C; Pd(PPh3)4, toluene, ethanol, Na2CO3, reflux) were not successful; the desired 20 was not observed in any case ().
Scheme 3. Synthesis of intermediate 20.
In an alternate approach, acetamide (7) was coupled with 19 under the Buchwald–Hartwig reaction condition to construct the adduct 21 in relatively low yield (37%). Hydrolysis of the latter with aqueous KOH gave nitroaniline (22), which in turn was subjected to hydrogenation under the Parr apparatus over Ra-Ni at 15 psi to produce diamine (23) in an overall yield of 31% from 7Citation27. Alternatively, intermediate 23 was also synthesized from the oxidation of 8 with trifluoroperacetic acid, generated in situ from hydrogen peroxide and trifluoroacetic acid, to produce dinitrobenzene (24), which was coupled with 19 under the Buchwald–Hartwig reaction condition at room temperature to produce adduct 25Citation28. Hydrogenation of intermediate 25, as described for the preparation of 23 from 22, finally produced compound 23 in an overall yield of 51% from 8. It is worth mentioning that reaction of 24 with 19 without the addition of catalyst at room temperature did not progress, the starting materials were intact or degraded over the course of longer reaction time, whereas gentle heating the reaction mixture to 40 °C resulted in a complex mixture of side products; the desired 19 was not observed. This was attributed to a thermal instability of 24 since gentle heating the reaction mixture under the Buchwald–Hartwig reaction condition did not yield the desired 19 either, only side products were observed. Reaction of 23 with CDI in THF at 80 °C yielded the desired intermediate 20, which was exposed to acid to produce the key intermediate 6 in 93% yield ().
Scheme 4. Synthesis of intermediate 6.
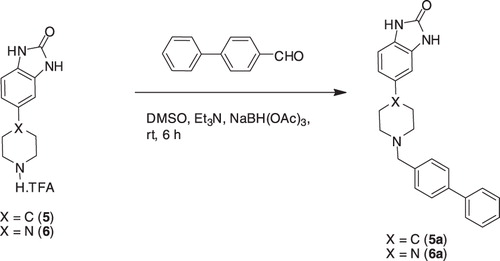
Having the desired intermediates 5 and 6 in hands, we next performed reductive amination of 5 and 6 with aldehydes (a–f)Citation21 in DMSO, using NaBH(OAc)3 as reducing agent to accomplish the synthesis of final compounds (5a–f) and (6a–f), respectively ().
Scheme 5. Synthesis of 5a and 6a, a representative example.
Biological results
SAR of D2 receptor binding
The SAR studies on D2 receptor binding of compounds revealed that compounds 6f, 5f and 5e were the most potent compared to others, which have moderate affinity (). Compound 5a (piperidine series) exhibited week affinity whereas an introduction of fluoro group at C-4′ in the biphenyl moiety, to produce 5b, improved its affinity to three-fold. However, introducing fluorine at C-4′ in biphenyl of 6a (piperazine series), to yield 6b, resulted in decrease in affinity. In case of compounds having phenylpyridine as a biaryl functionality, the introduction of fluoro group at C-4′ reduced the affinities both in piperidine and piperazine series. For instance, in case of piperidine series the affinity of 5d (Ki = 24.6 nM) was slightly reduced compared to its non-fluoro analogue 5c (Ki = 18.5 nM), whereas in case of piperazine series the affinity of 6c was reduced to six-fold when it was compared with 6d. The role of piperidine and piperazine moiety in the D2 receptor affinity was evident with the observations that affinity generally increased in compounds having piperazine moiety with biphenyl and phenylpyridine substituents as compared with piperidine counterpart (5a versus 6a and 5c versus 6c). However, the role of piperazine and piperidine moieties is reversed, i.e. affinity decreased, when these moieties bear 4-fluorobiphenyl and 4-fluorophenylpyridine substituents. For instance, the affinity was decreased to two-fold in case 5b versus 6b and four-fold in case of 5d versus 6d. As stated earlier that the most active compounds were 6f, 5f and 5e, which features cyclopentenylpyridine and cyclopentenylbenzene moieties in their structures. Such observation suggests that pyridine nitrogen of cyclopentenylpyridine may play a role in the high affinity of 6f and 5f. Also, relatively high affinity of 6f compared to 5f is indicative of important contribution of piperazine moiety.
SAR of 5-HT1A receptor binding
The compounds exhibited, in general, higher affinity to 5-HT1A receptor than that of D2. Compound 5e, being the most potent (Ki = 1.2 nM), reveals that piperidine with cyclopentenylbenzene is the right combination for optimal 5-HT1A affinity. This was also evident when 5e was compared with its counterpart piperazine analog 6e, which posses high 5-HT1A receptor affinity (Ki = 3.1 nM). Likewise compounds 5a, 5b, 6a and 6b exhibited moderate affinity. Both in piperidine and piperazine series with cyclopentenylbenzene moiety, the fluoro analogs of these compounds (5b and 6b) showed higher affinity than their non-fluoro counterparts (5a and 6a). However, in case of compounds bearing cyclopentenylpyridine moiety an opposite trend was observed. For instance, the affinity of 5c (Ki = 6.72 nM) was decreased almost half-fold when it was compared with its fluoro counterpart 5d (Ki = 9.42 nM); likewise the affinity was decreased to two-fold when 6c was compared with 6d. In addition, compounds with piperidine moiety (5a and 5b) were more active than compounds with piperazine moiety (6a and 6b). Furthermore, in the piperidine series, the presence of pyridine nitrogen of phenylpyridine moiety did not play a significant role in improving the binding affinity of 5-HT1A in contrast to that of D2; the affinity of 5e (Ki = 8.12 nM) was decreased as compared to 5f (Ki = 4.26 nM). However, in case of piperazine series, pyridine nitrogen improved the affinity of 6f (Ki = 3.1 nM) when it was compared with 6e (Ki = 10.4 nM).
Conclusion
A series of new 5-piperidinyl and 5-piperazinyl-1H-benzo[d]imidazol-2(3H)-ones have been synthesized. The evaluation of binding affinities for D2 and 5-HT1A receptors of these compounds revealed that, in general, compounds with cyclopentenylbenzene and cyclopentenylpyridine moieties have the higher affinities for both D2 and 5-HT1A receptors. In the piperidine series, 5f and 5e showed the highest affinities for D2 and 5-HT1A receptors, respectively. However, in case of piperazine series, 6f exhibited the highest affinity for both D2 and 5-HT1A receptors.
Declaration of interest
The financial support from King Abdulaziz City for Science and Technology (KACST) Project No: AR-28-38 is gratefully acknowledged.
Acknowledgements
The facilities provided by KFUPM are sincerely acknowledged.
Related Research Data
References
- Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Text revision. Washington, D.C.: American Psychiatric Association; 2000:297–343
- Arnt J, Bang-Andersen B, Dias R, Bogesø KP. Strategies for pharmacotherapy of schizophrenia. Drugs Future 2008;33:777–91
- Kapur S, Agid O, Mizrahi RLM. How antipsychotics work-from receptors to reality. NeuroRx 2006;3:10–21
- McCormick PN, Kapur S, Graff-Guerrero A, et al. The antipsychotics olanzapine, risperidone, clozapine, and haloperidol are D2-selective ex vivo but not in vitro. Neuropsychopharmacology 2010;35:1826–35
- Meltzer HY, Li Z, Kaneda YI. Serotonin receptors: their key role in drugs to treat schizophrenia. J Prog Neuropsychopharmacol Biol Psychiatry 2003;27:1159–72
- (a) Goff DC, Shader RI. Non-neurological side-effects of antipsychotic drugs. In: Hirsch SR, Weinberger D, eds. Schizophrenia. 2nd ed. Oxford: Blackwell Publishing; 2002:573–88; (b) Newman-Tancredi A, Chaput C, Verriele L, Millan MJ. Clozapine is a partial agonist at cloned, human serotonin 5-HT1A receptors. Neuropharmacology 1996;35:119–21; (c) Bartoszyk GD, Roos C, Ziegler H. 5-HT1A receptors are not involved in clozapine's lack of cataleptogenic potential. Neuropharmacology 1996;35:1645–6
- Meltzer HY. Clinical studies on the mechanism of action of clozapine: the dopamine-serotonin hypothesis of schizophrenia. Psychopharmacology 1989;99:S18–27
- Lindstrom LH. The effect of long-term treatment with clozapine in schizophrenia: a retrospective study in 96 patients treated with clozapine for up to 13 years. Acta Psychiatr Scand 1988;77:524–9
- Newman-Tancredi A, Cussac D, Depoortere R. Neuropharmacological profile of bifeprunox: merits and limitations in comparison with other third-generation antipsychotics. Curr Opin Investig Drugs 2007;8:539–54
- Jones CA, McCreary AC. Serotonergic approaches in the development of novel antipsychotics. Neuropharmacology 2008;55:1056–65
- (a) Prinssen EP, Colpaert FC, Koek W. 5-HT1A receptor activation and anti-cataleptic effects: high-efficacy agonists maximally inhibit haloperidol-induced catalepsy. Eur J Pharmacol 2002;453:217–21; (b) Depoortère R, Auclair AL, Bardin L, et al. F15063, a compound with D2/D3 antagonist, 5-HT1A agonist and D4 partial agonist properties. III. Activity in models of cognition and negative symptoms. Br J Pharmacol 2007;151:266–77
- Newman-Tancredi A, Assie MB, Leduc N, et al. Novel antipsychotics activate recombinant human and native rat serotonin 5-HT1A receptors: affinity, efficacy and potential implications for treatment of schizophrenia. Int J Neuropsychopharmacol 2005;8:341–56
- Assie MB, Ravailhe V, Faucillon V, Newman-Tancredi A. Contrasting contribution of 5-hydroxytryptamine 1A receptor activation to neurochemical profile of novel antipsychotics: frontocortical dopamine and hippocampal serotonin release in rat brain. J Pharmacol Exp Ther 2005;315:265–72
- Slot LAB, Vries LD, Newman-Tancredi A, Cussac D. Differential profile of antipsychotics at serotonin 5-HT1A and dopamine D2S receptors coupled to extracellular signal-regulated kinase. Eur J Pharmacol 2006;534:63–70
- Slot LAB, Kleven MS. Newman-Tancredi A. Effects of novel antipsychotics with mixed D2 antagonist/5-HT1A agonist properties on PCP-induced social interaction deficits in the rat. Neuropharmacology 2005;49:996–1006
- Kleven MS, Barret-Grevoz C, Slot LAB, Newman-Tancredi A. Novel antipsychotic agents with 5-HT1A agonist properties: role of 5-HT1A receptor activation in attenuation of catalepsy induction in rats. Neuropharmacology 2005;49:135–43
- Bantick RA, Deakin JFW, Grasby PM. The 5-HT1A receptor in schizophrenia: a promising target for novel atypical neuroleptics? J Psychopharmacol 2001;15:37–46
- McCreary AC, Glennon JC, Ashby Jr CR, et al. SLV313 (1-(2,3-dihydro-benzo[1,4]dioxin-5-yl)-4-[5-(4-fluoro-phenyl)-pyridin-3-ylmethyl]-piperazine monohydrochloride): a novel dopamine D2 receptor antagonist and 5-HT1A receptor agonist potential antipsychotic drug. Neuropsychopharmacology 2007;32:78–94
- Newman-Tancredi A. The importance of 5-HT1A receptor agonism in antipsychotic drug action: rationale and perspectives. Curr Opin Invest Drugs 2010;11:802–12
- Cuisiat S, Bourdiol N, Lacharme V, et al. Towards a new generation of potential antipsychotic agents combining D2 and 5-HT1A receptor activities. J Med Chem 2007;50:865–76
- Ullah N. Synthesis of new 1-aryl-4-(biarylmethylene) piperazinel ligands, structurally related to adoprazine (SLV313). Z Naturforsch 2012;67b:75–84
- Ullah N, Al-Shaheri AAQ. Synthesis of new 4-aryl-1-(biarylmethylene)piperidines structural analogs of adoprazine (SLV313). Z Naturforsch 2012;67b:253–62
- Scarselli M, Novi F, Schallmach E, et al. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J Biol Chem 2001;276:30308–14
- Fargin A, Raymond JR, Regan JW, et al. Effector coupling mechanisms of the cloned 5-HT1A receptor. J Biol Chem 1989;264:14848–52
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–108
- Evans KM, Slawin AMZ, Lebl T, et al. An improved understanding of the reaction of bis(bromomethyl)quinoxaline 1-N-oxides with amines using substituent effects. J Org Chem 2007;72:3186–93
- Galan AA, Chen J, Du H, et al. Preparation of 1H-imidazole-4,5-dicarboxamides as JAK-2 modulators. PCT Int. Appl. 2008042282
- Pelletier JC, Chengalvala M, Cottom J, et al. 2-phenyl-4-piperazinylbenzimidazoles: orally active inhibitors of the gonadotropin releasing hormone (GnRH) receptor. Bioorg Med Chem 2008;16:6617–40