
Abstract
Context: Natural strain variation and rapid resistance development makes development of broad spectrum hepatitis C virus (HCV) drugs very challenging and evaluation of inhibitor selectivity and resistance must account for differences in the catalytic properties of enzyme variants.
Objective: To understand how to study selectivity and relationships between efficacy and genotype or resistant mutants for NS3 protease inhibitors.
Materials and methods: The catalytic properties of NS3 protease from genotypes 1a, 1b and 3a, and their sensitivities to four structurally and mechanistically different NS3 protease inhibitors have been analysed under different experimental conditions.
Results: The optimisation of buffer conditions for each protease variant enabled the comparison of their catalytic properties and sensitivities to the inhibitors. All inhibitors were most effective against genotype 1a protease, with VX-950 having the broadest selectivity.
Discussion and conclusion: A new strategy for evaluation of inhibitors relevant for the discovery of broad spectrum HCV drugs was established.
Background
Hepatitis C virus (HCV) is today the leading cause of chronic liver disease worldwide, infecting around 2–3% of the population globallyCitation1,Citation2. The virus is classified into six major genotypes and a large number of subtypesCitation3. The most prevalent subtypes in Europe and North America are 1a and 1b, which together account for 60% of the infections worldwide. Genotype 3a is widely spread among intravenous drug users in several countries worldwide and is also predominant in India and Pakistan. Due to large natural strain variations and a predisposition for resistance development, it is challenging to develop drugs efficient against all clinically relevant variants of HCV.
The protease in the non-structural protein 3 (NS3) from HCV is an important target for anti-HCV drug discovery. Several NS3 protease inhibitors have reached clinical trials, but only two, boceprevir and telaprevir, have been approved. They are used in combination with ribavirin and pegylated interferon in patients infected with genotype 1. Although a sustained virological response is achieved in genotype 1 infected HCV patients, the treatment is associated with adverse side effects and is expected to give rise to resistance. Furthermore, it is not effective for all clinically relevant genotypes and subtypes. There is consequently a need for new drugs with improved potency, resistance profile and pharmacokinetic properties and it is therefore an area of high priority for pharmaceutical companiesCitation4.
The viral protease is part of the multifunctional non-structural protein 3 (NS3) of HCV. It is an attractive drug target since it is involved in the processing of the viral polyprotein, an essential step in viral replicationCitation5,Citation6. The protease is a serine protease with a chymotrypsin like fold. It is located in the N-terminal domain of NS3 (residues 1–180) and constitutes one-third of the 67 kDa NS3 proteinCitation7. In order for the protease to be fully folded and activated, NS3 needs to be in complex with Zn2+ and its cofactor NS4ACitation5. The central portion of this 54 residue peptide cofactor is integrated into the protease domain where it forms part of a β-sheet, stabilising the active conformation of the enzyme and contributing to the anchoring of the replication complex to the ER membraneCitation8. The larger C-terminal domain (residues 181–631) harbours an RNA-helicase and NTPase.
The protease and helicase domains are functionally independent of each other and truncated forms of the NS3 corresponding to the separate domains are often used as model systems for biochemical studies. However, we have shown that residues in the helicase domain affect inhibition of the proteaseCitation9 and there is evidence that also the protease domain and NS4A influences helicase functionalityCitation10. Hence, the use of full-length NS3 is advantageous if novel lead compounds with improved properties are to be discoveredCitation11, as has been illustrated in a recent fragment-based approach that has identified fragments that bind to an allosteric site in the interface region but not to the protease domain NS312.
This study was initiated since we were interested in identifying suitable NS3 model systems and improved experimental conditions for our anti-HCV drug design research. Our goal is to establish methods that would enable the design of inhibitors with improved properties, i.e. efficient broad spectrum HCV protease inhibitors with low propensity for resistance. It therefore involved the production of a set of enzyme variants, representing clinically relevant strains and a resistance mutation, and the subsequent evaluation of their sensitivity to selected inhibitors.
The panel of enzyme variants included full-length variants of genotype 1a, 1b and 3a, truncated variants of genotype 1a and 1b, co-constructs of full-length NS3/4 A from genotype 1a and 1b and the drug resistant variant R155K of full-length genotype 1a NS3 (). Four peptidomimetic NS3 inhibitors that all bind to the active site but vary in their molecular structure, inhibitory mechanism and potency were used for the comparison (). They include an early linear product-based inhibitor (compound 14 inCitation13, here denoted N-1725), and three more developed compounds which have reached or are currently in clinical trials. BILN-2061 (ciluprevir) is a macrocyclic product-based inhibitor and the first compound targeting NS3 protease to enter clinical trialsCitation14. It showed important proof-of-principle for this class of anti-HCV agents, but failed due to toxic effectsCitation15. ITMN-191 (danoprevir) is a slow tight binding inhibitorCitation16 that is structurally and mechanistically similar to BILN-2061. It is a competitive inhibitorCitation17 that exhibits a two step binding mechansim to NS3 proteaseCitation16 and is currently in phase II clinical trialsCitation18. VX-950 (telaprevir) is a linear mechanism-based inhibitor that forms a covalent but reversible bond with the active site serine residueCitation19. Telaprevir is used in combinational therapy against HCV genotype 1Citation20. The inhibitors used in this biochemical evaluation were chosen on the basis of their mechanistic and structural differences. Their pharmacokinetic properties or side effect profiles were hence not considered.
Figure 1. Schematic of the characterized enzyme constructs, with the protease domain (black), the helicase domain (dark grey), the co-factor NS4A (medium grey) and N-terminal His-tags (light grey).
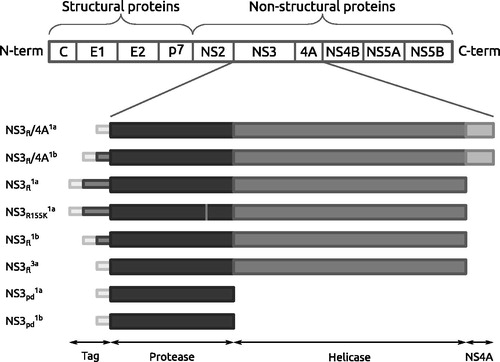
Figure 2. The four NS3 protease inhibitors evaluated and compared in this study.
Materials and methods
The different enzyme variants produced and characterized are schematically illustrated in . An alignment of the protein sequences is provided in Supplementary material (Figure S1). The DNA primers used for cloning are described in Supplementary material (Table S1).
Cloning of HCV NS3 variants from genotype 1a
Full-length NS3 protein from HCV genotype 1a () was previously cloned from total RNA extracted from serum of a patient infected with HCV strain 1aCitation21. For this study, full-length NS3 with full-length NS4A fused to the C-terminus (NS3fl/4A1a) was constructed using the same original material and a cDNA template encoding for residues 809–1725 (polyprotein sequence numbering). cDNA coding for the NS3fl/4A1a construct (amino acids 1027–1711) was amplified from the cDNA-template by PCR in two steps. In the first amplification, the PCR vial contained 1 µl cDNA-template, 400 nM each of forward and reverse primer (Supplementary material Table S1), 0.2 mM dNTPs, 2.5U Pfu DNA polymerase (Thermo Fisher Scientific, Waltham, MA), 0.02% dimethylsulfoxide (DMSO) in 50 µl Pfu buffer. In the second PCR step, the PCR product was used as the new template. The amplified DNA was purified by QIAquick PCR Purification Kit (QIAgen, Hilden, Germany) according to manufacturer’s protocol. The purified DNA was cloned into a pFastBac vector (Invitrogen, Carlsbad, CA), following standard procedures. The construct was used for transformation of Escherichia coli XL-1 Blue cells (Stratagene, La Jolla, CA) by heat shock. The purified clone was used as a template for further cloning of the NS3fl/4A1a construct into a pET-11a (Novagen, Merck KGaA, Darmstadt, Germany) expression vector to enable expression in E. coli.
An R155K variant (), in which the arginine (R) to lysine (K) mutation was introduced by PCR at position 155, was cloned in a similar way as has been described previouslyCitation22. A pET vector containing the same
as in the pBAD construct used here for expressionCitation21 was used as a template. The protease domain construct (
) was cloned from the pET-His-NS3fl/4A1a gene (see above).
Cloning of HCV NS3 variants from genotype 1b
HCV genotype 1b clones for full-length Citation23 and a full-length NS3–4 A fusion protein (NS3fl/4A1b)Citation24 were obtained from Raffaele De Francesco [National Institute in Molecular Genetics -- INGM, Milan, Italy]. A protease domain variant (
) was cloned with pET-11a–
as the template in the same way as for the genotype 1a variant (see above).
Cloning of HCV NS3 from genotype 3a
The gene for full-length NS3 from genotype 3a () was isolated and amplified by PCR from a patient isolate and cloned into a pET-11a expression vector. The blood samples were collected from different hospitals in Pakistan (a work done by the HCV Diagnostic services, PINUM Liver Center, Faisalabad, Pakistan). Total RNA was extracted from samples infected with HCV strain 3a using the QIAampViral RNA extraction kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions.
The cDNA was synthesized from the extracted RNA with gene specific primers (Supplementary material Table S1). The reaction vial contained 0.7 µg extracted RNA, 100 nM reverse primer/hexamer primers, 200 U RevertAid™ H Minus M-MuLV reverse transcriptase, 1 mM dNTPs in 20 µl reaction buffer [25 mM Tris–HCl pH 8.3, 25 mM KCl, 2 mM MgCl2, 5 mM dithiotreitol (DTT)]. The reaction was incubated at 42 °C for 1 h and the cDNA was amplified with PCR. The purified PCR product was cloned into a pET-11a expression vector that was transformed into E. coli XL1-Blue by heat shock. The plasmids were isolated, purified and sequenced.
Expression and purification of HCV NS3 variants
The expression and purification of all enzyme variants was performed according to previously published methodsCitation21,Citation23 but with some modifications. Briefly, the gene constructs were freshly transformed to heat competent BL21 (DE3; for the pET-11a gene constructs) or TOP10 cells (for the pBAD gene constructs). One colony was picked and grown in 10 ml of LB-media (90 µg/ml ampicillin). After 3–4 h of incubation at 37 °C, 1 ml was transferred to each of six culture flasks containing 500 ml LB-media (90 µg/ml ampicillin). Induction with isopropyl-β-D-thiogalactopyranoside (IPTG) was carried out at 11 °C for 22 h and the pellet was stored at −80 °C. Prior to purification, the pellet was thawed and resuspended in lysis buffer (25 mM HEPES pH 7.6, 20% glycerol, 0.5 M NaCl, 2.5 mM β-mercaptoethanol) and detergent (0.2% n-dodecyl β-d-maltoside (DDM) for ,
, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) for
, 0.1% Triton X-100 for NS3fl/4A1a and NS3fl/4A1b, 0.1% n-octyl-β-d-glucopyranoside (OGP) for
and
,
) followed by sonication and cell disruption. The insoluble material was pelleted at 13 000 rpm for 30 min.
The supernatant was loaded onto a 30 ml Chelating Sepharose FF column (GE Healthcare, Uppsala, Sweden) charged with Ni2+ and equilibrated with immobilized metal ion chromatography (IMAC) buffer (same as lysis buffer) according to the manufacturer’s instructions. After a two-step washing with IMAC buffer containing 5 mM and 50 mM imidazole, respectively, the protein was eluted with the IMAC buffer containing 200 mM imidazole. The eluted samples were exchanged into poly uracil [poly(U)] buffer (25 mM HEPES pH 7.6, 20% glycerol, 0.2 M NaCl, 3 mM DTT) and detergent (0.1% DDM for ,
, 0.1% OGP for
,
,
and NS3R155K1a, 0.1% Triton for NS3fl/4A1a and NS3fl/4A1b) using PD-10 columns (GE Healthcare, Uppsala, Sweden) and stored at −80 °C.
,
,
, NS3R155K1a, NS3fl/4A1a and NS3fl/4A1b were finally loaded onto a poly(U) Sepharose 4B affinity column (GE Healthcare, Uppsala, Sweden) equilibrated with poly(U) buffer. The protein was eluted in 2.5 ml fractions using poly(U) buffer containing 1 M NaCl. Protein concentrations were determined using a Bradford-based assay (Bio-Rad, Sundbyberg, Sweden). The fractions with the highest protein concentration were pooled and stored at −80 °C for later use.
Cofactor, inhibitors and FRET substrate
The NS4A cofactor peptide (NS4Apep, KKGSVVIVGRIVLSGK) was obtained from Gunnar Lindeberg, Department of Organic Pharmaceutical Chemistry, Uppsala University, Uppsala, Sweden. It corresponds to the central part of HCV NS4A for strain H77, with a C to S mutation and two N-terminal lysine residues added for increased solubility. BILN-2061 was obtained from Boehringer Ingelheim and VX-950 from the VIRGIL DrugPharm Team. ITMN-191 was synthesized according to published procedures (International Publication Number WO 2005/037214 A2, Compound AR00334191). Compound N-1725 was purchased from Bachem (Bubendorf, Switzerland). All inhibitors were freshly diluted in DMSO prior to use. The FRET substrate RET S1 (Ac-Asp-Glu-Asp(EDANS)-Glu-Glu-Abu-106 ψ-[COO]Ala-Ser-Lys(DABCYL)-NH2), which is based on the NS4A-4B cleavage site, was purchased from AnaSpec (San Jose, CA).
NS3 Protease activity and inhibition assay
A FRET-based steady-state enzyme activity assay was used for activity and inhibition measurements, as described previouslyCitation21. The data was inner filter effect corrected for high substrate concentrationsCitation25. All experiments were made in at least triplicates.
The catalytic properties of the different enzyme variants and the effects of different assay conditions (detergents, buffer and ionic strength) and inhibitors were characterized by pre-incubating 1 nM (5 nM for ) of NS3 enzyme with 25 µM NS4Apep (unless nothing else is stated) and inhibitor (when used), in a total volume of 290 µl, at 30 °C for 10 min. The reaction was started by adding 10 µl of substrate to a final concentration of 0.5 µM. For the determination of kinetic constants (Km and kcat), the substrate concentration was varied from 0 to 4 µM in an eight step interval.
The kinetic constants (Km, kcat and kcat/Km) were determined by fitting the Michaelis–Menten equation to the inner filter effect corrected initial rates by non-linear regression (Grafit 5.0.1, Erithacus Software, Staines, UK)Citation21. Similarly, the inhibition constant (Ki) was obtained by fitting an equation for competitive inhibition to the initial rates.
Vitality values
The vitality values were determined as described by Gulnik et al.Citation26 and used for comparisons of enzyme variants. The data for measured under optimized buffer conditions was used as the reference condition (i.e. used as “wild type” in the equation).
Results
Production of enzyme variants
The cloning, expression and purification of eight different variants of HCV NS3 representing three different strains (1a, 1b and 3a) and different forms of the protein (full-length NS3 ± full-length NS4A fused to the C-terminus, truncated protease domain and a resistant variant) was successful. The variants expressed in the PET expression system had a higher overall yield compared with the more tightly controlled pBAD expression system. Attempts to express the full-length wild-type variant from genotype 1a in a pET vector were unsuccessful. It resulted in higher overall expression levels but with insignificant yield of active enzyme. The enzyme is membrane-associated on its own and further optimization of the detergent conditions may result in higher expression levels of active enzyme in a pET expression system. The constructs with NS4A fused to NS3 led to both increased yield and higher stability of the over expressed proteins. No significant impurities were detected by SDS-PAGE analysis.
Identification of suitable buffers for the NS3 variants
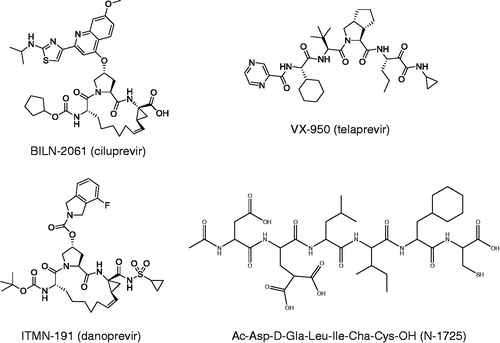
As a basis for the inhibition analysis of the different enzyme variants, it was essential to first determine suitable buffer conditions for the proteolytic activity of each variant and to characterize their catalytic properties under these conditions. The starting buffer was the one previously used for studies of Citation21. This “reference buffer” () was adjusted to better suit each enzyme variant, with respect to highest activity. To identify the most suitable detergent and detergent concentration, several detergents were tested at 0.1–0.8%. OGP was chosen for all NS3 variants except for the NS3fl/4A1a and NS3fl/4A1b for which Triton X-100 was found to be most suitable. A dependence of activity on the detergent conditions was seen for all enzyme variants (exemplified by
in ). Most of the variants had a maximal activity at 0.4% OGP, except for the truncated enzymes,
and
, which had their highest activity at 0.7 and 0.3%, respectively (data not shown). Interestingly, in the absence of added NS4Apep, the activities of the NS3fl/4A1a and NS3fl/4A1b co-constructs were very low in OGP but significantly higher in Triton X-100, for which 0.4% resulted in a maximal activity. Both co-constructs were completely inactive when DDM was present in the assay buffer, independent of DDM concentration, suggesting that detergent is the decisive factor for NS4A interaction.
Figure 3. Illustration of the relationship between the enzymatic activity of and the concentration of OGP.
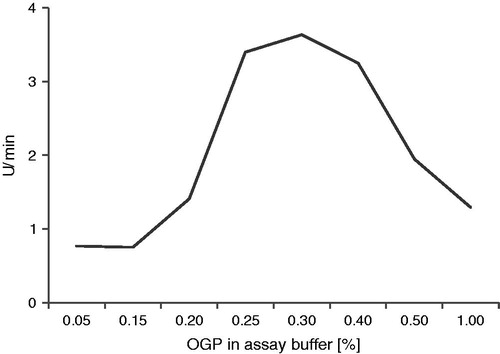
Table 1. Reference and optimized buffer compositions for analysis of proteolytic activity of NS3 variants.
Once the best detergent and detergent concentration had been identified for each enzyme variant, the effect of 5–50% glycerol and 0–300 mM NaCl was evaluated (). The enzyme variants exhibited very different dependence on these additives, with some variants being strongly dependent on glycerol and NaCl, while others being quite unaffected by the changes. In general, the highest activity was observed with the highest glycerol concentration (50%) and the lowest ionic strength (no NaCl added). Exceptions were the NS3fl/4A1a and NS3fl/4A1b co-constructs (without added NS4Apep) which had their highest activity at elevated ionic strengths.
Figure 4. Dependence of enzyme activity on detergent, glycerol and NaCl concentration for the studied enzyme variants. (a) in 0.4% OGP; (b)
in 0.4% OGP; (c)
in 0.4% OGP; (d) NS3fl/4A1a without NS4Apep in 0.4% Triton; (f) NS3 fl/4A1a with NS4Apep in 0.4% Triton; (e) NS4 fl/4A1b without NS4Apep in 0.4% Triton; (g) NS3 fl/4A1b with NS4Apep in 0.4% Triton; (h)
in 0.7% OGP; (i)
in 0.3% OGP; (j)
in 0.4% OGP.
The optimization of the buffer composition with respect to detergent, glycerol and ionic strength for each variant was critical since the enzymatic activities were non-detectable under many of the conditions and changes in the protein construct or strain changed the preferred buffer composition. Even so, for , the activity was very low at 1 nM, so 5 nM was used instead in order to obtain exploitable results. This should not affect the catalytic parameters, only the signal levels. The buffer composition that gave the highest proteolytic activity () defined the “optimized buffer” for each enzyme variant (). To better understand the consequences of buffer composition on the evaluation of inhibitors against different enzyme variants, the catalytic properties of each of the variants and their inhibition by the selected compounds was determined in both the reference and optimized buffers.
Effect of buffer conditions on catalytic parameters for NS3 variants
The catalytic parameters, kcat, Km and kcat/Km, for the different variants were determined in both the optimized and the reference buffers (). The catalytic efficiency, kcat/Km, was highest in the optimized buffer for most of the variants. In general, the increased catalytic efficiency can be attributed to both an increased kcat and a decreased Km. The increase in catalytic efficiency was as high as 14-fold for , although it was only 4-fold higher for the full-length variant from the same genotype. There was no significant effect on the catalytic efficiency upon a change in the buffer preference for the full-length enzyme from genotype 1a upon a R155K substitution, as can be expected since this residue is far away from the catalytic triad and is only involved in interactions with the substrate and not NS4A.
Table 2. Catalytic parameters of NS3 protease variants in reference and optimized buffers.
For almost all variants, kcat was approximately 2-fold higher in the optimized buffer compared to the reference buffer. The largest kcat difference was seen for , for which the kcat was more than 3-fold higher. Similarly, the Km was lower in the optimized buffer than in the reference buffer for almost all variants, except for the
mutant, for which the Km was almost 3-fold higher. For the
,
,
and
, the Km was only slightly decreased or unaffected in the optimized buffer.
In order to understand the potential role of the cofactor on the catalytic properties of NS3, the influence of NS4Apep on the catalytic constants of the NS3fl/4 A co-constructs from genotype 1a and 1b were examined by adding the peptide to the reference buffer while keeping the other buffer conditions constant. The kcat for NS3fl/4A1a and NS3fl/4A1b increased more than 2-fold when peptide was added. Km decreased 3-fold for both variants and consequently the catalytic efficiency increased six times.
Determination of inhibition constants
The inhibition constant, Ki, for the four inhibitors was determined for all enzyme variants using the optimized and reference buffer conditions (). The Ki was generally lower when using the optimized buffer, except for and
, for which the Ki was increased or almost unaffected, respectively. In the case of the
variant, the Ki for all inhibitors was considerably higher than the ones for the same construct from genotypes 1a and 1b.
Table 3. Inhibition constants (Ki) for NS3 protease variants in reference and optimized buffers.
For ITMN-191, the Ki was approximately 700 times higher in the optimized buffer for the than the same construct from genotype 1a. Compared with
, the Ki for ITMN-191 was 550 times higher with
. The
and
variants were inhibited by all inhibitors with a significant lower Ki than the truncated counterparts except for the N-1725 compound where the Ki was approximately three times higher for
. Fusing the NS4A peptide to the full-length constructs decreased the Ki significantly, for N-1725 as much 35 times. The resistance associated R155K substitution resulted in a significant increase of Ki for all inhibitors.
Resistance and selectivity analysis
Vitality values were used for analysis of the differences in resistance and selectivity of different enzyme variants towards the inhibitors. Since this metric account for the significant differences in their catalytic properties it provides a measure of the relative selective advantage between a standard/reference variant (typically the wild-type enzyme) and a mutant or homologueCitation26. In the current analysis, the data for measured under optimized conditions was used as the reference. The vitality values in show that all the inhibitors were less effective against the variant with a single mutation associated with resistance
(vitality values > 1). More significantly, the data also reveal that they are relatively more potent on the genotype 1a enzyme, with the genotype 1b and 3a enzymes being less compromised by the inhibition.
Table 4. Vitality values for the four inhibitors in this study under optimized conditions, where is used as reference.
Discussion
The large natural strain variations and rapid resistance development represent major challenges in the discovery of broad spectrum HCV drugs. It has long been a standard procedure for HCV protease inhibitor discovery to use truncated NS3, corresponding to the protease domain alone and genotype 1b isolates. These have been preferred because of their higher stability and ease of handling, compared to the full-length protein and enzyme from other strains. By extending the analysis to a panel of enzyme variants, it has later been a possibility to get a basic idea of inhibitor selectivity and resistance. But a meaningful evaluation must account also for differences in the catalytic properties of enzyme variants. The relevance of this for HCV protease inhibitor discovery has not been fully recognized or rigorously studied earlier. Here, we have not only addressed the significance of using relevant model systems, but also the importance of optimizing the experimental conditions for each enzyme variant and accounting for their catalytic differences.
Despite considerable experimental difficulties, our previous studies of NS3 protease have all been performed with a full-length construct from the less commonly studied genotype 1a and in the presence of a peptide corresponding to the central region of NS4A. In order to better understand the selectivity of different inhibitors, we have here also evaluated the characteristics of the protease from genotype 1b and 3a isolates. Furthermore, for the genotype 1a and 1b enzymes, three different variants were compared in order to understand the effect of the helicase domain and the NS4A cofactor protein. The latter was an extended construct with full-length NS4A attached to the C-terminus of NS3, as in the native polyprotein. Although it is cleaved off via auto proteolysis during preparation and handling, it remains in complex with the NS3 protein.
A comparative analysis of the relevance of using a full-length construct of the target was performed using genotypes 1a and 1b. It revealed that the truncated NS3 genotype 1a corresponding to the protease domain had similar catalytic properties as the full-length NS3 protein, once optimized conditions were identified. In contrast, there was a major difference in the catalytic properties for the full-length and truncated version of the genotype 1b enzyme, indicating that the proteolytic activity is more dependent on the helicase domain for genotype 1b than 1a. Moreover, the full-length NS3–NS4A co-construct had similar catalytic properties as the full-length NS3 in complex with only the central, activating part of NS4A, but the buffer conditions needed to be changed for the catalytic efficiency to reach the same level. This may be associated with the structural role that the rather hydrophobic NS4A has on the NS3 protein and that there is a difference when co-expressing the two proteins rather than adding the NS4A peptide separately in the assay. It could also be linked to our previous finding that the NS4A peptide is able to activate the enzyme, demonstrating the effect of NS4A on the structural dynamics of the enzyme, and may be related to more recent modelling studies identifying important structural features of the interactionCitation27,Citation28.
In the selectivity and resistance studies, where the inhibitor sensitivity of different NS3 protease variants was compared, it was immediately evident that the experimental conditions influenced the measured enzymatic properties to a degree that was problematic. It added a dimension to previous studies by us and others which have shown that the buffer composition has a major influence on the proteolytic activity of NS3. For example, a different dependency on ionic strength and glycerol concentration of the protease variants and characteristics of the full-length and truncated NS3 from genotype 1aCitation13,Citation21,Citation27,Citation29,Citation30. In the case of NS3, there were also rather large differences in expression levels and protein stability, influencing the active site concentration in a preparation. The underlying reasons for these effects are elusive since the NS3 protein is active as a complex and undergoes dynamic structural changes as part of its catalytic cycle. These environmental factors can involve a number of effects, including correct folding of improperly folded regions, increasing the affinities of the catalytically relevant interactions or simply positioning of catalytic residues.
Although it is well known that buffer conditions have a major impact on enzyme function, the optimal buffer for a certain enzyme is not trivial to define. In addition, the buffer resulting in the highest activity is not always optimal from a drug discovery perspective since other conditions may be more relevant for evaluation of the efficacy of inhibitors, as we have recently shown for evaluation of β-secretase inhibitorsCitation31. An additional difficulty arises when different variants of the same enzyme are to be studied since they may require different conditions for maximal activity. It is not obvious which conditions should be chosen for a relevant and informative comparative study. The buffer effects do not only have practical implication for the possibility of identifying an ideal buffer composition, but is also a more theoretical question. From a simple biochemical perspective, it can be assumed that each enzyme should be studied under conditions where it has the highest activity, while from a biological perspective it is more reasonable to use the same physiologically relevant conditions. The problem arises when not all enzyme variants are active under the same conditions. It is not simply a matter of increasing the enzyme concentration to overcome low activity since the amount of active protein should be as low as possible is required in order to get valid results.
The evaluation of the extensive exploration of experimental conditions for characterization of NS3 protease inhibitors showed that there were larger effects in the sensitivities for inhibitors than in the catalytic properties of the enzyme variants. This demonstrates that the structural features influencing the different steps of catalysis are different to those for inhibition, which are more complex than simply the bi-molecular interaction between the protein and the inhibitor which can be measured by other methods, e.g. surface plasmon resonance biosensor technologyCitation32. The chemodynamic analysis of these processes can reveal the dominating forces involved in inhibitor interactions with a target.
From an inhibitor comparison perspective, it revealed that all inhibitors are most effective against the wild-type 1a strain, if one accounts for the change in catalytic efficiency. This is the most evident for BILN-2061 and ITMN-191. These results can partly be validated by data from a replicon-based assayCitation14,Citation16,Citation33. Our data however suggests that VX-950 is slightly more selective for the 1a genotype as compared to the 1b genotype. This does not correlate with cell-based replicon data, for which the opposite is seen. This might be explained by the fact that this inhibitor is mechanism-based and that vitality values are determined from apparent Ki-values. To fully understand the inhibitor properties, it is therefore suggested to use alternative strategies for interaction analyses.
From a pragmatic drug discovery perspective, it may be sufficient to identify a model system and a set of conditions that identify potential lead compounds and then access the information required for lead optimization via other assays, e.g. using structural methods. In the case of HCV drug discovery, the availability of the replicon system makes it possible to relatively early on evaluate inhibitors in a physiological context. But, as for all cell-based systems, there are additional complexities that make interpretations more difficult. In addition, the system is not useful for analysis of weak inhibitors and therefore excludes the possibility of a fragment-based approach. A goal with the current analysis is therefore to identify biochemical procedures that correlate well with antiviral effect in a physiological system.
Conclusions
The current study has addressed one of the major challenges for HCV drug discovery, namely, the large natural genetic variation of the virus and the rapid evolution of resistant mutants. Although the analysis established that BILN-2061 and ITMN-191 were the most potent inhibitors against all enzyme variants and that the four inhibitors were most efficient in inhibiting full-length genotype 1a NS3 and less effective for the R155K mutant, from a perspective of having broad selectivity and low propensity for resistance, VX-950 was clearly superior to the other inhibitors. This study has thus provided a simple strategy for biochemical evaluation and comparison of inhibitors against multiple enzyme variants, here corresponding to different genotypes and resistance mutations. It is an alternative to the rather costly and time consuming cell-based replication and entry systems like replicons and cell culture producing virions that provide a means to study the viral life cycle and evaluate a wide range of therapeutic targets of HCV. The current approach is expected to be a good starting point for similar optimization of direct binding assays although these are not influenced by differences in catalytic activity. Such assays need to account for differences in basic functional characteristics in order to provide relevant kinetic and thermodynamic data, complementing the structural data required for design and optimization of lead compounds.
Declaration of interest
This project was supported by the Swedish Research Council (VR). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Supplementary materials online only - For review only at proofing stage
Supplementary Figures S1 and Table S1
Supplementary Material
Download PDF (52.7 KB)Acknowledgements
The clones for and NS3fl/4A1b were obtained from Raffaele De Francesco (National Institute in Molecular Genetics -- INGM, Milan, Italy). The NS4Apep was a kind gift from Gunnar Lindeberg, the Department of Organic Pharmaceutical Chemistry, Uppsala University, Uppsala, Sweden.
References
- Klenerman P, Gupta PK. Hepatitis C virus: current concepts and future challenges. QJM 2012;105:29–32
- WHO. Hepatitis C, 2012. Fact sheet N°164
- Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005;42:962–73
- Tse MT, Kirkpatrick P. 2012 in reflection. Nat Rev Drug Discov 2012;12:8–10
- Morikawa K, Lange CM, Gouttenoire J, et al. Nonstructural protein 3-4 A: the Swiss army knife of hepatitis C virus. J Viral Hepat 2011;18:305–15
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol 2007;5:453–63
- Raney KD, Sharma SD, Moustafa IM, Cameron CE. Hepatitis C virus non-structural protein 3 (HCV NS3): a multifunctional antiviral target. J Biol Chem 2010;285:22725–31
- Kim JL, Morgenstern KA, Lin C, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996;87:343–55
- Dahl G, Sandström A, Åkerblom E, Danielson UH. Effects on protease inhibition by modifying of helicase residues in hepatitis C virus nonstructural protein 3. FEBS J 2007;274:5979–86
- Beran RK, Lindenbach BD, Pyle AM. The NS4A protein of hepatitis C virus promotes RNA-coupled ATP hydrolysis by the NS3 helicase. J Virol 2009;83:3268–75
- Xue W, Wang M, Jin X, et al. Understanding the structural and energetic basis of inhibitor and substrate bound to the full-length NS3/4 A: insights from molecular dynamics simulation, binding free energy calculation and network analysis. Mol Biosyst 2012;8:2753–65
- Saalau-Bethell SM, Woodhead AJ, Chessari G, et al. Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nat Chem Biol 2012;8:920–5
- Ingallinella P, Altamura S, Bianchi E, et al. Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry 1998;37:8906–14
- Lamarre D, Anderson PC, Bailey M, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003;426:186–9
- Hinrichsen H, Benhamou Y, Wedemeyer H, et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 2004;127:1347–55
- Seiwert SD, Andrews SW, Jiang Y, et al. Preclinical characteristics of the hepatitis C virus NS3/4 A protease inhibitor ITMN-191 (R7227). Antimicrob Agents Chemother 2008;52:4432–41
- Rajagopalan R, Misialek S, Stevens SK, et al. Inhibition and binding kinetics of the hepatitis C virus NS3 protease inhibitor ITMN-191 reveals tight binding and slow dissociative behavior. Biochemistry 2009;48:2559–68
- Tong J, Wang Y-w, Lu Y-a. New developments in small molecular compounds for anti-hepatitis C virus (HCV) therapy. J Zhejiang Univ Sci B 2012;13:56–82
- Lin C, Lin K, Luong Y-P, et al. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J Biol Chem 2004;279:17508–14
- Mullard A. 2011 FDA drug approvals. Nat Rev Drug Discov 2012;11:91–4
- Poliakov A, Hubatsch I, Shuman CF, et al. Expression and purification of recombinant full-length NS3 protease-helicase from a new variant of Hepatitis C virus. Protein Expr Purif 2002;25:363–71
- Dahl G, Sandström A, Åkerblom E, Danielson UH. Resistance profiling of hepatitis C virus protease inhibitors using full-length NS3. Antivir Ther 2007;12:733–40
- Gallinari P, Brennan D, Nardi C, et al. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J Virol 1998;72:6758–69
- Gallinari P, Paolini C, Brennan D, et al. Modulation of hepatitis C virus NS3 protease and helicase activities through the interaction with NS4A. Biochemistry 1999;38:5620–32
- Liu Y, Kati W, Chen CM, et al. Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal Biochem 1999;267:331–5
- Gulnik SV, Suvorov LI, Liu B, et al. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 1995;34:9282–7
- Dahl G, Arenas OG, Danielson UH. Hepatitis C virus NS3 protease is activated by low concentrations of protease inhibitors. Biochemistry 2009;48:11592–602
- Zhu H, Briggs JM. Mechanistic role of NS4A and substrate in the activation of HCV NS3 protease. Proteins 2011;79:2428–43
- Misialek S, Rajagopalan R, Stevens SK, et al. Optimization of the multiple enzymatic activities of the hepatitis C virus NS3 protein. Analytical Biochem 2009;394:138–40
- Urbani A, Biasiol G, Brunetti M, et al. Multiple determinants influence complex formation of the hepatitis C virus NS3 protease domain with its NS4A cofactor peptide. Biochemistry 1999;38:5206–15
- Dominguez JL, Christopeit T, Villaverde MC, et al. Effect of the protonation state of the titratable residues on the inhibitor affinity to BACE-1. Biochemistry 2010;49:7255–63
- Geitmann M, Dahl G, Danielson UH. Mechanistic and kinetic characterization of hepatitis C virus NS3 protein interactions with NS4A and protease inhibitors. J Mol Recogn 2011;24:60–70
- Romano KP, Ali A, Aydin C, et al. The molecular basis of drug resistance against hepatitis C virus NS3/4 A protease inhibitors. PLoS Pathol 2012;8:e1002832