
Abstract
Because of the pivotal role of cyclooxygenase (COX) in the inflammatory processes, non-steroidal anti-inflammatory drugs (NSAIDs) that suppress COX activities have been used clinically for the treatment of inflammatory diseases/syndromes; however, traditional NSAIDs exhibit serious side-effects such as gastrointestinal damage and hyper sensitivity owing to their COX-1 inhibition. Also, COX-2 inhibition-derived suppressive or preventive effects against initiation/proliferation/invasion/motility/recurrence/metastasis of various cancers/tumours such as colon, gastric, skin, lung, liver, pancreas, breast, prostate, cervical and ovarian cancers are significant. In this study, design, synthesis and structure–activity relationship (SAR) of various novel {2-[(2-, 3- and/or 4-substituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues were investigated to seek and identify various chemotypes of potent and selective COX-2 inhibitors for the treatment of inflammatory diseases, resulting in the discovery of orally potent agents in the peripheral-inflammation model rats. The SARs and physicochemical properties for the analogues are described as significant findings. For graphical abstract: see Supplementary Material. (www.informahealthcare.com/enz)
Introduction
Cyclooxygenase (COX, or prostaglandin G/H synthase or endoperoxidase; EC 1.14.99.1) is a key rate-limiting enzyme for prostaglandin (PG) cascades that produce prostanoidsCitation1–3, and inhibition of COX activity is able to attenuate the levels of prostanoids that are involved in the inflammatory symptoms such as swelling, flare, heat and pain. Therefore, COX inhibition has been considered as a significant target of drug candidates for the treatment of inflammatory diseases/syndromes that include rheumatoid arthritis (RA) and osteoarthritis (OA)Citation4. Non-steroidal anti-inflammatory drugs (NSAIDs) that suppress COX activities have been used clinicallyCitation4–14; however, traditional NSAIDs that are non-selective inhibitors against COX isoforms, COX-1 (EC 1.14.99.1) and COX-2 (EC 1.14.99.1), exhibit serious side-effects such as gastrointestinal damageCitation15–17. COX-1 is predominantly expressed ubiquitously and constitutively and serves a housekeeping role in processes such as GI mucosa protection. COX-2 is absent or exhibits a low level of expression in most tissues, and is highly upregulated in response to inflammatory or tissue-injury stimuli/signals, growth factors, endotoxin, viruses, proinflammatory cytokines, activated T lymphocytes, carcinogens/tumour-promoters and hyperglycemia-induced mitochondrial reactive-oxygen species in the various types of organs, tissues and cellsCitation2,Citation6,Citation7,Citation10,Citation18–24(Ref. 25 by Hayashi et al.). Significantly, COX-2 is associated with prostaglandin E2 (PGE2) and prostacyclin (PGI2) production that evokes or sustains systemic or peripheral inflammatory-diseases and/or -symptomsCitation4,Citation5,Citation25 but it is not involved in the COX-1-mediated GI tract eventsCitation15–17. As well, the inhibitory actions of traditional NSAIDs that include aspirin (acetylsalicylic acid) against COX-1 can be crucial problems in clinical pharmacotherapy, such as respiratory- and cutaneous-hypersensitivities and cross-reactivitiesCitation26–28.
Furthermore, it has been elucidated that COX-2 has key and pivotal roles in (i) induction and proliferation of various types of cancers or tumours in various regions such as colon (colorectal)Citation29,Citation30, gastricCitation31,Citation32, esophagealCitation33,Citation34, skin (non-melanoma/melanoma)Citation35–37, lungCitation38–40, liverCitation41–44, pancreaticCitation45–47, bile ductCitation48, gallbladderCitation48,Citation49, urinary bladderCitation50, breastCitation51,Citation52, prostateCitation53,Citation54, cervical (uterine cervix)Citation55,Citation56, ovarianCitation57,Citation58, nasopharyngealCitation59 and oralCitation60 cancers, and in (ii) metastasis of cancers such as lymph nodeCitation61,Citation62, haematogenousCitation63,Citation64, gastricCitation65, lungCitation66, liverCitation67–69, melanomaCitation70–72 and boneCitation73,Citation74 metastases that are correlated with COX-2 upregulation in cancer/tumour cells by inflammatory- and mitogenic-stimuli and growth factorsCitation22,Citation38,Citation52,Citation61,Citation65,Citation75 and by other reasons (as mentioned below). In patients with various cancers, adverse prognosis, disease relapse and poor survival rates have been associated with overexpression of COX-230,Citation37–40,Citation49,Citation56,Citation60,Citation63,Citation69,Citation70,Citation76,Citation77, but not with expression of COX-130,Citation56,Citation69,Citation76. Also, it has been widely studied and demonstrated that COX-2 inhibition has great clinical potentials of chemotherapeutic and chemopreventive treatments, with its anti-inflammatory and mortality-reduction effects, for cancer/tumour patients, owing to its suppressive and preventive effects against COX-2-dependent (i) stimulation or increases in pro-mitogenic factors, carcinogenesis, tumour angiogenesis and lymph angiogenesis and (ii) promotion, growth, resistance to apotosis, proliferation, invasion, spreading, migration, drug-resistance, recurrence and metastasis of cancer/tumour cells/tissuesCitation29–31,Citation36–38,Citation40,Citation44–46,Citation48,Citation51–54,Citation57,Citation58,Citation60–69,Citation71–75,Citation78–82. Actually, the cancer/tumour progression mechanisms including initiation, proliferation, invasion, spreading, migration, drug-resistance and metastasis of cancer/tumour cells are caused by (i) COX-2-derived mediators including prostanoids such as PGE2, prostaglandin F2α (PGF2α) and thromboxane A2 (TXA2) and growth factorsCitation37,Citation38,Citation40,Citation43,Citation44,Citation53,Citation54,Citation60–62,Citation65,Citation69,Citation75,Citation79,Citation81,Citation83–87, by (ii) DNA oxidative damage resulting from COX-2 activityCitation54,Citation75,Citation88,Citation89 and/or by (iii) other COX-2-derived signal pathwaysCitation42,Citation45,Citation52,Citation57,Citation78, which are dependent on COX-2 upregulation and activation that result from (i) chronic inflammatory stimuli or carcinogens/tumour-promoters or from (ii) inflammatory- and mitogenic-stimuli in and growth-factors in cancer/tumour cells/tissues, as mentioned, or from (iii) other promoters/inducers, for example, increased/accumulated ultraviolet (UV) light (UVA, UVB and UVC) exposuresCitation35,Citation36,Citation71,Citation90,Citation91, radiation exposuresCitation92, various chronic infections such as Helicobacter pylori (H. pylori) infectionCitation32,Citation83, hepatitis B virus (HBV) or hepatitis C virus (HCV) infectionCitation41,Citation42 and human papillomavirus (HPV) infectionCitation55, cigarette (tobacco) smokeCitation23 and chronic morphine-treatmentCitation82, or from (iv) other cancer/tumour cells-derived mechanismsCitation40,Citation45,Citation53,Citation71,Citation72. COX-2 inhibitors can boost immune systems of patients and/or augment other immunotherapies; hence, immunotherapy effects of COX-2 inhibitors have been clinically important to treat cancer/tumour and to prevent metastasis as wellCitation93.
On the other hand, it has been reported that COX-2 inhibitors or traditional NSAIDs showed no or agent structure/character-dependent potential cardiovascular-risk in long-term clinical studies with attention to permanent blockade against COX-2Citation94–96, and that COX-2 selective inhibition improved endothelial function in coronary artery disease derived from vascular-inflammation and oxidative-stress in clinical studyCitation97, and attenuated cardiovascular-failure and liver-damage induced by endotoxin in rat model studyCitation98, which show importance of safety-index assessment for respective COX-2 inhibitors in long-term use and other clinical potential of COX-2 inhibitors for the treatment of cardiovascular- and liver-diseases as already mentioned in the previous report by Hayashi et al.Citation25
Accordingly, discovery of potent and selective diverse-chemotypes of COX-2 inhibitors has become increasingly important to meet the above various clinical needs with no or minimal/controllable adverse effects, and to reveal the molecular mechanisms of enzyme inhibition by COX-2 inhibitor in vitro and in vivo. As approaches for elucidation of ligand–COX-2 interaction mechanisms and for (further) drug-design of potent and selective COX-2 inhibitors, the studies of X-ray protein-crystallography and molecular-structure/-docking calculation for COX isozymes with or without ligands have been reportedCitation99–103. In the studies, the respective ligand structure-dependent various interactions with COX-2, that is, respective binding modes in the complexes, respective time-dependent enzyme inhibitions and respective conformational changes of the enzyme by the ligands have been displayed or proposed. To clarify further detailed molecular mechanisms, various design and SAR studies for potent and selective COX-2 inhibitors are highly needed.
Besides, it has been described already, design, synthesis and in vitro and in vivo structure–activity relationship (SAR) studies for novel [2-{[(4-substituted or 4,5-disubstituted)-pyridin-2-yl]carbonyl}-(5- or 6-substituted or 5,6-disubstituted)-1H-indol-3-yl]acetic acid analogues were investigated by Hayashi et al.Citation25,Citation104, which resulted in the discoveries of novel acid-types of COX-2 inhibitors as a new-class of orally potent anti-pyretic and/or anti-inflammatory drugs in vivo; also, it has been shown in the studies that the mechanisms of orally potent anti-pyretic effect and anti-oedematous effect in the in vivo models for the analogues were due to their respective highly potent and selective, intrinsic COX-2 inhibitions. In this study, design, synthesis and SAR of diverse novel {2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues were investigated to seek and identify various chemotypes of orally potent and selective COX-2 inhibitors as potential agents for the treatment of inflammatory diseases. Based on the results of these studies of in vitro and in vivo SARs and physicochemical properties for the analogues together with the previously reported studies by us, molecular mechanisms of actions for the in vivo anti-inflammatory efficacies of the present analogues as COX-inhibitors are also discussed herein.
Experimental
Chemistry
General
In general, reagents, solvents and other chemicals were used as purchased without further purification. All reactions with air- or moisture-sensitive reactants and solvents were carried out under nitrogen atmosphere unless noted otherwise. Flash column chromatography (medium pressure liquid chromatography) purifications were carried out using Merck silica gel 60 (230–400 mesh ASTM; Merck KGaA, Darmstadt, Land Hessen, Germany). Preparative thin-layer chromatography (PTLC) purifications were carried out on Merck silica gel 60 F254 pre-coated glass plates at a thickness of 0.5 or 1.0 mm (Merck KGaA). The structures of all isolated compounds were ensured by NMR, IR, MS or elementary analysis. 1H nuclear magnetic resonance (1H NMR) data were determined at 270 MHz on a JNM-LA 270 (JEOL Ltd., Akishima, Tokyo, Japan) spectrometer. Chemical shifts are expressed in δ (ppm). 1H NMR chemical shifts were determined relative to tetramethylsilane (TMS) as internal standard. NMR data are reported as follows: chemical shift, number of atoms, multiplicities (s, singlet; d, doublet; t, triplet; m, multiplet; dd, double doublet; ddd, double double doublet; br, broadened), and coupling constants. Infrared spectra were measured by an IR-470 (Shimadzu Co., Nakagyo-ku, Kyoto, Japan) infrared spectrometer. Low-resolution mass spectral data (EI) were obtained on an Automass 120 (JEOL Ltd.) mass spectrometer or an Integrity mass spectrometer (Waters Corporation, Milford, MA). Low-resolution mass spectral data (ESI) were obtained on a Quattro II (Micromass, Milford, MA) mass spectrometer–Agilent 1100 HPLC system (Agilent Technologies, Inc., Santa Clara, CA). Melting points were obtained using an Exstar 6000 (Seiko Instruments Inc., Chiba, Japan) and were uncorrected. All general chemicals were the highest available grade.
The detailed original-procedures and obtained data for the respective compounds are described as follows and in the Appendix of the Supplementary Material.
[6-Chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]acetic acid (8a): general procedure of Method A
Step 1. 6-Chloro-1-(phenylsulfonyl)-1H-indole (2). A mixture of 6-chloroindole (Lancaster Synthesis, Heysham, Lancashire, UK, 52.52 g, 346.4 mmol), n-Bu4NHSO4 (Wako Pure Chemical Industries, Ltd., Chuo-ku, Osaka, Japan, 11.76 g, 34.64 mmol), 50% aqueous KOH (Wako Pure Chemical Industries, Ltd., 200 mL) and benzene (Wako Pure Chemical Industries, Ltd., 500 mL) was stirred for 10 min at room temperature under N2, then cooled to 0 °C, benzenesulfonyl chloride (Tokyo Chemical Industry Co., Ltd., Chuo-ku, Tokyo, Japan, 66.3 mL, 519.5 mmol) was added dropwise at 0 °C under N2. The reaction mixture was allowed to room temperature, and stirred under N2 for 3 h. Some of the solvent were removed, and the resulting suspension was filtered with anhydrous Et2O (Wako Pure Chemical Industries, Ltd.). The organic layer of the filtrate was separated, and the aqueous layer was extracted with Et2O (Wako Pure Chemical Industries, Ltd., 200 mL × 3). The organic layers were combined, dried over anhydrous MgSO4 (Wako Pure Chemical Industries, Ltd.), filtered and concentrated in vacuo. The resulting brown crystalline solid was washed with EtOH (Wako Pure Chemical Industries, Ltd.) to afford 78.93 g of the title product 2 in 78% yield as a slight pinkish-white crystalline solid. 1H NMR (270 MHz, CDCl3) δ 8.03–8.02 (1 H, m), 7.89–7.86 (2 H, m), 7.57–7.41 (5 H, m), 7.20 (1 H, dd, J = 8.40 Hz, J = 1.81 Hz), 6.63 (1 H, d, J = 3.62 Hz).
Step 2. [6-Chloro-1-(phenylsulfonyl)-1H-indol-2-yl](2-methylphenyl)methanone (4a). To a stirred solution of 6-chloro-1-(phenylsulfonyl)-1H-indole 2 (2.000 g, 6.855 mmol) in anhydrous THF (Dojindo Laboratories, Mashikimachi, Kumamoto, Japan, 32.0 mL) was added dropwise tert-BuLi (Kanto Chemical Co., Inc., Chuo-ku, Tokyo, Japan, 1.64 M in pentane, 4.60 mL, 7.54 mmol) via gas-tight syringe at −78 °C under N2. The solution was then stirred at the same temperature under N2 for 45 min under N2. To the resulting yellow anion solution was added dropwise a solution of 2-toluoyl chloride (Wako Pure Chemical Industries, Ltd., 1.20 mL, 9.20 mmol) in anhydrous THF (5.0 mL) at −78 °C under N2Citation105. The reaction solution was stirred at the same temperature under N2 for 2 h, gradually warmed up to 0 °C, and then stirred for 45 min. After saturated aqueous NH4Cl (Wako Pure Chemical Industries, Ltd., 50 mL) was added to the reaction mixture at 0 °C, the resulting mixture was extracted with AcOEt (Wako Pure Chemical Industries, Ltd., 50 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 3.820 g of the title product 4a as a pale gray solid (crude). TLC: Rf = 0.34, AcOEt/hexane = (1:10) × 2 (hexane: Wako Pure Chemical Industries, Ltd.). 1H NMR (270 MHz, CDCl3) δ 8.19–6.79 (13 H, m), 2.54 (3 H, s).
Step 3. (6-Chloro-1H-indol-2-yl)(2-methylphenyl)methanone (5a). A mixture of [6-chloro-1-(phenylsulfonyl)-1H-indol-2-yl](2-methylphenyl)methanone 4a (crude, 1.170 g) and anhydrous K2CO3 (Wako Pure Chemical Industries, Ltd., 1.996 g, 14.44 mmol) in THF (Wako Pure Chemical Industries, Ltd., 16.0 mL)–MeOH (Wako Pure Chemical Industries, Ltd., 8.0 mL)–pure H2O (Wako Pure Chemical Industries, Ltd., 4.0 mL) was warmed up to reflux conditions using an oil bath, stirred under N2 for 14 h, then cooled to room temperature. The mixture was extracted with AcOEt (50 mL × 3). The combined extracts were washed with saturated aqueous NaHCO3 and brine (saturated aqueous NaCl) (NaHCO3, NaCl; Wako Pure Chemical Industries, Ltd.), dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt to afford 181.4 mg of the title product a white crystalline solid. The mother liquor was also repurified by flash column chromatography (silica gel, hexane/AcOEt = 10:1), and the fraction that contained desired product was recrystallised from AcOEt–hexane to afford 335.6 mg of the title product a white crystalline solid. In total, 517.0 mg of the title product 5a was obtained in 91% yield [theoretical yield of two steps from compound 2 calculated with the ratio for (obtained weights in step 2)/(used weights in step 3) of compound 4a]. 1H NMR (270 MHz, CDCl3) δ 9.37 (1 H, br s), 7.59–7.56 (2 H, m), 7.48 (1 H, m), 7.44–7.41 (1 H, m), 7.34–7.27 (2 H, m), 7.12 (1 H, dd, J = 8.56 Hz, J = 1.81 Hz), 6.87 (1 H, m), 2.44 (3 H, s).
Step 4. Diethyl 2-acetoxy-2-[6-chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]malonate (6a). A mixture of (6-chloro-1H-indol-2-yl)(2-methylphenyl)methanone 5a (455.3 mg, 1.69 mmol), diethyl malonate (Wako Pure Chemical Industries, Ltd., 1.29 mL, 8.45 mmol) and AcONa (Wako Pure Chemical Industries, Ltd., 693.2 mg, 8.45 mmol) in glacial AcOH (Wako Pure Chemical Industries, Ltd., 16.0 mL) was degassed then purged with N2 at room temperature, and then Mn(OAc)3 ·2H2O (Sigma-Aldrich, St. Louis, MO, 1.40 g, 5.22 mmol) was addedCitation106,Citation107. The reaction mixture was stirred at room temperature under N2 for 5 min, warmed to 80 °C, and stirred under N2. After several minutes, the dark-brown mixture turned to black. After being stirred for 1 h, more Mn(OAc)3 · 2H2O (1.40 g, 5.22 mmol) was added to the resulting brown mixture that contained several amount of starting material detected by TLC analysis. The reaction mixture was stirred at 80 °C for 1 h, cooled to room temperature, and concentrated in vacuo. The residue was suspended in AcOEt (50 mL)–brine (50 mL) with vigorously stirring, and then filtered. After the organic layer of the filtrate was separated, the aqueous layer was extracted with AcOEt (50 mL × 2). The organic layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 3:1) to afford 578.2 mg of the title product 6a in 70% yield as a brown amorphous solid. 1H NMR (270 MHz, CDCl3) δ 8.53 (1 H, br s), 7.78 (1 H, d, J = 8.88 Hz), 7.49–7.13 (6 H, m), 4.31–4.18 (4 H, m), 2.52 (3 H, s), 1.96 (3 H, s), 1.22 (6 H, t, J = 7.26 Hz).
Step 5. Diethyl [6-chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]malonate (7a). To a stirred solution of diethyl 2-acetoxy-2-[6-chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]malonate 6a (569.8 mg, 1.173 mmol) in dry CH2Cl2 (Wako Pure Chemical Industries, Ltd., 8.5 mL) was added TFA (Wako Pure Chemical Industries, Ltd., 108 µL, 1.41 mmol) followed by Et3SiH (Tokyo Chemical Industry Co., Ltd., 749 µL, 4.69 mmol) at room temperature under N2Citation107. The reaction solution was warmed up to reflux conditions using an oil bath, stirred under N2 for 17 h, then more TFA (451 µL, 5.85 mmol) was added, and stirred for 7 h. The reaction solution was cooled to 0 °C, ice-cooled saturated aqueous NaHCO3 (20 mL) was added, and the resulting mixture was stirred at 0 °C for 15 min. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (Wako Pure Chemical Industries, Ltd., 20 mL × 2). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 4:1) to afford 491.8 mg of the title product 7a in 98% yield as a brown oil. 1H NMR (270 MHz, CDCl3) δ 9.02 (1 H, br s), 7.74 (1 H, d, J = 8.88 Hz), 7.73–7.25 (5 H, m), 7.12 (1 H, dd, J = 8.75 Hz, J = 1.81 Hz), 4.90 (1 H, s), 4.20–4.08 (4 H, m), 2.35 (3 H, s), 1.19 (6 H, t, J = 7.10 Hz).
Step 6. [6-Chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]acetic acid (8a). A mixture of diethyl [6-chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]malonate 7a (491.8 mg, 1.15 mmol) and 2 N NaOH (Wako Pure Chemical Industries, Ltd., 2.00 mL, 4.00 mmol) in EtOH (13.0 mL) was warmed up to reflux conditions using an oil bath, and stirred under N2 for 4 h. The reaction solution was cooled to room temperature, concentrated in vacuo, then the residue was partitioned between 2 N HCl (Wako Pure Chemical Industries, Ltd., 50 mL)–Et2O (50 mL) with acidification. The organic layer was separated, and the aqueous layer was extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 178.0 mg of the title product 8a in 47% yield as a yellow crystalline solid. Mp: 150–152 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.68 (1 H, s), 7.68 (1 H, d, J = 8.72 Hz), 7.51–7.30 (5 H, m), 7.09 (1 H, dd, J = 8.72 Hz, J = 1.97 Hz), 3.55 (2 H, s), 2.23 (3 H, s). IR (KBr): 3321, 1717, 1624, 1602, 1568, 1531, 1431, 1319, 1249, 1230 cm−1. MS (EI direct) m/z: M+ 327. Anal. (C18H14ClNO3) C, H, N.
For the subsections below, see “Experimental S1” section in the Appendix of the Supplementary Material for the detailed procedures and obtained data for the respective compounds:
[6-Chloro-2-(3-methylbenzoyl)-1H-indol-3-yl]acetic acid (8b)
[6-Chloro-2-(4-methylbenzoyl)-1H-indol-3-yl]acetic acid (8c)
{6-Chloro-2-[4-(trifluoromethyl)benzoyl]-1H-indol-3-yl}acetic acid (8d)
[6-Chloro-2-(2-chlorobenzoyl)-1H-indol-3-yl]acetic acid (8e)
[6-Chloro-2-(2,4-dichlorobenzoyl)-1H-indol-3-yl]acetic acid (8f)
[6-Chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]acetic acid (14)
Step 1. [6-Chloro-1H-indol-2-yl](cyclohexyl)methanone (11). To a stirred solution of compound 2 (1.3133 g, 4.501 mmol) in anhydrous THF (8.0 mL) was added dropwise tert-BuLi (1.64 M in pentane, 3.30 mL, 5.41 mmol) via gas-tight syringe at −78 °C under N2. The resulting solution was then stirred at the same temperature under N2 for 20 min. The resulting yellow solution was slowly cannulated directing into a stirred solution of cyclohexanecarbonyl chloride (Wako Pure Chemical Industries, Ltd., 722 µL, 5.40 mmol) in anhydrous THF (3.0 mL) at −78 °C under N2 over 10 min. After being stirred at the same temperature under N2 for 2 h, the reaction mixture was poured into ice-cooled aqueous saturated NH4Cl (20 mL). The resulting mixture was extracted with AcOEt (20 mL × 3), and the combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 2.2561 g of [6-chloro-1-(phenylsulfonyl)-1H-indol-2-yl](cyclohexyl)methanone 10 as a brown solid (crude). TLC: Rf = 0.38, AcOEt/hexane = 1:4.
To a suspension of [6-chloro-1-(phenylsulfonyl)-1H-indol-2-yl](cyclohexyl)methanone 10 (crude, 2.2036 g) in EtOH (19.0 mL) was added 2 N NaOH (14.8 mL, 29.6 mmol) at room temperature under N2. The mixture was dissolved in THF (10.0 mL) at 40 °C. The resulting solution was warmed up to reflux conditions, and stirred under N2 for 30 min. The reaction solution was cooled to room temperature, and concentrated in vacuo. The residue was partitioned between H2O (30 mL) and AcOEt (30 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (30 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 1.3981 g of the title product 11 as a brown solid (crude). 1H NMR (270 MHz, CDCl3) δ 10.08 (1 H, br s), 7.59–7.03 (4 H, m), 3.21–3.11 (1 H, m), 2.28–1.20 (10 H, m).
Step 2. Diethyl [6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]malonate (13). A mixture of [6-chloro-1H-indol-2-yl](cyclohexyl)methanone 11 (crude, 1.370 g), diethyl malonate (3.60 mL, 23.7 mmol) and AcONa (1.846 g, 22.5 mmol) in glacial AcOH (44.0 mL) was degassed then purged with N2 at room temperature and then Mn(OAc)3 · 2H2O (3.73 g, 13.9 mmol) was added. The reaction mixture was stirred at room temperature under N2 for 5 min, warmed to 80 °C, stirred under N2 for 2 h, and then more Mn(OAc)3 · 2H2O (1.80 g, 6.71 mmol) was added. The reaction mixture was stirred at 80 °C for 4 h, cooled to room temperature, then poured into brine (30 mL). The resulting mixture was extracted with AcOEt (30 mL × 3). The organic layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was roughly purified by flash column chromatography (silica gel, hexane/AcOEt = 4:1) to afford 946.0 mg of diethyl 2-acetoxy-2-[6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]malonate 12 as a brown solid containing impurity.
To a stirred solution of diethyl 2-acetoxy-2-[6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]malonate 12 (containing impurity, 920.0 mg) in dry CH2Cl2 (33.0 mL) was added TFA (1.16 mL, 15.1 mmol) followed by Et3SiH (1.15 mL, 7.20 mmol) at room temperature under N2. The reaction solution was warmed up to reflux conditions using an oil bath, and stirred under N2 for 15 h. The reaction solution was cooled to 0 °C, ice-cooled saturated aqueous NaHCO3 (20 mL) was added, and the resulting mixture was stirred at 0 °C for 15 min. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (20 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 5:1) to afford 316.8 mg of the title product 13 in 18% yield [four steps from compound 2 calculated with the ratios for (obtained weights in the preparation step)/(used weights in the next step) of the respective intermediates] as a brown solid. 1H NMR (270 MHz, CDCl3) δ 8.91 (1 H, br s), 7.72 (1 H, d, J = 8.88 Hz), 7.35 (1 H, d, J = 1.81 Hz), 7.11 (1 H, dd, J = 8.88 Hz, J = 1.81 Hz), 5.70 (1 H, s), 4.30–4.16 (4 H, m), 3.03–2.93 (1 H, m), 1.96–1.22 (16 H, m, including 6 H at 1.23 ppm, t, 7.07 Hz).
Step 3. [6-Chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]acetic acid (14). A mixture of diethyl [6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]malonate 13 (302.7 mg, 0.7209 mmol) and 2 N NaOH (2.00 mL, 4.00 mmol) in EtOH (13.0 mL) was warmed up to reflux conditions using an oil bath, stirred under N2 for 14 h, cooled to room temperature and concentrated in vacuo. The residue was mixed with H2O (30 mL) and Et2O (30 mL), then the resulting mixture was vigorously stirred for 15 min. The organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (30 mL), acidified by adding 2 N HCl (5 mL), then extracted with AcOEt (30 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 73.0 mg of the title product 14 in 32% yields as a solid. Mp: 206–209 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.77 (1 H, s), 7.70 (1 H, d, J = 8.56 Hz), 7.45 (1 H, d, J = 1.81 Hz), 7.07 (1 H, dd, J = 8.56 Hz, J = 1.81 Hz), 4.00 (2 H, s), 3.20 (1 H, m), 1.89–1.13 (10 H, m). IR (KBr): 3314, 2924, 2856, 1734, 1650, 1537, 1396, 1248 cm−1. Anal. (C17H18ClNO3) C, H, N.
[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B
Step 1. 6-Chloro-1-(phenylsulfonyl)-1H-indole-2-carboxylic acid (15). To a stirred solution of dry iso-Pr2NH (Wako Pure Chemical Industries, Ltd., 23.5 mL, 168 mmol) in anhydrous THF (150 mL) was added dropwise n-BuLi (Kanto Chemical Co., Inc., 1.54 M in hexane, 109 mL, 168 mmol) below −60 °C under N2. The resulting solution was stirred at 0 °C under N2 for 1.5 h, then cooled to −78 °C. To a stirred solution of compound 2 (refer Step 1 of “[6-Chloro-2-(2-methylbenzoyl)-1H-indol-3-yl]acetic acid (8a): general procedure of Method A” section; 43.77 g, 150.0 mmol) in anhydrous THF (440 mL) was added the above cooled solution of lithium diisopropylamide (LDA) through cannula at −78 °C under N2 for 15 min. After being stirred at the same temperature under N2 for 1 h, CO2 gas was bubbled into the reaction solution at −78 °C for 1.5 h. Saturated aqueous NH4Cl (300 mL) that was cooled in an ice-cooled water bath was added to the stirred reaction solution at −78 °C, and the resulting mixture was allowed to warm to room temperature with stirring, and then H2O (600 mL) was added. The resulting mixture was extracted with Et2O (800 mL). The ethereal layer was separated, and the aqueous layer was acidified with 2 N HCl (30 mL), then extracted with Et2O (400 mL). The ethereal layers were combined, extracted with 2 N NaOH (250 mL × 3). The aqueous layers were combined, acidified with 2 N HCl (900 mL), then extracted with Et2O (900 mL × 1, then 500 mL × 1). The ethereal layers were combined, then AcOEt (300 mL) was added to dissolve solid parts of the product in the organic extracts. The resulting solution was washed with brine (400 mL × 2), dried over anhydrous MgSO4, filtered, then concentrated in vacuo to afford 42.44 g of the title product 15 in 84% yield as a white crystalline solid. TLC: Rf = 0.27, CH2Cl2/MeOH = 5:1. 1H NMR (270 MHz, DMSO-d6) δ 8.01–7.38 (10 H, m).
Step 2. 6-Chloro-1H-indole-2-carboxylic acid (16). A mixture of 6-chloro-1-(phenylsulfonyl)-1H-indole-2-carboxylic acid 15 (35.83 g, 106.7 mmol), 2 N NaOH (500 mL, 1.00 mol) and EtOH (500 mL) was warmed up to reflux conditions using an oil bath, and stirred under N2 for 11 h. The resulting solution was concentrated in vacuo to a half-volume. The resulting white suspension was partitioned between H2O (750 mL) and Et2O (750 mL) with shaking vigorously. The aqueous layer was separated, acidified with 2 N HCl (750 mL) and then extracted with Et2O (1 L). The organic layer was washed with brine (300 mL), dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 19.74 g of the title product 16 in 95% yield as a white crystalline solid. TLC: Rf = 0.17, CH2Cl2/MeOH = 5:1. 1H NMR (270 MHz, DMSO-d6) δ 11.94 (1 H, s), 7.69 (1 H, d, J = 8.56 Hz), 7.50 (1 H, m), 7.16 (1 H, m), 7.12–7.09 (1 H, m).
Step 3. 6-Chloro-N-methoxy-N-methyl-1H-indole-2-carboxamide (17). To a stirred suspension of 6-chloro-1H-indole-2-carboxylic acid 16 (7.00 g, 35.8 mmol) in SOCl2 (Wako Pure Chemical Industries, Ltd., 30 mL) was added dropwise dry DMF (Wako Pure Chemical Industries, Ltd., 1.0 mL) at room temperature under N2, with heating by handy-drier several times. The resulting solution was stirred at room temperature under N2 for 30 min, then concentrated in vacuo. The residue was dissolved in dry CH2Cl2 (100 mL) at room temperature under N2, N,O-dimethylhydroxylamine hydrochloride (Sigma-Aldrich, 6.98 g, 71.6 mmol) was added with stirring, cooled to 0 °C, then dry pyridine (Wako Pure Chemical Industries, Ltd., 15.0 mL, 185 mmol) was addedCitation108. After being stirred at 0 °C under N2 for 2 h, the mixture was quenched with H2O (100 mL), and then extracted with CH2Cl2 (150 mL × 2). The combined organic extracts were washed with 2 N HCl (100 mL), saturated aqueous NaHCO3 (100 mL) and brine (100 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 8.20 g of the title product 17 in 96% yield as a yellow solid. 1H NMR (CDCl3) δ 9.48 (1 H, br s), 7.60 (1 H, d, J = 8.6 Hz), 7.46–7.41 (1 H, m), 7.22–7.18 (1 H, m), 7.11 (1 H, dd, J = 8.6 Hz, J = 1.8 Hz), 3.85 (3 H, s), 3.44 (3 H, s).
Step 4. (6-Chloro-1H-indol-2-yl)(3-fluorophenyl)methanone (19a). To a stirred solution of 6-chloro-N-methoxy-N-methyl-1H-indole-2-carboxamide 17 (1.193 g, 5.000 mmol) and 1-bromo-3-fluorobenzene (Wako Pure Chemical Industries, Ltd., 1.68 mL, 15.0 mmol) in anhydrous THF (40.0 mL) was added n-BuLi (1.54 M in hexane, 9.74 mL, 15.0 mmol) at −78 °C under N2Citation109,Citation110. The reaction solution was stirred at the same temperature under N2 for 2 h. The reaction solution was poured into ice-cooled saturated aqueous NH4Cl (40 mL), then extracted with AcOEt (40 mL × 3). The combined extracts were washed with 2 N HCl (40 mL) followed by saturated aqueous NaHCO3 (40 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 12:1) to afford 943.0 mg of the title product 19a in 69% yield as a yellow solid. 1H NMR (270 MHz, CDCl3) δ 9.28 (1 H, br s), 7.79–7.75 (1 H, m), 7.68–7.63 (2 H, m), 7.56–7.48 (2 H, m), 7.36–7.30 (1 H, m), 7.17–7.14 (2 H, m).
Step 5. Diethyl 2-acetoxy-2-[6-chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]malonate (20a). A mixture of (6-chloro-1H-indol-2-yl)(3-fluorophenyl)methanone 19a (930.9 mg, 3.401 mmol), diethyl malonate (2.58 mL, 17.0 mmol) and AcONa (1.395 g, 17.0 mmol) in glacial AcOH (34.0 mL) was degassed then purged with N2 at room temperature, and then Mn(OAc)3 · 2H2O (2.82 g, 10.5 mmol) was added. The reaction mixture was stirred at room temperature under N2 for 5 min, warmed to 80 °C, stirred under N2 for 3 h, and then more Mn(OAc)3 · 2H2O (1.41 g, 5.26 mmol) was added. The reaction mixture was stirred at 80 °C for 2.5 h, cooled to room temperature, then poured into brine (50 mL). The resulting mixture was extracted with AcOEt (50 mL × 3). The combined extracts were washed with brine (50 mL), dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 3:1) to afford 1.273 g of the title product 20a in 76% yield as a yellow crystalline solid. 1H NMR (270 MHz, CDCl3) δ 9.15 (1 H, br s), 8.83 (1 H, d, J = 8.72 Hz), 7.66–7.27 (5 H, m), 7.17 (1 H, dd, J = 8.72 Hz, J = 2.00 Hz,), 4.25–4.13 (4 H, m), 1.75 (3 H, s), 1.19 (6 H, t, J = 7.07 Hz).
Step 6. Diethyl [6-chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]malonate (21a). To a stirred solution of diethyl 2-acetoxy-2-[6-chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]malonate 20a (1.233 g, 2.517 mmol) in dry CH2Cl2 (26.0 mL) was added TFA (1.360 mL, 17.65 mmol) followed by Et3SiH (1.610 mL, 10.08 mmol) at room temperature under N2. The reaction solution was warmed up to reflux conditions using an oil bath, and stirred under N2 for 8 h. The reaction solution was cooled to 0 °C, and ice-cooled saturated aqueous NaHCO3 (40 mL) was added. The resulting mixture was stirred at 0 °C for 15 min. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 5:1) to afford 784.5 mg of the title product 21a in 72% yield as a yellow viscosity oil. 1H NMR (270 MHz, CDCl3) δ 8.91 (1 H, br s), 7.77–7.12 (7 H, m), 5.21 (1 H, s), 4.25–4.11 (4 H, m), 1.22 (6 H, t, J = 7.07 Hz).
Step 7. [6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a). A mixture of diethyl [6-chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]malonate 21a (753.4 mg, 1.74 mmol) and 2 N NaOH (3.00 mL, 6.00 mmol) in EtOH (20.0 mL) was warmed up to reflux conditions using an oil bath, and stirred under N2 for 16 h. The reaction solution was concentrated in vacuo, then the residue was partitioned between 2 N HCl (40 mL) and AcOEt (40 mL) with acidification. The organic layer was separated, and the aqueous layer was extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 446.7 mg of the title product 22a in 77% yield as a yellow crystalline solid. Mp: 278–281 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.79 (1 H, br s), 7.76–7.48 (6 H, m), 7.13 (1 H, dd, J = 8.56 Hz, J = 1.97 Hz), 3.80 (2 H, s). IR (KBr): 3385, 1697, 1638, 1583, 1541, 1508, 1420, 1400, 1315, 1261, 1236 cm−1. MS (EI direct) m/z: M+ 331. Anal. (C17H11ClFNO3) C, H, N.
For the subsections below, see “Experimental S1” section in the Appendix of the Supplementary Material for the detailed procedures and obtained data for the respective compounds:
[6-Chloro-2-(4-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22b)
{6-Chloro-2-[3-(trifluoromethyl)benzoyl]-1H-indol-3-yl}acetic acid (22c)
[2-(4-Chlorobenzoyl)-5-fluoro-1H-indol-3-yl]acetic acid (29)
Step 1. 5-Fluoro-N-methoxy-N-methyl-1H-indole-2-carboxamide (24). According to the procedure described in step 3 of general procedure of Method B (refer “[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B” section), the title product 24 was prepared from 5-fluoro-1H-indole-2-carboxylic acid (Sigma-Aldrich) instead of 6-chloro-1H-indole-2-carboxylic acid 16 in the procedure. 1H NMR (270 MHz, CDCl3) δ 10.15 (1 H, br s), 7.39 (1 H, dd, J = 8.88 Hz, J = 4.62 Hz), 7.32 (1 H, dd, J = 9.23 Hz, J = 2.48 Hz), 7.20 (1 H, dd, J = 2.13 Hz, J = 0.81 Hz), 7.05 (1 H, ddd, J = 9.23 Hz, J = 9.05 Hz, J = 2.48 Hz), 3.84 (3 H, m), 3.47 (3 H, s).
Step 2. (4-Chlorophenyl)(5-fluoro-1H-indol-2-yl)methanone (26). According to the procedure described in step 4 of general procedure of Method B (refer “[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B” section), 474 mg of the title product 26 was prepared in 64% yield as a slight yellow solid from 5-fluoro-N-methoxy-N-methyl-1H-indole-2-carboxamide 24 (600 mg, 2.70 mmol) and 1-bromo-4-chlorobenzene (Tokyo Chemical Industry Co., Ltd., 1.55 g, 8.10 mmol). 1H NMR (270 MHz, CDCl3) δ 9.27 (1 H, br s), 7.94 (2 H, d, J = 8.4 Hz), 7.52 (2 H, d, J = 8.4 Hz), 7.45–7.40 (1 H, m), 7.37–7.33 (1 H, m), 7.21–7.12 (1 H, m), 7.10–7.09 (1 H, m).
Step 3. Diethyl 2-acetoxy-2-[2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]malonate (27). According to the procedure described in step 5 of general procedure of Method B (refer “[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B” section), 500 mg of the title product 27 was prepared in 59% yield as a yellow solid from (4-chlorophenyl)(5-fluoro-1H-indol-2-yl)methanone 26 (474 mg, 1.73 mmol). 1H NMR (270 MHz, CDCl3) δ 9.01 (1 H, br s), 7.80–7.77 (2 H, m), 7.58–7.54 (1 H, m), 7.45–7.41 (2 H, m), 7.36–7.27 (1 H, m), 7.12–7.01 (1 H, m), 4.29–4.15 (4 H, m), 1.74 (3 H, s), 1.28–1.17 (6 H, m).
Step 4. Diethyl [2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]malonate (28). According to the procedure described in step 6 of general procedure of Method B (refer “[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B” section), 361 mg of the title product 28 was prepared in 84% yield as a slight yellow solid from diethyl 2-acetoxy-2-[2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]malonate 27 (486 mg, 0.992 mmol). 1H NMR (270 MHz, CDCl3) δ 8.98 (1 H, br s), 7.75 (2 H, ddd, J = 8.72 Hz, J = 2.13 Hz, J = 1.97 Hz), 7.49 (2 H, ddd, J = 8.75 Hz, J = 2.16 Hz, J = 1.97 Hz), 7.40 (1 H, dd, J = 9.72 Hz, J = 2.48 Hz), 7.16 (1 H, dd, J = 8.88 Hz, J = 4.13 Hz), 7.00 (1 H, ddd, J = 8.99 Hz, J = 8.88 Hz, J = 2.48 Hz), 5.29 (1 H, s), 4.25–4.07 (4 H, m), 1.24 (6 H, t, J = 7.10 Hz).
Step 5. [2-(4-Chlorobenzoyl)-5-fluoro-1H-indol-3-yl]acetic acid (29). According to the procedure described in step 7 of general procedure of Method B (refer “[6-Chloro-2-(3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (22a): general procedure of Method B” section), 240 mg of the title product 29 was prepared in 87% yield as a yellow crystalline solid from diethyl [2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]malonate 28 (360 mg, 0.834 mmol). Mp: 233–238 °C. 1H NMR (270 MHz, DMSO-d6) δ 12.21 (1 H, br s), 11.73 (1 H, br s), 7.79–7.74 (2 H, m), 7.67–7.63 (2 H, m), 7.52–7.44 (2 H, m), 7.22–7.19 (1 H, ddd, J = 9.23 Hz, J = 9.05 Hz, J = 2.48 Hz), 3.84 (2 H, s). IR (KBr): 3317, 1707, 1624, 1609, 1587, 1526, 1458, 1408, 1344, 1263, 1242 cm−1. MS (EI direct) m/z: M+ 331. Anal. (C17H11ClFNO3) C, H, N.
[2-(4-Chlorobenzoyl)-6-fluoro-1H-indol-3-yl]acetic acid (44a): general procedure of Method C
Step 1. Methyl [2-(4-chlorobenzoyl)-6-fluoro-1H-indol-3-yl]acetate (43a). A mixture of methyl (2 E)-3-(4-fluoro-2-{[(4-methylphenyl)sulfonyl]amino}phenyl)acrylate 30a {(181.7 mg, 0.520 mmol); that was synthesised by us at Pfizer Global Research & Development, Nagoya Laboratories, Pfizer Japan Inc. (Aichi, Japan) according to the reported procedure by Hayashi et al.Citation104, 1H NMR (270 MHz, CDCl3) δ 7.64 (1 H, d, J = 8.40 Hz), 7.46–7.19 (6 H, m), 6.94–6.87 (1 H, m), 6.77 (1 H, br s), 6.13 (1 H, d, J = 15.8 Hz), 3.79 (3 H, s), 2.38 (3 H, s)}, 4-chlorophenacyl bromide (Tokyo Chemical Industry Co., Ltd., 182.1 mg, 0.780 mmol) and anhydrous K2CO3 (718.7 g, 5.20 mmol) in dry acetone (Wako Pure Chemical Industries, Ltd., 5.2 mL) was stirred at room temperature under N2 for 22.5 h, then 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU; Wako Pure Chemical Industries, Ltd., 233 µL, 1.56 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 17 h, then concentrated in vacuo. The residue was partitioned between H2O (40 mL) and AcOEt (40 mL). The organic layer was separated and the aqueous layer was extracted with AcOEt (40 mL × 4). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 6:1) to afford 152.4 mg of the title product 43a in 85% yield as a yellow solid. 1H NMR (270 MHz, CDCl3) δ 9.10 (1 H, br s), 7.76–7.44 (5 H, m), 7.02–6.90 (2 H, m), 3.81 (2 H, s), 3.66 (3 H, s).
Step 2. [2-(4-Chlorobenzoyl)-6-fluoro-1H-indol-3-yl]acetic acid (44a). To a stirred mixture of methyl [2-(4-chlorobenzoyl)-6-fluoro-1H-indol-3-yl]acetate 43a (152.4 mg, 0.441 mmol) in MeOH (10.0 mL)–THF (20.0 mL) was added 2 N NaOH (1.10 mL, 2.20 mmol) at room temperature under N2. The reaction mixture was warmed up to reflux conditions using an oil bath, stirred under N2 for 2.5 h, cooled to room temperature, then concentrated in vacuo. The residue was partitioned between H2O (40 mL) and Et2O (40 mL), and the organic layer was separated to remove impurity. The aqueous layer was acidified by adding 2 N HCl (3.0 mL), then extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 114.0 mg of the title product 44a in 78% yield as a brownish-yellow solid. Mp: 214 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.73 (1 H, brs), 7.78–7.72 (3 H, m), 7.66–7.63 (2 H, m), 7.17 (1 H, dd, J = 9.72 Hz, J = 2.30 Hz), 7.19 (1 H, ddd, J = 9.72 Hz, J = 8.96 Hz, J = 2.48 Hz), 3.84 (2 H, s). IR (KBr): 3335, 1699, 1618, 1605, 1587, 1531, 1425, 1327, 1267, 1231, 1134, 1094, 1001 cm−1. MS (EI direct) m/z: M+ 331. Anal. (C17H11ClFNO3) C, H, N.
For the subsections below, see “Experimental S1” section in the Appendix of the Supplementary Material for the detailed procedures and obtained data for the respective compounds:
[5-(tert-Butyl)-2-(4-chlorobenzoyl)-1H-indol-3-yl]acetic acid (44b)
[2-(4-Chlorobenzoyl)-6-methoxy-1H-indol-3-yl]acetic acid (44c)
[2-(4-Chlorobenzoyl)-5-methoxy-1H-indol-3-yl]acetic acid (44d)
[2-(4-Chlorobenzoyl)-5-(trifluoromethoxy)-1H-indol-3-yl]acetic acid (44e)
[6-Chloro-2-(4-chloro-3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (44f)
Step 1. 1-(4-Chloro-3-fluorophenyl)ethanone (34). A solution of n-BuLi (Kanto Chemical Co., Inc., 1.55 M in hexane, 6.77 mL, 10.5 mmol) was added to a stirring solution of 1-bromo-4-chloro-3-fluorobenzene (Fluorochem Ltd., Hadfield, Derbyshire, UK, 2.09 g, 9.98 mmol) in anhydrous Et2O (12.0 mL) at −78 °C under N2 over 5 min, warmed to −20 °C, then stirred for 45 min. To the above solution, a solution of dry N,N-dimethylacetamide (DMA; Wako Pure Chemical Industries, Ltd., 1.04 mL, 11.2 mmol) in anhydrous Et2O (1.5 mL) was added at the same temperature under N2 over 5 min. The reaction solution was stirred at the temperature for 1 h, allowed to room temperature, and stirred for 3 h. The reaction solution was poured into ice-cooled saturated aqueous NH4C1 (20 mL), then extracted with Et2O (30 mL × 3). The organic layers were combined, washed with 2 N HCl (20 mL), saturated aqueous NaHCO3 (20 mL) and brine (20 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 1.6203 g of the title product 34 as a yellow oil in 94% yield (that was calculated with the gross yields containing a small amount of impurity). This compound was used for the next step without further purification. 1H NMR (270 MHz, CDCl3) δ 7.74–7.66 (2 H, m), 7.53–7.47 (1 H, m), 2.59 (3 H, s).
Step 2. 2-Bromo-1-(4-chloro-3-fluorophenyl)ethanone (35). A mixture of 1-(4-chloro-3-fluorophenyl)ethanone 34 (1.0355 g, 6.00 mmol, estimated as 100% purity) and tetrabutylammonium tribromide (Tokyo Chemical Industry Co., Ltd., 3.182 g, 6.60 mmol) in dry CH2Cl2 (18.0 mL)–dry MeOH (9.0 mL) was stirred at room temperature under N2 for 19 hCitation111,Citation112. The reaction solution was poured into H2O (20 mL), then extracted with Et2O (30 mL × 2). The combined extracts were washed with H2O (30 mL × 2), dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was washed with iso-Pr2O (Wako Pure Chemical Industries, Ltd.) and AcOEt to afford 224.0 mg of the title product 35 in 15% yield as a colourless crystalline solid. 1H NMR (270 MHz, CDCl3) δ 7.80–7.71 (2 H, m), 7.55 (1 H, dd, J = 8.24 Hz, J = 6.94 Hz), 4.38 (2 H, s).
Step 3. Methyl [6-chloro-2-(4-chloro-3-fluorobenzoyl)-1H-indol-3-yl]acetate (43f). A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f {279.8 mg, 0.795 mmol; that was synthesised by us at Pfizer Global Research & Development, Nagoya Laboratories, Pfizer Japan Inc. according to the reported procedure by Hayashi et al.Citation25, 1H NMR (270 MHz, CDCl3) δ 7.75–7.72 (2 H, m), 7.58–7.36 (6 H, m), 7.20 (1 H, dd, J = 8.56 Hz, J = 2.13 Hz), 7.14 (1 H, br s), 6.15 (1 H, d, J = 15.8 Hz), 3.78 (3 H, s)}, 2-bromo-1-(4-chloro-3-fluorophenyl)ethanone 35 (220.0 mg, 0.875 mmol) and anhydrous K2CO3 (329.8 mg, 2.39 mmol) in dry acetone (6.0 mL) was stirred at room temperature under N2 for 16 h, then DBU (357 µL, 2.39 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 11 h, then cooled to 0 °C. The reaction mixture was poured into ice-cooled 2 N HCl (20 mL), then the resulting mixture was extracted with AcOEt (20 mL × 3). The organic layers were combined, washed with 2 N HCl (20 mL × 2), saturated aqueous NaHCO3 (20 mL) and brine (20 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 6:1) followed by recrystallisation from 2-propanol (Wako Pure Chemical Industries, Ltd.) –hexane to afford 150.5 mg of the title product 43f in 50% yield as a yellow solid. 1H NMR (270 MHz, CDCl3) δ 8.87 (1 H, br s), 7.66–7.35 (5 H, m), 7.25–7.12 (1 H, m), 3.80 (2 H, s), 3.68 (3 H, s).
Step 4. [6-Chloro-2-(4-chloro-3-fluorobenzoyl)-1H-indol-3-yl]acetic acid (44f). A mixture of methyl [6-chloro-2-(4-chloro-3-fluorobenzoyl)-1H-indol-3-yl]acetate 43f (150.5 mg, 0.396 mmol) and 2 N NaOH (1.00 mL, 2.00 mmol) in EtOH (7.0 mL) was warmed up to reflux conditions using an oil bath, and stirred under N2 for 1.5 h. The reaction mixture was cooled to room temperature, 2 N HCl (1.0 mL) was added, and the resulting mixture was concentrated in vacuo. The residue was dissolved in AcOEt, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 126.5 mg of the title product 44f in 87% yield as a yellow solid. Mp: 179–182 °C. 1H NMR (270 MHz, DMSO-d6) δ 12.28 (1 H, br s), 11.79 (1 H, br s), 7.83–7.71 (3 H, m), 7.58 (1 H, dd, J = 8.40 Hz, J = 1.81 Hz), 7.46 (1 H, d, J = 1.81 Hz), 7.12 (1 H, dd, J = 8.56 Hz, J = l.81 Hz), 3.83 (2 H, s). MS (EI direct) m/z: M+ 365. Anal. (C17H10Cl2FNO3) C, H, N.
(6-Chloro-2-{4-[(methylsulfonyl)amino]benzoyl}-1H-indol-3-yl)acetic acid (44g)
Step 1. N-(4-Acetylphenyl)methanesulfonamide (37). To a stirred solution of 4′-aminoacetophenone (Wako Pure Chemical Industries, Ltd., 1.3517 g, 10.0 mmol) in dry CH2Cl2 (60.0 mL) was added dry pyridine (34.0 mL) at room temperature under N2. The solution was cooled to 0 °C, then methanesulfonyl chloride (Wako Pure Chemical Industries, Ltd., 0.851 mL, 11.0 mmol) was added dropwise under N2. The reaction mixture was allowed to room temperature, and stirred for 23 h. The volatiles were then removed in vacuo, azeotroping with H2O. The residue was partitioned between 1 N HCl (Wako Pure Chemical Industries, Ltd., 100 mL) and AcOEt (100 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (100 mL × 2). The combined extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 2.14 g of the title product 37 in 100% yield. 1H NMR (270 MHz, CDCl3) δ 7.97 (2 H, d, J = 8.75 Hz), 7.26 (2 H, d, J = 8.75 Hz), 6.75 (1 H, br s), 3.10 (3 H, s), 2.59 (3 H, s).
Step 2. N-[4-(2-Bromoacetyl)phenyl]methanesulfonamide (38). A mixture of N-(4-acetylphenyl)methanesulfonamide 37 (1.0663 g, 5.00 mmol) and tetrabutylammonium tribromide (2.652 g, 5.50 mmol) in dry CH2Cl2 (15.0 mL)–dry MeOH (7.5 mL) was stirred at room temperature under N2 for 19 h. The reaction solution was poured into H2O (15 mL), then extracted with Et2O (40 mL × 2). The combined extracts were washed with H2O (30 mL × 2), dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 873.8 mg of the title product 38 in 60% yield as a white solid. 1H NMR (270 MHz, CDCl3) δ 8.01 (2 H, d, J = 8.72 Hz), 7.28 (2 H, d, J = 8.72 Hz), 6.60 (1 H, br s), 4.40 (2 H, s), 3.13 (3 H, s).
Step 3. Methyl (6-chloro-2-{4-[(methylsulfonyl)amino]benzoyl}-1H-indol-3-yl)acetate (43g). A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f (351.8 mg, 1.00 mmol), N-[4-(2-bromoacetyl)phenyl]methanesulfonamide 38 (350.6 mg, 1.20 mmol) and anhydrous K2CO3 (414.6 mg, 3.00 mmol) in dry acetone (7.0 mL) was stirred at room temperature under N2 for 4 h, then DBU (449 µL, 3.00 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 24 h. The reaction mixture was diluted with AcOEt (30 mL), washed with 2 N HCl (20 mL × 2), saturated aqueous NaHCO3 (20 mL × 2) and brine (20 mL × 2), then dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 2:1 to 1:1) to afford 182.0 mg of the title product 43g in 43% yield as a yellow solid. 1H NMR (270 MHz, CDCl3) δ 8.86 (1 H, br s), 7.82 (2 H, d, J = 8.72 Hz), 7.58 (1 H, d, J = 8.72 Hz), 7.42 (1 H, d, J = 1.49 Hz), 7.30 (2 H, d, J = 8.72 Hz), 7.17 (1 H, dd, J = 8.72 Hz, J = 1.81 Hz), 6.80 (1 H, br s), 3.83 (2 H, s), 3.67 (3 H, s), 3.14 (3 H, s).
Step 4. (6-Chloro-2-{4-[(methylsulfonyl)amino]benzoyl}-1H-indol-3-yl)acetic acid (44g). A mixture of methyl (6-chloro-2-{4-[(methylsulfonyl)amino]benzoyl}-1H-indol-3-yl)acetate 43g (174.7 mg, 415 mmol) and 2 N NaOH (1.00 mL, 2.00 mmol) in EtOH (7.0 mL) was warmed up to reflux conditions using an oil bath, stirred under N2 for 20 h, cooled to room temperature and concentrated in vacuo. The residue was partitioned between H2O (30 mL) and Et2O (30 mL), then the organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (30 mL × 3), acidified by adding 2 N HCl (30 mL), then extracted with AcOEt (30 mL × 4). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 124.0 mg of the title product 44g in 73% yield as a yellow solid. Mp: 207–210 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.72 (1 H, br s), 7.75 (2 H, d, J = 8.88 Hz), 7.70 (1 H, d, J = 8.56 Hz), 7.45 (1 H, J = 1.65 Hz), 7.33 (1 H, J = 8.72 Hz), 7.11 (1 H, dd, J = 8.72 Hz, J = 1.65 Hz), 3.80 (2 H, s), 3.12 (3 H, s). IR (KBr): 3333, 3248, 1715, 1603, 1570, 1529, 1508, 1394, 1323, 1259, 1231, 1159, 1061 cm−1. MS (EI direct) m/z: M+ 406. Anal. (C18H15ClN2O5S) C, H, N.
{6-Chloro-2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-1H-indol-3-yl}acetic acid (44h)
Step 1. 2-Bromo-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone (40). To a stirred solution of 1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone (Sigma-Aldrich, 2.138 g, 12.00 mmol) in dry CHCl3 (Wako Pure Chemical Industries, Ltd., 10.0 mL) was added 25% HBr/AcOH (Wako Pure Chemical Industries, Ltd., 25.0 mL) at room temperature under N2. The reaction mixture was cooled to 0 °C, then a solution of bromine (Wako Pure Chemical Industries, Ltd., 0.68 mL, 13.2 mmol) in glacial AcOH (5.4 mL) was added dropwise at 0 °C under N2 over 15 min. The reaction mixture was allowed to room temperature, stirred under N2 for 1.5 h and concentrated in vacuo. The residue was diluted with Et2O (80 mL), then basified by adding saturated aqueous 2 N NaOH. The ethereal layer was separated and the aqueous layer was extracted with Et2O (50 mL × 2). The ethereal layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 3.42 g of the title product 40 as a pale-green solid (crude). 1H NMR (270 MHz, CDCl3) δ 7.64–7.51 (2 H, m), 6.96–6.92 (1 H, m), 4.37 (2 H, s), 4.36–4.28 (4 H, m).
Step 2. Methyl {6-chloro-2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-1H-indol-3-yl}acetate (43h). A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f (211.1 mg, 0.600 mmol), bromo-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone 40 (crude, 348.5 mg) and anhydrous K2CO3 (1.106 g, 8.00 mmol) in dry acetone (7.0 mL) was stirred at room temperature under N2 for 11 h, then DBU (269 µL, 1.80 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 14 h, and concentrated in vacuo. The residue was partitioned between H2O (30 mL) and AcOEt (40 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (30 mL × 3). The organic layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 2:1) to afford 97.3 mg of the title product 43h in 42% yield as an orange-yellowish white solid. 1H NMR (270 MHz, CDCl3) δ 8.85 (1 H, br s), 7.56 (1 H, d, J = 8.56 Hz), 7.41–7.34 (3 H, m), 7.15 (1 H, dd, J = 8.56 Hz, J = 1.81 Hz), 6.98–6.94 (1 H, m), 4.38–4.29 (4 H, m), 3.88 (2 H, s), 3.68 (3 H, s).
Step 3. {6-Chloro-2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-1H-indol-3-yl}acetic acid (44h). To a stirred solution of methyl {6-chloro-2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-1H-indol-3-yl}acetate 43h (97.3 mg, 0.252 mmol) in MeOH (5.0 mL)–THF (5.0 mL) was added 2 N NaOH (0.600 mL, 1.20 mmol) at room temperature under N2. The reaction mixture was warmed up to reflux conditions using an oil bath, stirred under N2 for 6 h, cooled to room temperature and concentrated in vacuo. The residue was partitioned between H2O (20 mL)–Et2O (20 mL), and the organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (20 mL), acidified by adding 2 N HCl, and then extracted with AcOEt (30 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 61.0 mg of the title product 44h in 65% yield as a pale-yellow solid. Mp: 233 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.71 (1 H, br s), 7.68 (1 H, d, J = 8.75 Hz), 7.46 (1 H, d, J = 1.65 Hz), 7.29 (1 H, dd, J = 8.40 Hz, J = 2.00 Hz), 7.24 (1 H, d, J = 1.97 Hz), 7.11 (1 H, dd, J = 8.72 Hz, J = 1.97 Hz), 7.02 (1 H, d, J = 8.40 Hz), 4.36–4.29 (4 H, m), 3.80 (2 H, s). IR (KBr): 3321, 1707, 1612, 1568, 1429, 1317, 1288, 1263, 1225, 1119, 1065, 1005, 922, 895 cm−1. MS (EI direct) m/z: M+ 371. Anal. (C19H14ClNO5) C, H, N.
{2-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonyl)-5-(trifluoromethyl)-1H-indol-3-yl}acetic acid (44i)
Step 1. Methyl {2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-5-(trifluoromethyl)-1H-indol-3-yl}acetate (43i). A mixture of methyl (2 E)-3-{2-[(phenylsulfonyl)amino]-5-(trifluoromethyl)phenyl}acrylate 30g {536.6 mg, 1.39 mmol; that was synthesised by us at Pfizer Global Research & Development, Nagoya Laboratories, Pfizer Japan Inc. according to the reported procedure by Hayashi et al.Citation25, 1H NMR (CDCl3, 270 MHz) δ 7.79–7.75 (2 H, m), 7.66–7.54 (5 H, m), 7.50–7.44 (2 H, m), 7.18 (1 H, br s), 6.26 (1 H, d, J = 15.8 Hz), 3.81 (3 H, s)}, bromo-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone 40 (1.548 g, 6.00 mmol) and anhydrous K2CO3 (2.948 g, 21.3 mmol) in dry acetone (20.0 mL) was stirred at room temperature under N2 for 11 h, then DBU (718 µL, 4.80 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 14 h, then concentrated in vacuo. The residue was partitioned between H2O (30 mL) and AcOEt (40 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (30 mL × 3). The organic layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 2:1) to afford 119.3 mg of the title product 43i in 20% yield as a yellowish-white solid. 1H NMR (270 MHz, CDCl3) δ 9.19 (1 H, br s), 7.95 (1 H, m), 7.58–7.47 (2 H, m), 7.38 (1 H, s), 7.37 (1 H, dd, J = 7.75 Hz, J = 2.16 Hz), 6.98–6.95 (1 H, m), 4.37–4.29 (4 H, m), 3.91 (2 H, s), 3.70 (3 H, s).
Step 2. {2-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonyl)-5-(trifluoromethyl)-1H-indol-3-yl}acetic acid (44i). To a stirred solution of methyl {2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl)-5-(trifluoromethyl)-1H-indol-3-yl}acetate 43i (119.3 mg, 0.284 mmol) in MeOH (7.0 mL)–THF (7.0 mL) was added 2 N NaOH (0.700 mL, 1.40 mmol) at room temperature under N2. The reaction mixture was warmed up to reflux conditions using an oil bath, stirred under N2 for 5 h, cooled to room temperature and concentrated in vacuo. The residue was partitioned between H2O (30 mL) and Et2O (30 mL), and the organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (30.0 mL), acidified by adding 2 N HCl and then extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 90.4 mg of the title product 44i in 79% yield as a pale-yellow solid. Mp: >250 °C. 1H NMR (270 MHz, DMSO-d6) δ 12.03 (1 H, br s), 8.11 (1 H, s), 7.64–7.52 (2 H, m), 7.33–7.26 (2 H, m), 7.03 (1 H, d, J = 8.40 Hz), 4.37–4.29 (4 H, m), 3.88 (2 H, s). IR (KBr): 3304, 1701, 1624, 1578, 1508, 1333, 1288, 1236, 1165, 1109, 1049, 1011, 893, 820 cm−1. MS (EI direct) m/z: M+ 405. Anal. (C20H14F3NO5) C, H, N.
[6-Chloro-2-(2,3-dihydrobenzofuran-5-carbonyl)-1H-indol-3-yl]acetic acid (44j)
Step 1. 2-Bromo-1-(2,3-dihydrobenzofuran-5-yl)ethanone (42). To a stirred solution of 1-(2,3-dihydrobenzofuran-5-yl)ethanone (Lancaster Synthesis, 486.6 mg, 3.00 mmol) in dry CHCl3 (6.0 mL) was added 25% HBr/AcOH (6.3 mL) at room temperature under N2. The reaction mixture was cooled to 0 °C, then a solution of bromine (155 µL, 3.01 mmol) in glacial AcOH (1.4 mL) was added dropwise at 0 °C under N2 over 10 min. The reaction mixture was allowed to room temperature, stirred under N2 for 1.5 h and concentrated in vacuo. The residue was diluted with Et2O (30 mL), then basified by adding saturated aqueous 2 N NaOH. The ethereal layer was separated and the aqueous layer was extracted with Et2O (30 mL × 4). The ethereal layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 1.108 g of the title product 42 as a green oil (crude). 1H NMR (270 MHz, CDCl3) δ 7.98–6.81 (3 H, m), 4.73–4.67 (2 H, m), 4.39 (2 H, s), 3.32–3.25 (2 H, m).
Step 2. Methyl [6-chloro-2-(2,3-dihydrobenzofuran-5-carbonyl)-1H-indol-3-yl]acetate (43j). A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f (361.6 mg, 1.03 mmol), 2-bromo-1-(2,3-dihydrobenzofuran-5-yl)ethanone 42 (crude, 1.108 g) and anhydrous K2CO3 (4.15 g, 30.0 mmol) in dry acetone (20.0 mL) was stirred at room temperature under N2 for 20 h, then DBU (1.00 mL, 6.69 mmol) was added. The reaction mixture was stirred at the same temperature under N2 for 22 h, then concentrated in vacuo. The residue was partitioned between H2O (50 mL) and AcOEt (50 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (50 mL × 3). The organic layers were combined, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/AcOEt = 7:3) to afford 154.1 mg of the title product 43j in 40% yield as a pale-yellow solid. 1H NMR (270 MHz, CDCl3) δ 9.03 (1 H, br s), 7.73–7.64 (2 H, m), 7.55 (1 H, d, J = 8.72 Hz), 7.39 (1 H, d, J = 1.65 Hz), 7.13 (1 H, dd, J = 8.72 Hz, J = 1.81 Hz), 6.84 (1 H, d, J = 8.40 Hz), 4.69 (2 H, t, J = 8.72 Hz), 3.85 (2 H, s), 3.67 (3 H, s), 3.26 (2 H, t, J = 8.72 Hz).
Step 3. [6-Chloro-2-(2,3-dihydrobenzofuran-5-carbonyl)-1H-indol-3-yl]acetic acid (44j). To a stirred solution of methyl [6-chloro-2-(2,3-dihydrobenzofuran-5-carbonyl)-1H-indol-3-yl]acetate 43j (154.1 mg, 0.417 mmol) in MeOH (10.0 mL)–THF (10.0 mL) was added 2 N NaOH (1.00 mL, 2.00 mmol) at room temperature under N2. The reaction mixture was warmed up to reflux conditions using an oil bath, stirred under N2 for 5 h, cooled to room temperature and concentrated in vacuo. The residue was partitioned between H2O (40 mL) and Et2O (40 mL), and the organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (40 mL × 3), acidified by adding 2 N HCl and then extracted with AcOEt (50 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 35.6 mg of the title product 44j in 24% yield as a yellow solid. Mp: 217 °C. 1H NMR (270 MHz, DMSO-d6) δ 11.73 (1 H, br s), 7.70–7.59 (3 H, m), 7.47–7.46 (1 H, m), 7.12 (1 H, dd, J = 8.59 Hz, J = 1.81 Hz), 6.92 (1 H, d, J = 8.24 Hz), 4.67 (2 H, t, J = 8.72 Hz), 3.78 (2 H, s), 3.26 (2 H, t, J = 8.72 Hz). IR (KBr): 3368, 1699, 1618, 1605, 1578, 1570, 1541, 1329, 1265, 1250, 1097, 1059, 939 cm−1. MS (EI direct) m/z: M+ 355. Anal. (C19H14ClNO4) C, H, N.
2-(6-Chloro-2-cyclobutanecarbonyl-1H-indol-3-yl)acetic acid (48a)
Step 1. Methyl (6-chloro-2-cyclobutanecarbonyl-1H-indol-3-yl)acetate (47a). A mixture of cyclobutyl methyl ketone (Tokyo Chemical Industry Co., Ltd., 1.09 mL, 10.0 mmol) and tetrabutylammonium tribromide (5.30 g, 11.0 mmol) in dry CH2Cl2 (15.0 mL)–dry MeOH (7.5 mL) was stirred at room temperature under N2 for 30 h, then concentrated in vacuo. The residue was partitioned between H2O (30 mL) and Et2O (30 mL). The ethereal layer was separated, and the aqueous layer was extracted with Et2O (30 mL). The ethereal layers were combined, washed with H2O (30 mL × 2), dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 1.608 g of 2-bromo-1-cyclobutylethanone 46a as an oil (crude). TLC: Rf = 0.65, AcOEt/hexane = 1:2.
A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f (492.5 mg, 1.40 mmol), 2-bromo-1-cyclobutylethanone 46a (crude, 424.9 mg) and anhydrous K2CO3 (580.5 mg, 4.20 mmol) in dry acetone (12.0 mL) was stirred at room temperature under N2 for 6 h, then DBU (628 µL, 4.20 mmol) was added. The reaction mixture was stirred under N2 at the same temperature under N2 for 24 h, then cooled to 0 °C. The reaction mixture was partitioned between AcOEt (30 mL) and 2 N HCl (30 mL). The organic layer was separated and the aqueous layer was extracted with AcOEt (30 mL). The organic layers were combined, washed with 2 N HCl (30 mL × 2), saturated aqueous NaHCO3 (30 mL) and brine (30 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 440.7 mg of the title product 47a as a yellow solid (crude). 1H NMR (270 MHz, CDCl3) δ 9.35 (1 H, br s), 7.53 (1 H, d, J = 8.56 Hz), 7.19 (1 H, d, J = 1.81 Hz), 7.07 (1 H, dd, J = 8.56 Hz, J = 1.81 Hz), 4.10 (2 H, s), 3.77 (3 H, s), 3.77–3.71 (1 H, m), 2.44–1.86 (6 H, m).
Step 2. 2-(6-Chloro-2-cyclobutanecarbonyl-1H-indol-3-yl)acetic acid (48a). A mixture of methyl (6-chloro-2-cyclobutanecarbonyl-1H-indol-3-yl)acetate 47a (crude, 440.7 mg) and 2 N NaOH (3.50 mL, 7.00 mmol) in EtOH (24.5 mL) was warmed up to reflux conditions using an oil bath, and stirred under N2 for 20 h. The reaction mixture was cooled to room temperature, and concentrated in vacuo. The residue was partitioned between H2O (40 mL) and Et2O (40 mL), then the organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (40 mL × 3), acidified by adding 2 N HCl (30 mL) and then extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 221.4 mg of the title product 48a in 54% yield (two steps from compound 30f) as a yellow solid. Mp: 225–228 °C. 1H NMR (270 MHz, DMSO-d6) δ 12.26 (1 H, br s), 11.63 (1 H, br s), 7.71 (1 H, d, J = 8.72 Hz), 7.46 (1 H, d, J = 1.97 Hz), 7.09 (1 H, dd, J = 8.72 Hz, J = 1.97 Hz), 4.04 (2 H, s), 4.07–3.95 (1 H, m), 2.30–1.78 (6 H, m). IR (KBr): 3303, 2954, 1705, 1632, 1564, 1529, 1437, 1412, 1335, 1242, 1213, 1188, 1157, 1056, 1024 cm−1. MS (EI direct) m/z: M+ 291. Anal. (C15H14ClNO3) C, H, N.
(6-Chloro-2-cyclopropanecarbonyl-1H-indol-3-yl)acetic acid (48b)
Step 1. Methyl (6-chloro-2-cyclopropanecarbonyl-1H-indol-3-yl)acetate (47b). A mixture of cyclopropyl methyl ketone (Tokyo Chemical Industry Co., Ltd., 1.20 mL, 12.1 mmol) and tetrabutylammonium tribromide (6.365 g, 13.2 mmol) in dry CH2Cl2 (18.0 mL)–dry MeOH (9.0 mL) was stirred at room temperature under N2 for 30 h, then concentrated in vacuo. The residue was partitioned between H2O (30 mL) and Et2O (30 mL), then the ethereal layer was separated. The aqueous layer was extracted with Et2O (30 mL). The ethereal layers were combined, washed with H2O (30 mL × 2), dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 2.879 g of 2-bromo-1-cyclopropylethanone 46b as an oil (crude). TLC: Rf = 0.57, AcOEt/hexane = 1:2.
A mixture of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f (492.5 mg, 1.40 mmol), 2-bromo-1-cyclopropylethanone 46b (crude, 391.2 mg) and anhydrous K2CO3 (580.5 mg, 4.20 mmol) in dry acetone (12.0 mL) was stirred at room temperature under N2 for 6 h, then DBU (628 µL, 4.20 mmol) was added. The reaction mixture was stirred under N2 at the same temperature under N2 for 24 h. The reaction mixture was partitioned between AcOEt (30 mL) and 2 N HCl (30 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (30 mL). The organic layers were combined, washed with 2 N HCl (30 mL × 2), saturated aqueous NaHCO3 (30 mL) and brine (30 mL), then dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 384.4 mg of the title product 47b in 94% yield (from compound 30f) as an oil. 1H NMR (270 MHz, CDCl3) δ 9.55 (1 H, br s), 7.58 (1 H, d, J = 8.56 Hz), 7.26 (1 H, d, J = 1.65 Hz), 7.10 (1 H, dd, J = 8.56 Hz, J = 1.65 Hz), 4.17 (2 H, s), 3.73 (3 H, s), 2.58–2.49 (1 H, m), 1.30–l.25 (2 H, m), 1.09–1.02 (2 H, m). {Recrystallisation study: Compound 47b was recrystallised from hexane to afford a crystalline solid. 1H NMR spectrum of the crystalline solid was the same as that of the above compound 47b. MS (ESI positive) m/z: [M+H]+ 291.9. MS (ESI negative) m/z: [M–H]− 289.9. MS (EI direct) m/z: M+ 291.10. Mp: 158.0 °C. Anal. (C15H14ClNO3) C, H, N}.
Step 2. (6-Chloro-2-cyclopropanecarbonyl-1H-indol-3-yl)acetic acid (48b). A mixture of methyl (6-chloro-2-cyclopropanecarbonyl-1H-indol-3-yl)acetate 47b (384.4 mg, 1.318 mmol) and 2 N NaOH (3.20 mL, 6.40 mmol) in EtOH (22.4 mL) was warmed up to reflux conditions using an oil bath, stirred under N2 for 20 h, cooled to room temperature and concentrated in vacuo. The residue was partitioned between H2O (40 mL) and Et2O (40 mL). The organic layer was separated to remove impurity. The aqueous layer was further washed with Et2O (40 mL × 3), acidified by adding 2 N HCl (30 mL) and then extracted with AcOEt (40 mL × 3). The combined extracts were dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was recrystallised from AcOEt–hexane to afford 147.3 mg of the title product 48b in 40% yield as a pale brownish-yellow solid. Mp: 249–252 °C. 1H NMR (270 MHz, DMSO-d6) δ 12.22 (1 H, br s), 11.98 (1 H, br s), 7.75 (1 H, d, J = 8.72 Hz), 7.48 (1 H, d, J = 1.81 Hz), 7.11 (1 H, dd, J = 8.72 Hz, J = l.81 Hz), 4.08 (2 H, s), 2.73 (1 H, quintet, J = 6.24 Hz), 1.07 (4 H, d, J = 6.24 Hz). IR (KBr): 3304, 3013, 1709, 1624, 1566, 1443, 1414, 1387, 1340, 1286, 1248, 1217, 1200, 1157, 1057, 1045, 1022 cm−1. MS (EI direct) m/z: M+ 277. Anal. (C14H12ClNO3) C, H, N.
Biology
Materials
Human umbilical vein endothelial cells (HUVECs) were purchased from Morinaga Institute of Biological Science (Yokohama, Kanagawa, Japan). Recombinant human interleukin-1β (IL-1β) was purchased from R&D Systems (Minneapolis, MN). A23187 (calcium ionophore) and indomethacin were purchased from Sigma-Aldrich. Thromboxane B2 (TXB2), 6-keto-prostaglandin F1α (6-keto-PGF1α) and prostaglandin E2 (PGE2) were purchased from Cayman Chemical (Ann Arbor, MI). λ-Carrageenan (Picnin-A) was purchased from Zushikagaku (Zushi, Kanagawa, Japan). {2-[(2-, 3- or 4-Substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acids 8a–f, 14, 22a–c, 29, 44a–j and 48a,b were used for pharmacological evaluations as free acids, respectively. All other chemicals were obtained from Wako Pure Chemical Industries, Ltd. unless stated otherwise. All common chemicals were analytical or the highest available grade.
In vitro studies
Human cell-based COX-1 assay
Platelets were prepared from human peripheral blood obtained from healthy adult volunteers under informed consent. Fresh blood was collected into vacutainers (Becton Dickinson, Franklin Lakes, NJ) containing 1/10 volume of anticoagulant solution (3.8% sodium citrate), and centrifuged at 200 × g for 10 min. The supernatant (platelet-rich plasma) was mixed with 50% volume of 0.14 M NaCl containing 12 mM Tris–HCl, 1.2 mM EDTA, pH 7.4. This mixture was centrifuged at 750 × g for 10 min, and the pellet was suspended in platelet buffer (Ca2+-free Hanks buffer containing 20 mM HEPES, pH 7.4 and 0.2% BSA). After centrifugal washing with the platelet buffer, the resulting pellet, referred to as HWPs, was resuspended in the platelet buffer at a cell concentration of 2.85 × 108 cells/mL, then stored at room temperature until use. Immediately prior to assay, 10 µL of 12.6 mM CaCl2 was added to 70 µL HWPs suspension (2.0 × 107 cells/mL in a 96-well U bottom plate). Platelets were pre-incubated in the absence or presence of 10 µL of test compound dissolved in DMSO at final concentrations varying 0.01–10 µM (final concentration, less than 0.1%) for 20 min before stimulation with 10 µL of A23187 (final concentration, 10 µM). After further 15 min incubation at 37 °C with A23187, the reaction was stopped by the addition of EDTA (final concentration, 7.7 mM), and the reaction medium was quantitated for TXB2 by a radioimmunoassay (RIA) kit (Amersham, Little Chalfont, Buckinghamshire, UK) according to the manufacturer’s procedureCitation25,Citation104,Citation113.
Human cell-based COX-2 assay
The human cell-based COX-2 assay was carried out as previously describedCitation25,Citation104,Citation114. Confluent HUVECs (2 × 104 cells/well) in a 96-well plate were washed with 100 µL of RPMI-1640 containing 2% foetal calf serum (FCS), and incubated with 300 U/mL of recombinant human IL-1β for 24 h at 37 °C for induction of COX-2. After washing with Hanks buffer containing 20 mM HEPES, pH 7.4 and 0.2% BSA, HUVECs were pre-incubated in Hanks buffer containing 20 mM HEPES, pH 7.4 and 0.2% BSA, with or without 10 µL of test compound dissolved in DMSO at final concentrations varying 1 nM to 1 µM (final concentration, less than 0.1%) for 20 min before stimulation with 30 µM of A23187. After further 15 min incubation at 37 °C with A23187, the reaction medium was quantitated for 6-keto-PGF1α, that is, spontaneously degraded stable form of prostacyclin (PGI2), by a RIA kit (Amersham).
In vivo studies
General
Animal experiment was conducted according to the guideline of animal care and use, and its procedure was approved by the Animal Ethics Committee in Pfizer Global Research & Development, Nagoya Laboratories. The animal work was also approved by the Pfizer Institutional Animal Care and Use Committee (IACUC).
Carrageenan-induced foot-oedema formation in rats
Specific pathogen free and virus antibody free (SPF/VAF) male Sprague–Dawley (SD) rats [Crj:CD(SD), 5 weeks old, 110–130 g, Charles River Laboratories, Japan Inc., Hino, Tokyo, Japan] that were fasted overnight were injected intraplantarly with 0.1 mL of a 1% (w/v) λ-carrageenan (Picnin-A) suspension in saline into the right hind paw as previously reportedCitation25,Citation104,Citation115–117. Either the vehicle [5% (v/v) Tween 80 in distilled water] or a test compound was dosed orally in a volume of 1 mL/100 g body weight at 1 h before carrageenan injection. Foot volume was measured by a water displacement plethysmometer (Unicom Co., Yachiyo, Chiba, Japan) before and 3 h after carrageenan injection. The increases in foot volume for 3 h were calculated. Foot oedema was compared with vehicle-control group, and the percent inhibition was calculated taking the values in the control group as 0%.
PGE2 production in carrageenan-induced oedema site of rats
Determination of PGE2 synthesised in the inflammatory site was carried out essentially according to a previously described methodCitation25,Citation118. Foot oedema in SPF/VAF male SD rats [Crj:CD(SD), 5 weeks old, 110–130 g] was induced by intraplantar injection of a 1% (w/v) λ-carrageenan suspension. The measurement of inhibitory activity against carrageenan-induced foot oedema for the test compound, that was administrated orally at 1 h before carrageenan injection, was performed at 3 h after carrageenan injection [refer “Carrageenan-induced foot-oedema formation in rats” section]. And then immediately, the animals were euthanised and sacrificed by cervical dislocation. The foot was amputated, frozen in liquid nitrogen and stored at −80 °C until analysis. The frozen foot was crushed, mixed with 7 mL of ethanol containing 10 µg/mL of indomethacin to inhibit PGE2 production during further handling, pulverised in a Waring blender and clarified by centrifugation at 2000 × g for 10 min at 4 °C. PGE2 was extracted by a Sep-Pak C18 cartridge (Waters Corporation), and dried in vacuum. Samples were diluted to a final volume of 0.5 mL with assay buffer [50 mM Tris/HCl, pH 7.4 containing 0.9% NaCl, 0.01% Triton X-100 and 0.1% (w/v) bactogelatin] and the levels of PGE2 were determined by RIA according to the manufacture’s direction (RIA kit, Amersham). Either the vehicle [5% (v/v) Tween 80 in distilled water] or the test compound was dosed orally in a volume of 1 mL/100 g body weight 1 h before carrageenan injection as already described in “Carrageenan-induced foot-oedema formation in rats” section. The mean values of compound-treated group were compared to that of vehicle-treated control group to determine the percent inhibition of PGE2 production as the following equation:
Physicochemistry
The lipophilicity values of COX-2 inhibitors were estimated as values of ACD log D7.4, the octanol–water distribution coefficient for ionizable compounds at pH 7.4, calculated by ACD software, ACD/Laboratories 9.0 (Advanced Chemistry Development, Inc., Ontario, Canada).
Results and discussion
Chemistry
A synthetic method for {6-chloro-2-[(2-, 3- or 4-substituted or 2,4-disubstituted)-benzoyl]-1H-indol-3-yl}acetic acid derivatives using indole derivatives and aroyl chlorides as starting materials is depicted in (Method A). Thus, commercially available 6-chloroindole 1 was converted into 6-chloro-1-(phenylsulfonyl)-1H-indole 2 with benzenesulfonyl chloride, then arylcarbonylated with various (2-, 3- or 4-substituted or 2,4-disubstituted)-benzoyl chlorides 3a–f via lithiation with tert-BuLi at 2-position of the indole core, that was attached with the 2-carbanion-stabilising (phenylsulfonyl)amino-group, to afford aryl[6-chloro-1-(phenylsulfonyl)-1H-indol-2-yl]methanones 4a–f, respectivelyCitation105, followed by hydrolysis of the phenylsulfonyl portion to afford aryl(6-chloro-1H-indol-2-yl)methanones 5a–f, respectively. Next, 3-position of indole core, which was adjacent to the electron-withdrawing aroyl group on the core, was acetoxymalonylated by oxidative radical condition with diethyl malonate and Mn(III) in AcOHCitation106,Citation107 to afford diethyl 2-acetoxy-2-[2-(arylcarbonyl)-6-chloro-1H-indol-3-yl]malonates 6a–f, respectively, followed by reductive deacetoxylation with Et3SiH in TFACitation107 to give diethyl indolylmalonates 7a–f, respectively. Finally, hydrolysis and decarboxylation of the diethyl-malonate moietyCitation106,Citation107 afforded the requisite {6-chloro-2-[(2-, 3- or 4-substituted or 2,4-disubstituted)-benzoyl]-1H-indol-3-yl}acetic acid derivatives 8a–f as sole products in good yields, respectively.
Scheme 1. Synthesis of {6-chloro-2-[(2-, 3- or 4-substituted or 2,4-disubstituted)-benzoyl]-1H-indol-3-yl}acetic acids 8a–f. Reagents and conditions: (a) n-Bu4NHSO4, 50% aqueous KOH, benzene, room temp to 0 °C; then benzenesulfonyl chloride, 0 °C to room temp; (b) tert-BuLi/pentane, THF, −78 °C to 0 °C, or −78 °C; (c) K2CO3/THF–MeOH–H2O or 2 N NaOH/EtOH–THF, up to reflux; (d) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (e) TFA, Et3SiH, CH2Cl2, room temp to reflux; (f) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.
![Scheme 1. Synthesis of {6-chloro-2-[(2-, 3- or 4-substituted or 2,4-disubstituted)-benzoyl]-1H-indol-3-yl}acetic acids 8a–f. Reagents and conditions: (a) n-Bu4NHSO4, 50% aqueous KOH, benzene, room temp to 0 °C; then benzenesulfonyl chloride, 0 °C to room temp; (b) tert-BuLi/pentane, THF, −78 °C to 0 °C, or −78 °C; (c) K2CO3/THF–MeOH–H2O or 2 N NaOH/EtOH–THF, up to reflux; (d) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (e) TFA, Et3SiH, CH2Cl2, room temp to reflux; (f) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.](/cms/asset/444134cf-f06e-409b-8493-a310d704fbbd/ienz_a_864650_f0001_b.jpg)
As well, [6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]acetic acid 14 was prepared as a sole product in good yield by way of cyclohexanecarbonylation at 2-position of compound 2 with cyclohexanecarbonyl chloride 9 under similar conditions as used for the synthesis of compounds 4a–f, and subsequent process in the same way as described in method A ().
Scheme 2. Synthesis of [6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]acetic acid 14. Reagents and conditions: (a) tert-BuLi/pentane, THF, −78 °C; (b) 2 N NaOH, EtOH–THF, up to reflux; (c) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (d) TFA, Et3SiH, CH2Cl2, room temp to reflux; (e) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.
![Scheme 2. Synthesis of [6-chloro-2-(cyclohexanecarbonyl)-1H-indol-3-yl]acetic acid 14. Reagents and conditions: (a) tert-BuLi/pentane, THF, −78 °C; (b) 2 N NaOH, EtOH–THF, up to reflux; (c) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (d) TFA, Et3SiH, CH2Cl2, room temp to reflux; (e) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.](/cms/asset/11ddb25b-e065-4357-b5cb-665cf458b43d/ienz_a_864650_f0002_b.jpg)
In case 1-bromo-(3 or 4-substituted)-benzenes were available as starting materials, {6-chloro-2-[(3- or 4-substituted)-benzoyl]-1H-indol-3-yl}acetic acid derivatives were prepared as depicted in (Method B). Thus, after introduction of carboxylic acid group onto 2-position of the above compound 2 with lithium diisopropylamide (LDA) and carbon dioxide (CO2) gas, the resulting 6-chloro-1-(phenylsulfonyl)-1H-indole-2-carboxylic acid 15 was converted into 6-chloro-1H-indole-2-carboxylic acid 16 by hydrolysis of the phenylsulfonyl moiety, and then the resulting acid was led to 6-chloro-N-methoxy-N-methyl-1H-indole-2-carboxamide 17 via the corresponding acid chloride. The N-methoxy-N-methylamide moiety was substituted by (3- or 4-substituted)-benzenes with (substituted phenyl)lithium reagents that were prepared from 1-bromo-(3- or 4-substituted)-benzenes 18a–c and n-BuLi in situ by bromo-group–metal exchangeCitation108–110. The resulting (6-chloro-1H-indol-2-yl)[(3- or 4-substituted)-phenyl]methanones 19a–c were converted into the final products in a similar way for compounds 8a–f from compounds 5a–f in Method A. Thus, radical acetoxymalonylation for the aryl(1 H-indol-2-yl)methanones was performed to afford diethyl 2-acetoxy-2-[2-(arylcarbonyl)-6-chloro-1H-indol-3-yl]malonates 20a–c, followed by reductive deacetoxylation to form respective diethyl indolylmalonates 21a–c, respectively. Subsequent hydrolysis and decarboxylation of the diethyl malonates afforded requisite {6-chloro-2-[(3- or 4-substituted)-benzoyl]-1H-indol-3-yl}acetic acids 22a–c as sole products in good yields, respectively. Utilising the same manner, [2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]acetic acid 29 was also prepared from commercially available 5-fluoro-1H-indole-2-carboxylic acid 23 as shown in .
Scheme 3. Synthesis of {6-chloro-2-[(3- or 4-substituted)-benzoyl]-1H-indol-3-yl}acetic acids 22a–c. Reagents and conditions: (a) LDA, THF, −78 °C; then CO2 (gas), −78 °C; (b) 2 N NaOH, EtOH, reflux; (c) i) SOCl2, DMF, heating several times; then concentrated in vacuo. ii) N,O-dimethylhydroxylamine hydrochloride, CH2Cl2, room temp; then pyridine, 0 °C; (d) n-BuLi/hexane, THF, −78 °C; (e) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (f) TFA, Et3SiH, CH2Cl2, room temp to reflux; (g) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.
![Scheme 3. Synthesis of {6-chloro-2-[(3- or 4-substituted)-benzoyl]-1H-indol-3-yl}acetic acids 22a–c. Reagents and conditions: (a) LDA, THF, −78 °C; then CO2 (gas), −78 °C; (b) 2 N NaOH, EtOH, reflux; (c) i) SOCl2, DMF, heating several times; then concentrated in vacuo. ii) N,O-dimethylhydroxylamine hydrochloride, CH2Cl2, room temp; then pyridine, 0 °C; (d) n-BuLi/hexane, THF, −78 °C; (e) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (f) TFA, Et3SiH, CH2Cl2, room temp to reflux; (g) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.](/cms/asset/19af7d34-22fd-44ba-b717-9831e0ecac8e/ienz_a_864650_f0003_b.jpg)
Scheme 4. Synthesis of [2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]acetic acid 29. Reagents and conditions: (a): (i) SOCl2, DMF, heating several times; then concentrated in vacuo. (ii) N,O-dimethylhydroxylamine hydrochloride, CH2Cl2, room temp; then pyridine, 0 °C; (b) n-BuLi/hexane, THF, −78 °C; (c) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (d) TFA, Et3SiH, CH2Cl2, room temp to reflux; (e) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.
![Scheme 4. Synthesis of [2-(4-chlorobenzoyl)-5-fluoro-1H-indol-3-yl]acetic acid 29. Reagents and conditions: (a): (i) SOCl2, DMF, heating several times; then concentrated in vacuo. (ii) N,O-dimethylhydroxylamine hydrochloride, CH2Cl2, room temp; then pyridine, 0 °C; (b) n-BuLi/hexane, THF, −78 °C; (c) diethyl malonate, AcONa, Mn(OAc)3 · 2H2O, AcOH, room temp to 80 °C; (d) TFA, Et3SiH, CH2Cl2, room temp to reflux; (e) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.](/cms/asset/f1d75744-b68e-4cc3-b11a-012d72121816/ienz_a_864650_f0004_b.jpg)
Alternatively, {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid derivatives were prepared by convergent synthesis consisted of three independent parts, that is, (i) the synthesis of methyl 3-{2-[(arylsulfonyl)amino]phenyl}acrylates, (ii) the synthesis of 2-bromo-1-(aryl, bicyclic heterocycloalkanophenyl or cycloalkyl)ethanones and (iii) the synthesis of the desired {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid derivatives via indole-skeleton construction (Method C), similar to the already-described synthesis of [2-{[(4-substituted or 4,5-disubstituted)-pyridin-2-yl]carbonyl}-(5- or 6-substituted or 5,6-disubstituted)-1H-indol-3-yl]acetic acid derivatives by Hayashi et al.Citation25,Citation104
First, various methyl 3-{(4- or 5-substituted)-2-[(arylsulfonyl)amino]phenyl}acrylates 30a–g were synthesised via Heck–Mizoroki reactionCitation119 or via known procedureCitation120, respectively, as described already by Hayashi et al.Citation25,Citation104
Second, except commercially available 4-chlorophenacyl bromide 31, various 2-bromo-1-[(4-substituted or 3,4-disubstituted)phenyl or bicyclic heterocycloalkanophenyl]ethanones were synthesised as depicted in . In the case of 4-chloro-3-fluorophenyl derivative, N,N-dimethylacetamide 32 was coupled with (4-chloro-3-fluorophenyl)lithium, that was generated from 1-bromo-4-chloro-3-fluorobenzene 33 in situ with n-BuLi, to form 1-(4-chloro-3-fluorophenyl)ethanone 34. For 4-[(methylsulfonyl)amino]phenyl derivative, 4′-aminoacetophenone 36 was converted into N-(4-acetylphenyl)methanesulfonamide 37 with methanesulfonyl chloride. Then the acetyl moieties of the above 1-arylethanones 34 and 37 were brominated with tetrabutylammonium tribromideCitation111,Citation112 to afford 2-bromo-1-(4-chloro-3-fluorophenyl)ethanone 35 and N-[4-(2-bromoacetyl)phenyl]methanesulfonamide 38, respectively. For bicyclic-heterocycloalkanophenyl derivatives, 2-bromo-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone 40 and 2-bromo-1-(2,3-dihydrobenzofuran-5-yl)ethanone 42 were also synthesised from the corresponding (bicyclic heterocycloalkanophenyl)ethanones 39 and 41 by bromination with bromine in HBr/AcOHCitation25,Citation104, respectively.
Scheme 5. Synthesis of 2-bromo-1-[(4-substituted or 3,4-disubstituted)-phenyl or bicyclic-heterocycloalkanophenyl]ethanones 35, 38, 40 and 42. Reagents and conditions: (a) i) n-BuLi/hexane, Et2O, −78 °C to −20 °C; then DMA/Et2O, −20 °C to room temp; (b) tetrabutylammonium tribromide, CH2Cl2–MeOH; (c) methanesulfonyl chloride, pyridine, CH2Cl2, 0 °C to room temp; (d) Br2, HBr/AcOH, CHCl3, 0 °C to room temp.
![Scheme 5. Synthesis of 2-bromo-1-[(4-substituted or 3,4-disubstituted)-phenyl or bicyclic-heterocycloalkanophenyl]ethanones 35, 38, 40 and 42. Reagents and conditions: (a) i) n-BuLi/hexane, Et2O, −78 °C to −20 °C; then DMA/Et2O, −20 °C to room temp; (b) tetrabutylammonium tribromide, CH2Cl2–MeOH; (c) methanesulfonyl chloride, pyridine, CH2Cl2, 0 °C to room temp; (d) Br2, HBr/AcOH, CHCl3, 0 °C to room temp.](/cms/asset/4a9402fa-451e-4789-ae91-58509063691a/ienz_a_864650_f0005_b.jpg)
Third, the above methyl 3-{(4- or 5-substituted)-2-[(arylsulfonyl)amino]phenyl}acrylates 30a–g and the above bromides 31, 35, 38, 40 and 42 were converted into indoline skeletons by Michael addition reaction with K2CO3, and subsequent one-pot dephenylsulfonation assisted-aromatisation with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded methyl {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetates 43a–j, respectively. Then hydroxylation of the methyl acetates gave requisite {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid derivatives 44a–j, respectively ().
Scheme 6. Synthesis of {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acids 44a–j. Reagents and conditions: (a) K2CO3, acetone; then DBU; (b) 2 N NaOH, MeOH–THF or MeOH or EtOH, reflux; then acidified by 2 N HCl, room temp. Note: Refer text in this article and Refs. 25 and 104 by Hayashi et al. for the preparation of methyl 3-{(4- or 5-substituted)-2-[(arylsulfonyl)amino]phenyl}acrylates 30a–g.
![Scheme 6. Synthesis of {2-[(4-substituted or 3,4-disubstituted)-benzoyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acids 44a–j. Reagents and conditions: (a) K2CO3, acetone; then DBU; (b) 2 N NaOH, MeOH–THF or MeOH or EtOH, reflux; then acidified by 2 N HCl, room temp. Note: Refer text in this article and Refs. 25 and 104 by Hayashi et al. for the preparation of methyl 3-{(4- or 5-substituted)-2-[(arylsulfonyl)amino]phenyl}acrylates 30a–g.](/cms/asset/a9b8857d-4a6c-4e80-84d2-80b8c22c57ab/ienz_a_864650_f0006_b.jpg)
Furthermore, (6-chloro-2-cycloalkanecarbonyl-1H-indol-3-yl)acetic acid derivatives were prepared using the above convergent synthesis as depicted in . Thus, cyclobutyl methyl ketone 45a and cyclopropyl methyl ketone 45b were brominated by tetrabutylammonium tribromide to afford the corresponding 2-bromo-1-cyclobutylethanone 46a and 2-bromo-1-cyclopropylethanone 46b, respectively. Subsequent Michael addition reaction of methyl 3-{4-chloro-2-[(phenylsulfonyl)amino]phenyl}acrylate 30f and the above bromides 46a,b with K2CO3, followed by aromatisation in situ with DBU gave methyl (6-chloro-2-cycloalkanecarbonyl-1H-indol-3-yl)acetates 47a,b, respectively, which were hydrolysed to afford requisite (6-chloro-2-cyclobutanecarbonyl-1H-indol-3-yl)acetic acid 48a and (6-chloro-2-cyclopropanecarbonyl-1H-indol-3-yl)acetic acid 48b, respectively.
Scheme 7. Synthesis of [2-cycloalkanecarbonyl-(5- or 6-substituted)-1H-indol-3-yl]acetic acids 48a,b. Reagents and conditions: (a) tetrabutylammonium tribromide, CH2Cl2–MeOH; (b) K2CO3, acetone; then DBU; (c) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.
![Scheme 7. Synthesis of [2-cycloalkanecarbonyl-(5- or 6-substituted)-1H-indol-3-yl]acetic acids 48a,b. Reagents and conditions: (a) tetrabutylammonium tribromide, CH2Cl2–MeOH; (b) K2CO3, acetone; then DBU; (c) 2 N NaOH, EtOH, reflux; then acidified by 2 N HCl, room temp.](/cms/asset/e1e076fa-c44e-4c3b-ac32-177c9fb274e1/ienz_a_864650_f0007_b.jpg)
The detailed procedures and obtained data for the above respective compounds are described in the “Experimental” section and in the Appendix of the Supplementary Material.
All of these final test compounds {2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid derivatives 8a–f, 14, 22a–c, 29, 44a–j and 48a,b were used for SAR studies with pharmacological evaluations as free acids, respectively.
Structure–activity relationships of COX-2 inhibitors
In vitro activities and selectivities against COX isoforms
Actually, design, synthesis and SAR of various novel {2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues were investigated to seek and identify various chemotypes of orally potent and selective COX-2 inhibitors as potential agents for the treatment of inflammatory diseases.
At first, the in vitro inhibitory activities and selectivities for the compounds against enzymatic activities of COX isozymes, inducible COX-2 and constitutional COX-1, in human cells were evaluated with our reported proceduresCitation25,Citation104. In brief, the activities of the COX isozymes were determined (i) by 6-keto-prostaglandin F1α (6-keto-PGF1α, spontaneously degraded stable-form of PGI2) production in human umbilical vein endothelial cells (HUVECs) that were expressing COX-2 induced by interleukin-1β (IL-1β) for COX-2, and (ii) by thromboxane B2 (TXB2, spontaneously degraded stable-form of TXA2) production in human washed platelets (HWPs) for COX-1, respectivelyCitation113,Citation114. For the selectivity indices of human COX-2 inhibitors in this study in vitro, COX-2-inhibition selectivities were estimated as IC50 against COX-2 over IC50 against COX-1 in the human cellular studies for the compounds. As described alreadyCitation25, the above 6-keto-PGF1α production in HUVEC in our study condition was established as a COX-2 induction-dependent response.
In this in vitro design and SAR study using human cellular assays, the acid-type novel analogues gave significant findings regarding potency and selectivity of their COX-2 inhibition dependent on the 5- or 6-substituent of the indole core and on the 2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl] portion, indeed. The results of the in vitro SAR study for the analogues are summarised in and .
Table 1. In vitro structure−activity relationships of inhibitory effects against COX-2/COX-1 activities in human cellular assays for {2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues 8a–f, 22a–c, 29 and 44a–g.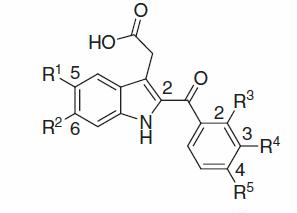
Table 2. In vitro structure−activity relationships of inhibitory effects against COX-2/COX-1 activities in human cellular assays for {2-[cycloalkanecarbonyl or (bicyclic heterocycloalkanophenyl)carbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues 14, 44h–j and 48a,b.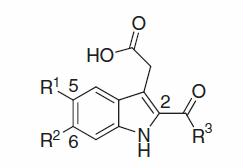
First, substituted-phenyl group effects at 2-benzoyl portion of {6-chloro-2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl]-1H-indol-3-yl}acetic acid analogues were studied in vitro. Thus, meta- or para-toluoyl analogues 8b and 8c, meta- or para-trifluoromethylphenyl analogues 22c and 8d, and meta- or para-fluorophenyl analogues 22a and 22b exhibited potent COX-2 inhibitions, with showing respective patterns of regioselectivities by their substituents for the inhibitory activities. Specifically, para-methyl and -trifluoromethyl groups (8c and 8d) on the phenyl core of the analogues gave more potent activities in vitro (i.e. the IC50 values were 0.192 µM and 0.209 µM for compounds 8c and 8d, respectively) than the corresponding meta-methyl and -trifluoromethyl groups (8b and 22c) (i.e. the IC50 values were 0.248 µM and 0.340 µM for compounds 8b and 22c, respectively); while meta- and para-fluoro groups gave almost the same activities each other (i.e. the IC50 values were 0.229 µM and 0.228 µM for compounds 22a and 22b, respectively). On the other hand, compared with the meta- and para-toluoyl analogues 8b and 8c, ortho-toluoyl analogue 8a reduced activity (IC50 = 3.31 µM). Furthermore, meta-fluoro-para-chlorophenyl analogue 44f retained potent activity (IC50 = 0.904 µM), whereas ortho-chlorophenyl analogue 8e and ortho,para-dichlorophenyl analogue 8f much reduced or lacked activity. Overall, compared with the ortho-substituent, the meta- and para-substituents were favourable for COX-2 inhibition. Besides, para-methanesulfonylamidophenyl analogue 44g was inactive, which would show limitation of the size or volume for the substituent effect at this position owing to the steric hindrance against binding-site of the protein.
Also, the selectivity study of the [6-chloro-2-(2-, 3- or 4-substituted)-benzoyl-1H-indol-3-yl]acetic acid analogues for their COX-2 inhibition over COX-1 inhibition in the human cellular assays demonstrated the significance of electron-density distribution and bulkiness for the substituted-phenyl portion within some ranges. Notably, COX-2 selectivity of para-trifluoromethylphenyl analogue 8d was greater than 47.6-fold, which was considerably higher than that of meta- and para-toluoyl analogues 8b and 8c, meta-fluorophenyl analogue 22a, and meta-fluoro-para-chlorophenyl analogue 44f, that is, 7.75-, 3.19-, 1.87- and 4.49-fold for compounds 8b, 8c, 22a and 44f, respectively. In detail, as substituent effects on the phenyl core, (i) more electron-negative para-trifluoromethyl group (8d) was much prefer to less electron-negative (rather electron-donating) para-methyl group (8c), (ii) larger meta-methyl group (8b) was better than smaller meta-fluoro group (22a) and (iii) a pair of meta-fluoro and para-chloro groups (44f), whose para-group is larger and more electron-negative, was more favourable compared with a pair of meta-fluoro and para-hydrogen groups (22a), whose para-group is smaller and less electron-negative, respectively.
Second, substituent effects at 5- or 6-position on the indole core of [2-(4-chlorobenzoyl)-(5- or 6-substituted)-1H-indol-3-yl]acetic acid analogues were studied in vitro. In this study, 5- or 6-fluoro group (29 or 44a) and 5-trifluoromethoxy group (44e), thus (relatively) electron-negative groups, were effective to gain potent or highly potent COX-2 inhibitory activities in vitro, that is, the IC50 values were 0.264, 0.0156 and 0.493 µM for compounds 29, 44a and 44e, respectively. But, replacement by 5- or 6-methoxy group (44d or 44c) or 5-tert-butyl group (44b), thus rather electron donating and/or bulky group, led to loss or reduction of the activity. Importantly, the above potent or highly potent 5- or 6-substituted indolyl analogues 29, 44a and 44e exhibited much high or high selectivities of COX-2 inhibition over COX-1 inhibition, that is, 509-fold for 6-fluoro analogue 44a, 24.2-fold for 5-fluoro analogue 29 and 14.0-fold for 5-trifluoromethoxy analogue 44e, respectively. As described above, the structural and physicochemical properties of the substituents are significant for the present potent and selective COX-2 inhibitors; and the strong effects of 5- and/or 6-substituents located on the indole core in vitro were also demonstrated in the design and SAR studies of various novel [2-{[(4-substituted or 4,5-disubstituted)-pyridin-2-yl]carbonyl}-(5- or 6-substituted or 5,6-disubstituted)-1H-indol-3-yl]acetic acid analogues by Hayashi et al.Citation25,Citation104, which achieved highly potent and selective COX-2 inhibitory activities for the analogues with respective manners as reported.
Third, SAR of {2-(bicyclic heterocycloalkanophenyl)carbonyl-(6-chloro or 5-trifluoromethyl)-1H-indol-3-yl}acetic acid analogues was studied in vitro. Indeed, (6-chloro-1H-indol-3-yl)acetic acid analogue 44h and (5-trifluoromethyl-1H-indol-3-yl)acetic acid analogue 44i as 2-(2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl) derivatives and [6-chloro-2-(2,3-dihydrobenzofuran-5-carbonyl)-1H-indol-3-yl]acetic acid analogue 44j exhibited potent or highly potent COX-2 inhibitory activities in vitro, that is, the IC50 values were 0.148, 0.389 and 0.0566 µM for compounds 44h, 44i and 44j, respectively. Furthermore, these 2-(bicyclic heterocycloalkanophenyl)carbonyl analogues exhibited high selectivities of COX-2 inhibition over COX-1 inhibition, that is, greater than 67.6-fold, greater than 25.7-fold, and 52.4-fold for compounds 44h, 44i and 44j, respectively. In comparison of compounds 44h and 44i, the 6- or 5-substituent apparently affected the respective in vitro activities and selectivities, similar to the cases of 6- or 5-substituents on indole core of the above 2-(monocyclic aroyl) analogues as described. Besides, the bicyclic-group effects for their potent and selective COX-2 inhibitions were similar to that of our previously reported study by Hayashi et al. for 2-[(bicyclic cycloalkanopyridinyl)carbonyl] analogue, that is, [6-chloro-2-(5,6,7,8-tetrahydroisoquinolin-3-ylcarbonyl)-1H-indol-3-yl]acetic acid whose IC50 value of COX-2 inhibition was 0.0192 µM and selectivity of the COX-2 inhibition over COX-1 inhibition was greater than 521-fold in the human cellular assaysCitation104. These studies demonstrates the profitability of the present and previous analogues that have bicyclic-group portion with some degree of bulkiness and volume at 2-carbonyl group, for potent and selective interactions between COX-2 and the molecules in the enzyme protein.
Fourth, SAR of (6-chloro-2-cycloalkanecarbonyl-1H-indol-3-yl)acetic acid analogues was studied in vitro. Notably, 2-cyclohexanecarbonyl analogue 14 exhibited highly potent COX-2 inhibitory activity with COX-2 inhibition selectivity over COX-1 inhibition in vitro, that is, the IC50 value was 0.0730 µM and the selectivity was 15.9-fold. Hence, the incorporation of 2-cyclohexanecarbonyl group instead of the above 2-aroyl groups was well effective in potent and selective COX-2 inhibition as well. Besides, compared with compound 14, other 2-cycloalkanecarbonyl analogues with smaller rings showed weaker activities dependent on the degree of reduction of the respective ring-size. Indeed, the rank of the orders of COX-2 inhibitory activities for the cycloalkyl analogues in vitro was cyclohexyl analogue 14 (IC50 = 0.0730 µM) > cyclobutyl analogue 48a (IC50 = 2.84 µM) > cyclopropyl analogue 48b (IC50 = greater than 10.0 µM). Overall, as especially indicated by the lacking- or reducing-aromaticity effect on six-membered ring, the aromaticity of this portion attached with 2-carbonyl group of the indole skeleton was unnecessary or not so important for potent and selective COX-2 inhibition on the corresponding interaction-site in COX-2 protein as far as the size of this portion was appropriate or sufficient for the potent and selective interaction between the compound and COX-2, which shows a potential of further analogue-design around them.
COX-2 has approximately 60% similarity to COX-1 in coding-sequence comparison between the isozymes, also as a structural difference between COX-2 and COX-1 originated from the amino acid sequence regarding shape and size of the cyclooxygenase active-sites, COX-2 has valine 523 and COX-1 has isoleucine 523 in the corresponding active-sites, hence the active site of COX-2 is larger than that of COX-14,Citation18,Citation99–103,Citation121. Actually, the present in vitro SAR study of the diverse novel acid-types of analogues clarified the variety of effects of 5- or 6-substituent on the indole core and of 2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl] portion, owing to the respective chemotypes and structural features that include shapes, sizes/volumes, positions and electronic states, for their potent and selective COX-2 inhibition processes in vitro as critical and key factors of highly organised multisite interaction/coordination with pharmacophores in the enzyme protein, which determine dynamic molecular interaction-mechanisms, -strengths and -modes (position and orientation) of the ligands. Interestingly, bulkier size/substituent effects for COX-2/COX-1 selectivities, such as profitablities of bicyclic group, bulkier mono- or disubstituent(s) bearing-(hetero)aryl group and relatively large-ring group at 2-carbonyl portion on the indole core of the analogues, have been demonstrated as results of this study and the previous design and SAR studies by Hayashi et al.Citation25,Citation104 for the respective series of COX-2 inhibitors within the appropriate ranges of positions, shapes and sizes for whole/local molecular structures as described, which would be associated with the differences of shape, size and interaction-/coordination-mode of the ligand-interaction/-coordination sites between COX-2 and COX-1 proteins; whereas appropriate electronic effects/states for the substituents and for π-electron densities on the 2-carbonyl(hetero)aryl portion have been also important for the COX-2 selective inhibitions in the present study and in the previous SAR studies in vitro by Hayashi et al.Citation104, respectively. All together, the present and the previously reported SAR studies in vitro and the respective diverse analogues per se have provided unique and significant information to recognise/understand potent and selective interaction between COX-2 and the artificial inhibitors in vitro for further COX-2 inhibitor study regarding molecular mechanisms of actions, and have demonstrated various useful directions for further design of potent and selective COX-2 inhibitors, which can be exploited to elucidate further detailed underlying the mechanisms and to discover/develop further COX-2 inhibitors.
In vivo oral activities in the rats and physicochemical properties
Encouraged by the potent and selective COX-2 inhibitory activities of the novel acid-types of analogues in the above study in vitro, SAR of representative novel {2-[(3- or 4-substituted)-benzoyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues for oral anti-inflammatory efficacies in peripheral-inflammation model were investigated in vivo as the next step of this study. Thus, in vivo inhibitory effects against oedema-swelling stimulated by intraplantarly injected-carrageenan in the hind paw of specific pathogen free and virus antibody free (SPF/VAF) male Sprague–Dawley (SD) rats, and in vivo suppressive effects against PGE2 production stimulated by carrageenan in the foot of the SPF/VAF SD rats, that was the oedema-formation site in the above peripheral-inflammation model, were evaluated for the compounds per os (po)Citation25,Citation104,Citation115–118. As well, physicochemical properties of the compounds were estimated to consider key factors of the in vivo oral activities, together with in vitro activities for the analogues. For example, lipophilicity values for the whole structures of the compounds were calculated as ACD log D at pH 7.4 (ACD log D7.4). The results of the SAR studies of oral activities in vivo and physicochemical properties for COX-2 inhibitors as potential anti-inflammatory drugs are summarised in .
Table 3. In vivo structure−activity relationships of oral inhibitory effects against oedema formation and prostaglandin E2 production in peripheral-inflammation model rats and physicochemical properties for COX-2 inhibitors 8d, 14, 22b, 22c and 29.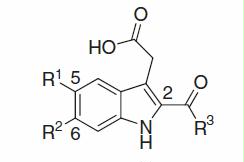
Actually, in this in vivo SAR study with the analogues for oral inhibitory effects against carrageenan-induced oedema-swelling in the foot of SPF/VAF SD rats, 6-chloro-2-[4-(trifluoromethyl)benzoyl]-1H-indolyl analogue 8d, 6-chloro-2-(cyclohexanecarbonyl)-1H-indolyl analogue 14, 6-chloro-2-(4-fluorobenzoyl)-1H-indolyl analogue 22b and 2-(4-chlorobenzoyl)-5-fluoro-1H-indolyl analogue 29 exhibited 25.6–43.0% inhibitions at 10 mg/kg per os. Especially, compound 29 demonstrated the most potent inhibitory efficacy in the model among these compounds (10 mg/kg, po). These orally potent anti-oedematous efficacies of the compounds would be due to their potent COX-2 inhibitions in vitro (IC50 = 0.0730–0.264 µM), which are in accordance with our previous reports by Hayashi et al.Citation25,Citation104 for [2-{[(4-substituted or 4,5-disubstituted)-pyridin-2-yl]carbonyl}-(5- or 6-substituted or 5,6-disubstituted)-1H-indol-3-yl]acetic acid analogues that exhibited highly potent and selective COX-2 inhibitions in vitro and orally potent anti-oedematous efficacies in the peripheral-inflammation model. In contrast, 6-chloro-2-[3-(trifluoromethyl)benzoyl]-1H-indol-3-yl analogue 22c exhibited less potent in vivo anti-oedematous efficacy (7.40% at 10 mg/kg, po) owing to or contributed by its weaker COX-2 inhibition in vitro (IC50 = 0.340 µM) and less appropriate physicochemical property compared with the above compounds as discussed later.
Furthermore, SAR of these COX-2 inhibitors for in vivo oral suppressive effects against carrageenan-induced PGE2 synthesis in the foot of the SPF/VAF SD rats was studied. Thus, the in vivo oral suppressive effects of the above compounds 8d, 14, 22b, 22c and 29 against PGE2 production in the peripheral-inflammation site were 33.3–91.1% inhibitions at 10 mg/kg per os. Significantly, compound 29 exhibited the most potent inhibitory activity in the model among them (10 mg/kg, po). As well, the tendency of the degree of the in vivo oral suppressive effects among most of the above compounds against PGE2 production in the site (10 mg/kg, po) were similar to that of the degree of in vivo oral anti-oedematous effects among the compounds in the peripheral-inflammation model rats (10 mg/kg, po), although the respective values of the percent inhibitions for the oral suppressive effects on PGE2 production and for the oral anti-oedematous effects were influenced by the respective in vivo endpoints-dependent sensitivities per os for the analogues.
In the viewpoints of molecular characteristics, the above orally potent compounds 8d, 14, 22b and 29 in the peripheral-inflammation model rats possess favourable physicochemical properties and appropriate structural features to achieve orally potent anti-inflammatory activities and high plasma-to-brain selectivities at the oral (systemic)-administration conditions in the SD rats as our previously described COX-2 inhibitor studies by Hayashi et al.Citation25,Citation104. Indeed, the above present compounds 8d, 14, 22b and 29 have far low lipophilicities for the respective whole structures under physiological plasma condition (ACD log D7.4 = –0.36 to 0.37), appropriate ranges or values of other properties such as molecular weight (MW; 319.78–381.73 Da), topological polar surface area (TPSA; 70.16 Å2), the numbers of hydrogen bond donor (HBD; two) and hydrogen bond acceptor (HBA; three), and hydrophilic functionalities such as HBDs at plural sites of the respective molecules. In contrast, compared with these compounds, 6-chloro-2-[3-(trifluoromethyl)benzoyl]-1H-indol-3-yl analogue 22c, that had weaker potency in vitro as mentioned and higher lipophilicity (ACD log D7.4 = 0.47), showed lower anti-inflammatory effect and lower suppressive effect on PGE2 production in vivo (po), although other physicochemical properties and structural features (e.g. MW, TPSA and multisite HBD functionalities) that related to the oral activities are within the above appropriate-ranges in common with all these compounds.
As reported alreadyCitation4,Citation5,Citation122–126, mechanisms of peripheral-inflammatory oedema-formation by carrageenan comprise multiphasic processes in vivo that include the production of PGE2 and PGI2 (as inflammatory mediators) owing to COX-2 induction/upregulation and COX-2 activity in the peripheral-inflammatory sites. Therefore, the mechanisms of actions of the oral anti-inflammatory effects for the present series of novel {2-[(3- or 4-substituted)-benzoyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues would be explicable by their inhibitory effects against PGE2 and PGI2 productions in the peripheral oedema-sites, directly depended on their intrinsic inhibitory potencies against COX-2, their physicochemical properties and their structural features, which is in accordance with the detailed mechanism-study for the previously reported analogues by Hayashi et al.Citation25,Citation104.
Taken together, the present study demonstrates that (i) the novel acid-types of COX-2 inhibitors 8d, 14, 22b and 29 were orally potent potential anti-inflammatory drugs in vivo, and (ii) the SARs of the analogues regarding in vitro and in vivo inhibitory activities against inflammatory-mediator productions derived from COX-2 with the physicochemical properties and structural-features of the analogues clarified the essential and key factors to achieve the orally potent anti-oedematous efficacies in the peripheral-inflammation model of rats in vivo, which led to the in vivo mechanisms of actions for the analogues, as unique and significant findings. These results also show possibility of the present series of various novel acid-types of analogues, as well as other/further compounds around the analogues that have potent and selective COX-2 inhibitory activities and appropriate physicochemical-properties/structural-features, as orally potent anti-inflammatory drugs in the in vivo model. Moreover, the present in vitro and in vivo SARs and the diverse COX-2 inhibitors per se, together with our previously reported COX-2 inhibitor studies and the diverse COX-2 inhibitors thereinCitation25,Citation104, might be helpful/useful to investigate further COX-2 inhibitor study in vivo for the treatment of inflammatory diseases/syndromes, injuries/traumas, infections, and/or chronic inflammation-related disorders such as RA and OA, and/or for chemotherapy or chemoprevention of cancers/tumours, and to elucidate underlying further molecular mechanisms of actions for the orally potent agents in the in vitro and in vivo environments.
Conclusions
Through this drug-discovery study, various novel {2-[(2-, 3- or 4-substituted, or 2,4- or 3,4-disubstituted)-benzoyl, (bicyclic heterocycloalkanophenyl)carbonyl or cycloalkanecarbonyl]-(5- or 6-substituted)-1H-indol-3-yl}acetic acid analogues were designed, synthesised and biologically evaluated to seek and identify various chemotypes of potent and selective COX-2 inhibitors for the treatment of inflammatory diseases. In fact, diverse acid-types of novel potent and selective COX-2 inhibitors were discovered in vitro, showing a variety of indispensable structural features/factors that are responsible for the potencies and the selectivities in vitro. Furthermore, novel COX-2 inhibitors 8d, 14, 22b and 29 exhibited orally potent anti-oedematous efficacies in the carrageenan-induced peripheral-inflammation model of SPF/VAF male SD rats with orally potent inhibitory activities against PGE2 production in the peripheral-inflammation sites of the rats, which were dependent on the intrinsic COX-2 inhibitory activities, physicochemical properties and structural features of the respective molecules. These unique and significant findings that include the in vitro and in vivo SAR results demonstrate that the representative compounds 8d, 14, 22b and 29 per se are orally potent potential anti-inflammatory drugs in vivo with unique and promising potential in or around the present acid-types of analogues, which would be quite useful to elucidate or understand underlying potent and selective interaction between COX-2 and COX-2 inhibitors in vitro. Moreover, the present study and the findings might be helpful to investigate further COX-2 inhibitor study in vivo for the treatment of inflammatory diseases/syndromes, injuries/traumas, infections, OA and RA, and/or for chemotherapy or chemoprevention of cancers/tumours, and to clarify underlying further molecular mechanisms of actions for the orally potent agents in vivo, together with our previously described unique and significant studies of orally potent novel [2-{[(4-substituted or 4,5-disubstituted)-pyridin-2-yl]carbonyl}-(5- or 6-substituted or 5,6-disubstituted)-1H-indol-3-yl]acetic acid analogues as highly potent and selective COX-2 inhibitors by Hayashi et al.Citation25,Citation104, and with the highly organised physicochemical properties and structural features of the molecules for dynamic enzyme-inhibition processes in vitro and in vivo as described in this and the previous studies.
Declaration of interest
We report no declarations of interest.
Supplementary material available online
Appendix. Supplementary Material
Supplementary Material
Download PDF (283.1 KB)References
- Funk CD, Funk LB, Kennedy ME, et al. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J 1991;5:2304–12
- Patrignani P, Panara MR, Greco A, et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther 1994;271:1705–12
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthase (cyclooxygenase)-1 and -2. J Biol Chem 1996;271:33157–60
- Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 2004;56:387–437. [Review]
- Zeilhofer HU. Prostanoids in nociception and pain. Biochem Pharmacol 2007;73:165–74
- Kojima F, Kapoor M, Kawai S, Crofford LJ. New insights into eicosanoid biosynthetic pathways: implication for arthritis. Expert Rev Clin Immunol 2006;2:277–91. [Review]
- Kang RY, Freire-Moar J, Sigal E, Chu C-Q. Expression of cyclooxygenase-2 in human and an animal model of rheumatoid arthritis. Br J Rheumatol 1996;35:711–18
- Blatteis CM. Endotoxic fever: new concepts of its regulation suggest new approaches to its management. Pharmacol Ther 2006;111:194–223. [Review]
- Ivanov AI, Pero RS, Scheck AC, Romanovsky AA. Prostaglandin E2-synthesizing enzymes in fever: differential transcriptional regulation. Am J Physiol Regul Integr Comp Physiol 2002;283:R1104–17
- Romanovsky AA, Almeida MC, Aronoff DM, et al. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front Biosci 2005;10:2193–216
- Steiner AA, Ivanov AI, Serrats J, et al. Cellular and molecular bases of the initiation of fever. PLoS Biol 2006;4:1517–24
- Li S, Wang Y, Matsumura K, et al. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2−/–, but not in cyclooxygenase-1−/– mice. Brain Res 1999;825:86–94
- Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res. 2003;110:255–8 . [Review]
- Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther 107;2005:139–54. [Review]
- Laine L. GI risk and risk factors of NSAIDs. J Cardiovasc Pharmacol 2006;47(Suppl. 1):S60–6
- Radi ZA, Khan NK. Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp Toxicol Pathol 2006;58:163–73
- Warner TD, Giuliano F, Vojnovic I, et al. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci USA 1999;96:7563–8
- Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA. 1992;89:7384–8
- O’Neill GP, Ford-Hutchinson AW. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett 1993;330:156–60
- Beiche F, Scheuerer S, Brune K, et al. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Lett 1996;390:165–9
- Steer SA, Corbett JA. The role and regulation of COX-2 during viral infection. Viral Immunol 2003;16:447–60. [Review]
- Duque J, Díaz-Muñoz MD, Fresno M, Iñiguez MA. Up-regulation of cyclooxygenase-2 by interleukin-1β in colon carcinoma cells. Cell Signal 2006;18:1262–9
- Kelley DJ, Mestre JR, Subbaramaiah K, et al. Benzo[a]pyrene up-regulates cyclooxygenase-2 gene expression in oral epithelial cells. Carcinogenesis 1997;18:795–9
- Kiritoshi S, Nishikawa T, Sonoda K, et al. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells. Potential role in diabetic nephropathy. Diabetes 2003;52:2570–7
- Hayashi S, Sumi Y, Ueno N, et al. Discovery of a novel COX-2 inhibitor as an orally potent anti-pyretic and anti-inflammatory drug: design, synthesis, and structure–activity relationship. Biochem Pharmacol 2011;82:755–68
- Kowalski ML, Makowska J. Use of nonsteroidal anti-inflammatory drugs in patients with aspirin hypersensitivity. Safety of cyclo-oxygenase-2 inhibitors. Treat Respir Med 2006;5:399–406
- Sánchez-Borges M. NSAID hypersensitivity (respiratory, cutaneous, and generalized anaphylactic symptoms). Med Clin North Am 2010;94:853–64
- Harrington LS, Lucas R, McMaster SK, et al. COX-1, and not COX-2 activity, regulates airway function: relevance to aspirin-sensitive asthma. FASEB J 2008;22:4005–10
- Reddy BS, Rao CV. Novel approaches for colon cancer prevention by cyclooxygenase-2 inhibitors. J Environ Pathol Toxicol Oncol 2002;21:155–64
- Tsuji M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA 1997;94:3336–40
- Wu K, Nie Y, Guo C, et al. Molecular basis of therapeutic approaches to gastric cancer. J Gastroenterol Hepatol 2009;24:37–41
- Jüttner S, Cramer T, Wessler S, et al. Helicobacter pylori stimulates host cyclooxygenase-2 gene transcription: critical importance of MEK/ERK-dependent activation of USF1/-2 and CREB transcription factors. Cell Microbiol 2003;5:821–34
- Ku GY, Ilson DH. Esophagogastric cancer: targeted agents. Cancer Treat Rev 2010;36:235–48
- Saadi A, Shannon NB, Lao-Sirieix P, et al. Stromal genes discriminate preinvasive from invasive disease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proc Natl Acad Sci USA 2010;107:2177–82
- Lee JL, Mukhtar H, Bickers DR, et al. Cyclooxygenases in the skin: pharmacological and toxicological implications. Toxicol Appl Pharmacol 2003;192:294–306
- Camp WL, Turnham JW, Athar M, Elmets CA. New agents for prevention of ultraviolet-induced nonmelanoma skin cancer. Semin Cutan Med Surg 2011;30:6–13
- Mao Y, Poschke I, Wennerberg E, et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res 2013;73:3877–87
- Krysan K, Reckamp KL, Sharma S, Dubinett SM. The potential and rationale for COX-2 inhibitors in lung cancer. Anti-Cancer Agents Med Chem 2006;6:209–20
- Yuan A, Yu C-J, Shun C-T, et al. Total cyclooxygenase-2 mRNA levels correlate with vascular endothelial growth factor mRNA levels, tumor angiogenesis and prognosis in non-small cell lung cancer patients. Int J Cancer 2005;115:545–55
- Sato M, Shames DS, Hasegawa Y. Emerging evidence of epithelial-to-mesenchymal transition in lung carcinogenesis. Respirology 2012;17:1048–59
- Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 2011;26(Suppl. 1):144–52
- Fernández-Martínez A, Mollá B, Mayoral R, et al. Cyclo-oxygenase 2 expression impairs serum-withdrawal-induced apoptosis in liver cells. Biochem J 2006;398:371–80
- Li G, Han C, Xu L, et al. Cyclooxygenase-2 prevents Fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology 2009;50:834–43
- Zhao Q-T, Yue S-Q, Cui Z, et al. Potential involvement of the cyclooxygenase-2 pathway in hepatocellular carcinoma-associated angiogenesis. Life Sci 2007;80:484–92
- Mohammed A, Qian L, Janakiram NB, et al. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48Cre/+ LSL-KrasG12D/+ mice. Int J Cancer 2012;131:1951–62
- Kokawa A, Kondo H, Gotoda T, et al. Increased expression of cyclooxygenase-2 in human pancreatic neoplasms and potential for chemoprevention by cyclooxygenase inhibitors. Cancer 2001;91:333–8
- Niijima M, Yamaguchi T, Ishihara T, et al. Immunohistochemical analysis and in situ hybridization of cyclooxygenase-2 expression in intraductal papillary-mucinous tumors of the pancreas. Cancer 2002;94:1565–73
- Hayashi N, Yamamoto H, Hiraoka N, et al. Differential expression of cyclooxygenase-2 (COX-2) in human bile duct epithelial cells and bile duct neoplasm. Hepatology 2001;34:638–50
- Kawamoto T, Shoda J, Asano T, et al. Expression of cyclooxygenase-2 in the subserosal layer correlates with postsurgical prognosis of pathological tumor stage 2 carcinoma of the gallbladder. Int J Cancer 2002;98:427–34
- Shirahama T, Arima J, Akiba S, Sakakura C. Relation between cyclooxygenase-2 expression and tumor invasiveness and patient survival in transitional cell carcinoma of the urinary bladder. Cancer 2001;92:188–93
- Hu M, Peluffo G, Chen H, et al. Role of COX-2 in epithelial–stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc Natl Acad Sci USA 2009;106:3372–7
- Barnes NLP, Warnberg F, Farnie G, et al. Cyclooxygenase-2 inhibition: effects on tumour growth, cell cycling and lymphangiogenesis in a xenograft model of breast cancer. Br J Cancer 2007;96:575–82
- Jain S, Chakraborty G, Kundu GC. The crucial role of cyclooxygenase-2 in osteopontin-induced protein kinase C α/c-Src/IκB kinase α/β-dependent prostate tumor progression and angiogenesis. Cancer Res 2006;66:6638–48
- Badawi AF. The role of prostaglandin synthesis in prostate cancer. BJU Int 2000;85:451–62
- Lee S, Kim J-H, Kim H, et al. Activation of the interleukin-32 pro-inflammatory pathway in response to human papillomavirus infection and over-expression of interleukin-32 controls the expression of the human papillomavirus oncogene. Immunology 2010;132:410–20
- Kim YB, Kim GE, Cho NH, et al. Overexpression of cyclooxygenase-2 is associated with a poor prognosis in patients with squamous cell carcinoma of the uterine cervix treated with radiation and concurrent chemotherapy. Cancer 2002;95:531–9
- Uddin S, Ahmed M, Hussain A, et al. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int J Cancer 2010;126:382–94
- Xin B, Yokoyama Y, Shigeto T, et al. Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer 2007;110:791–800
- Murono S, Inoue H, Tanabe T, et al. Induction of cyclooxygenase-2 by Epstein–Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci USA 2001;98:6905–10
- Urade M. Cyclooxygenase (COX)-2 as a potent molecular target for prevention and therapy of oral cancer. Jpn Dent Sci Rev 2008;44:57–65
- Iwata C, Kano MR, Komuro A, et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res 2007;67:10181–9
- Liu H, Yang Y, Xiao J, et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via VEGF-C. Anat Rec 2009;292:1577–83
- Tomozawa S, Tsuno NH, Sunami E, et al. Cyclooxygenase-2 overexpression correlates with tumour recurrence, especially haematogenous metastasis, of colorectal cancer. Br J Cancer 2000;83:324–8
- Tomozawa S, Nagawa H, Tsuno N, et al. Inhibition of haematogenous metastasis of colon cancer in mice by a selective COX-2 inhibitor, JTE-522. Br J Cancer 1999;81:1274–9
- Chen J-H, Wu C-W, Kao H-L, et al. Effects of COX-2 inhibitor on growth of human gastric cancer cells and its relation to hepatocyte growth factor. Cancer Lett 2006;239:263–70
- Kobayashi H, Gonda T, Uetake H, et al. JTE-522, a selective COX-2 inhibitor, interferes with the growth of lung metastases from colorectal cancer in rats due to inhibition of neovascularization: a vascular cast model study. Int J Cancer 2004;112:920–6
- Yao M, Lam EC, Kelly CR, et al. Cyclooxygenase-2 selective inhibition with NS-398 suppresses proliferation and invasiveness and delays liver metastasis in colorectal cancer. Br J Cancer 2004;90:712–19
- Nagatsuka I, Yamada N, Shimizu S, et al. Inhibitory effect of a selective cyclooxygenase-2 inhibitor on liver metastasis of colon cancer. Int J Cancer 2002;100:515–19
- Chen W-S, Wei S-J, Liu JM, et al. Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer 2001;91:894–9
- Mougiakakos D, Johansson CC, Trocme E, et al. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer 2010;116:2224–33
- Kim H-S, Kim M-J, Kim EJ, et al. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharm 2012;83:385–94
- Said N, Theodorescu D. Permissive role of endothelin receptors in tumor metastasis. Life Sci 2012;91:522–7
- Valsecchi ME, Pomerantz SC, Jaslow R, Tester W. Reduced risk of bone metastasis for patients with breast cancer who use COX-2 inhibitors. Clin Breast Cancer 2009;9:225–30
- Ono K, Akatsu T, Murakami T, et al. Involvement of cyclo-oxygenase-2 in osteoclast formation and bone destruction in bone metastasis of mammary carcinoma cell lines. J Bone Miner Res 2002;17:774–81
- Wu KK. Differential cyclooxygenase-2 transcriptional control in proliferating versus quiescent fibroblasts. Prostaglandins Other Lipid Mediat 2007;83:175–81
- Denkert C, Winzer K-J, Müller B-M, et al. Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer 2003;97:2978–87
- Ali-Fehmi R, Che M, Khalifeh I, et al. The effect of cyclooxygenase-2 expression on tumor vascularity in advanced stage ovarian serous carcinoma. Cancer 2003;98:1423–9
- Dormond O, Foletti A, Paroz C, Rüegg C. NSAIDs inhibit αVβ3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat Med 2001;7:1041–7
- Connolly EM, Harmey JH, O'Grady T, et al. Cyclo-oxygenase inhibition reduces tumour growth and metastasis in an orthotopic model of breast cancer. Br J Cancer 2002;87:231–7
- Rozic JG, Chakraborty C, Lala PK. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer 2001;93:497–506
- Chang S-H, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA 2004;101:591–6
- Farooqui M, Li Y, Rogers T, et al. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumour growth, metastasis and mortality, without compromising analgesia. Br J Cancer 2007;97:1523–31
- Oshima H, Oshima M. Mouse models of gastric tumors: Wnt activation and PGE2 induction. Pathol Int 2010;60:599–607
- Qualtrough D, Kaidi A, Chell S, et al. Prostaglandin F2α stimulates motility and invasion in colorectal tumor cells. Int J Cancer 2007;121:734–40
- Sales KJ, Maldonado-Pérez D, Grant V, et al. Prostaglandin F2α-F-prostanoid receptor regulates CXCL8 expression in endometrial adenocarcinoma cells via the calcium–calcineurin–NFAT pathway. Biochim Biophys Acta 2009;1793:1917–28
- Sakai H, Suzuki T, Takahashi Y, et al. Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett 2006;580:3368–74
- Nie D, Lamberti M, Zacharek A, et al. Thromboxane A2 regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun 2000;267:245–51
- Cline SD, Riggins JN, Tornaletti S, et al. Malondialdehyde adducts in DNA arrest transcription by T7 RNA polymerase and mammalian RNA polymerase II. Proc Natl Acad Sci USA 2004;101:7275–80
- Mao H, Schnetz-Boutaud NC, Weisenseel JP, et al. Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc Natl Acad Sci USA 1999;96:6615–20
- Flockhart RJ, Diffey BL, Farr PM, et al. NFAT regulates induction of COX-2 and apoptosis of keratinocytes in response to ultraviolet radiation exposure. FASEB J 2008;22:4218–27
- Fritsche E, Schäfer C, Calles C, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci USA 2007;104:8851–6
- Zhou H, Ivanov VN, Gillespie J, et al. Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci USA 2005;102:14641–6
- Hussain M, Javeed A, Ashraf M, et al. Non-steroidal anti-inflammatory drugs, tumour immunity and immunotherapy. Pharmacol Res 2012;66:7–18
- White WB, Faich G, Borer JS, Makuch RW. Cardiovascular thrombotic events in arthritis trials of the cyclooxygenase-2 inhibitor celecoxib. Am J Cardiol 2003;92:411–18
- Hinz B, Renner B, Brune K. Drug insight: cyclo-oxygenase-2 inhibitors—a critical appraisal. Nat Clin Pract Rhum 2007;3:552–60
- Oitate M, Hirota T, Murai T, et al. Covalent binding of rofecoxib, but not other cyclooxygenase-2 inhibitors, to allysine aldehyde in elastin of human arota. Drug Metab Dispos 2007;35:1846–52
- Chenevard R, Hürlimann D, Béchir M, et al. Selective COX-2 inhibition improves endothelial function in coronary artery disease. Circulation J Am Heart Assoc 2003;107:405–9
- Höcherl K, Dreher F, Kurtz A, Bucher M. Cyclooxygenase-2 inhibition attenuates lipopolysaccharide-induced cardiovascular failure. Hypertension J Am Heart Assoc 2002;40:947–53
- Limongelli V, Bonomi M, Marinelli L, et al. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc Natl Acad Sci USA 2010;107:5411–16
- Rimon G, Sidhu RS, Lauver DA, et al. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc Natl Acad Sci USA 2010;107:28–33
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8
- Gierse JK, McDonald JJ, Hauser SD, et al. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem 1996;271:15810–14
- Luong C, Miller A, Barnett J, et al. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol 1996;3:927–33
- Hayashi S, Ueno N, Murase A, et al. Novel acid-type cyclooxygenase-2 inhibitors: design, synthesis, and structure–activity relationship for anti-inflammatory drug. Eur J Med Chem 2012;50:179–95
- Katritzky AR, Akutagawa K. Carbon dioxide: a reagent for the protection of nucleophilic centres and the simultaneous activation of alternative locations to electrophilic attack part I. A new synthetic method for the 2-substitution of 1-unsubstituted indoles. Tetrahedron Lett 1985;26:5935–8
- Baciocchi E, Muraglia E. Mn(III)-promoted homolytic methylmalonylation of pyrrole and indole derivatives. A simple route to α-methyl-2-pyrrole- and α-methyl-3-indoleacetic acids. J Org Chem 1993;58:7610–12
- Artis DR, Cho I-S, Muchowski JM. Radical-based syntheses of 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylic acid (ketorolac). Can J Chem 1992;70:1838–42
- Nahm S, Weinreb SM. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett 1981;22:3815–18
- Moyer MP, Shiurba JF, Raporport H. Metal–halogen exchange of bromoindoles. A route to substituted indoles. J Org Chem 1986;51:5106–10
- Frye SV, Johnson MC, Valvano NL. Synthesis of 2-aminobenzophenones via rapid halogen–lithium exchange in the presence of a 2-amino-N-methoxy-N-methylbenzamide. J Org Chem 1991;56:3750–2
- Giordano C, Coppi, L. Br3− vs Br2: opposite diastereoselectivity in the bromination of enantiomerically pure ketals. J Org Chem 1992;57:2765–6
- Bellucci G, Bianchini R, Vecchiani S. Comparison of molecular bromine and tribromide ion as brominating reagents. 2. Kinetic and product investigation of the bromination of 3-substituted cylohexenes. J Org Chem 1986;51:4224–32
- Riendeau D, Percival MD, Boyce S, et al. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol 1997;121:105–17
- Moore PF, Larson DL, Otterness IG, et al. Tenidap, a structurally novel drug for the treatment of arthritis: antiinflammatory and analgesic properties. Inflammation Res 1996;45:54–61
- Winter CA, Risley EA, Nuss GV. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 1962;111:544–7
- Lombardino JG, Otterness IG, Wiseman EH. Acidic anti-inflammatory agents-correlations of some physical, pharmacological and clinical data. Arzneim Forsch 1975;25:1629–35
- Gracia Leme J, Hamamura L, Leite MP, Rocha e Silva M. Pharmacological analysis of the acute inflammatory process induced in the rat’s paw by local injection of carrageenin and by heating. Br J Pharmacol 1973;48:88–96
- Opas EE, Dallob A, Herold E, et al. Pharmacological modulation of eicosanoid levels and hyperalgesia in yeast-induced inflammation. Biochem Pharmacol 1987;36:547–51
- Patel BA, Ziegler CB, Cortese NA, et al. Palladium-catalyzed vinylic substitution reactions with carboxylic acid derivatives. J Org Chem 1977;42:3903–7
- Carling RW, Leeson PD, Moore KW, et al. 3-Nitro-3,4-dihydro-2(1H)-quinolones. Excitatory amino acid antagonists acting at glycine-site NMDA and (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. J Med Chem 1993;36:3397–408
- Xie WL, Chipman JG, Robertson DL, et al. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci USA 1991;88:2692–6
- Sugimoto Y, Narumiya S, Ichikawa A. Distribution and function of prostanoid receptors: studies from knockout mice. Prog Lipid Res 2000;39:289–314
- Romano M, Faggioni R, Sironi M, et al. Carrageenan-induced acute inflammation in the mouse air pouch synovial model. Role of tumour necrosis factor. Mediat Inflamm 1997;6:32–8
- Padi SSV, Jain NK, Singh S, Kulkarni SK. Pharmacological profile of paracoxib: a novel, potent injectable selective cyclooxygenase-2 inhibitor. Eur J Pharmacol 2004;491:69–76
- Posadas I, Bucci M, Roviezzo F, et al. Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br J Pharmacol 2004;142:331–8
- Claudino RF, Kassuya CAL, Ferreira J, Calixto JB. Pharmacological and molecular characterization of the mechanisms involved in prostaglandin E2-induced mouse paw edema. J Pharmacol Exp Ther 2006;318:611–18