
Abstract
A series of benzenesulfonamides incorporating aroylhydrazone, piperidinyl, sulfone, [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenyl-methylene)-1,3,4-thiadiazol-3(2H)-yl moieties was investigated as inhibitors of four α-carbonic anhydrases (CAs, EC 4.2.1.1), the human (h) isoforms hCA I, II (cytosolic, offtarget enzymes) and hCA IX and XII (transmembrane, tumor-associated isoforms). Low nanomolar activity was observed against hCA II (KIs of 0.56–17.1 nM) with these sulfonamides, whereas the slow cytosolic isoform hCA I was less inhibited by these compounds (KIs of 86.4 nM–32.8 µM). Most of these sulfonamides significantly inhibited CA IX, with KIs in the range of 4.5–47.0 nM, although some of the derivatives incorporating bulkier bicyclic moieties, as well as 2-thienyl fragments, showed a weaker activity against this isoform (KIs in the range 50.1–553 nM). All the investigated compounds also inhibited CA XII with KIs in the range 0.85–376 nM. The best inhibitors were those incorporating bulky [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl moieties and 1,3,4-thiadiazol-3(2H)-yl groups.
Introduction
The primary sulfonamides, RSO2NH2, are the classical inhibitors of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation1–4. They have been widely used for almost 60 years as diuretic or systemically acting antiglaucoma drugs, since the introduction of acetazolamide (AAZ) in clinical use in 1954Citation5–9. However, AAZ and the other clinically used sulfonamides/sulfamate CA inhibitors (CAIs), such as methazolamide and ethoxzolamideCitation1,Citation4,Citation6, target all mammalian CA isoforms (16 of them are known to date in vertebrates)Citation1,Citation8–11, and as thus, they show a range of undesired side effectsCitation1,Citation4,Citation6,Citation11,Citation12, motivating the continuous search of novel such agents with a selective inhibition profile against the desired isoform(s)Citation13–20.
These enzymes are also versatile catalysts of other hydrolytic reactions except the hydration of CO2 to bicarbonate and protons, being esterases with a range of carboxylic acid esters, phosphate and sulfate estersCitation1,Citation3,Citation13,Citation14.
Recently, a large of number of sulfonamides and some of their derivatives started to be obtained which showed a good selectivity level for the inhibition of various CA isoformsCitation4,Citation15–28, some of which are cytosolic (e.g. CA I, II, III, VII and XIII, as well as the acatalytic isoforms CA VIII, X and XI)Citation1,Citation3,Citation6,Citation21, membrane associated/transmembrane (CA IV, IX, XII, XIV and XV)Citation1,Citation2,Citation8,Citation25, mitochondrial (CA VA and VB)Citation1,Citation4, 17 or secreted (CA VI)Citation1,Citation4,Citation15,Citation20.
The tumor associated isoform CA IX is highly overexpressed in many cancer types by the hypoxia inducible factor-1α (HIF-1α) cascadeCitation1–4. Around 70% of the hypoxic tumors overexpress CA IX and show a bad response to classical chemo- and radiotherapiesCitation2. CA IX was shown to significantly contribute to the extracellular acidification of the tumor environment, by means of the protons resulted from the catalysis of carbon dioxide hydrationCitation1–4. This leads to the acquisition of metastasic phenotypes and chemoresistance towards many anticancer drugs. Ultimately, targeted structure-based drug design campaigns of CAIs against this novel target led to a range of interesting derivatives with a significant activity, selectivity and promising in vivo action against several types of tumorsCitation29,Citation30. CA XII is also a tumor-associated CA and its inhibition was also shown to lead to anticancer effectsCitation31.
Continuing our interest in the design of CA IX and XII inhibitors of the sulfonamide type, here we report that a series of benzenesulfonamides incorporating aroylhydrazone, [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenyl-methylene)-1,3,4-thiadiazol-3(2H)-yl moieties, reported earlier by us for the inhibition of bacterial enzymes from extremophilesCitation26, shows interesting inhibition profile against the two tumor-associated isoforms CA IX and XII.
Materials and methods
Chemistry
Compounds 3–10 investigated here were reported in a previous study from our groupCitation26.
Enzymology
hCA I, II, IX and XII were recombinant enzymes obtained in-house as described earlierCitation1–7.
CA catalytic and inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation27. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM HEPES (pH 8.4) and 20 mM NaBF4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver–Burk plots, as reported earlierCitation12–17,Citation28, and represent the mean from at least three different determinations.
Results and discussion
Chemistry
Sulfonamides investigated here were obtained from oxo-N′-(4-sulfamoylphenyl)propanehydrazonoyl chloride (1)Citation26 which was reacted with aroylhydrazines 2 to form the key intermediates of type 3 (Scheme 1). The 2-(2-arylhydrazono)-N′-(4-sulfamoylphenyl)propanehydrazonoyl chlorides 3a–d were then reacted with piperidine in ethanol led to the formation of piperidine derivatives 4a–d or with sodium benzenesulfinate to afford the corresponding sulfones 5a–d, respectively (Scheme 1)Citation26.
Scheme 1. Synthesis of compounds 3a–d, 4a–d and 5a–d.
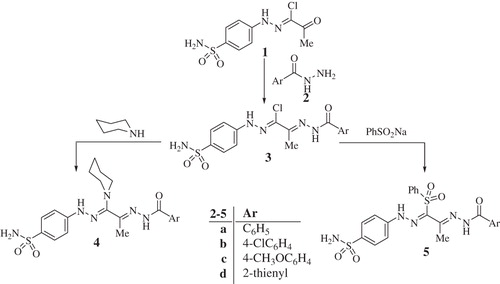
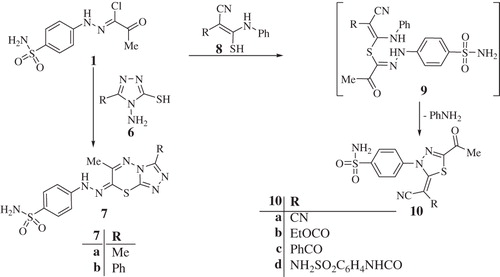
Alternatively, treatment of 1 with 4-amino-5-(methyl/phenyl)-4H-1,2,4-triazole-3-thiol 6a, b gave 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines 7a, b (Scheme 2). In another approach, 1 was reacted with thioanilide derivatives 8a–d in the presence of triethylamine, which afforded the 1,3,4-thiadiazoles 10a–d, as depicted in Scheme 2.
Scheme 2. Synthesis of compounds 7a, b and 10a–d.
All these compounds have been characterized by physico-chemical and spectral techniques confirming the proposed structures (see ref.Citation26 for details).
CA inhibition
The inhibition studies against four mammalian CA isoforms of this series of benzenesulfonamides containing the interesting tails of the [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenyl-methylene)-1,3,4-thiadiazol-3(2H)-yl type (3–10), i.e. hCA I, II, IX and XII are shown in .
Table 1. Inhibition data against the human isoforms hCA I, II, IX and XII with sulfonamides 3–10 by a stopped-flow, CO2 hydrase assayCitation27.
As seen from data of , sulfonamides 3a–10d (and acetazolamide, 5-acetamido-1,3,4-thiadiazole-2-sulfonamide, AAZ, as standard inhibitor) showed interesting inhibitory properties against all four investigated CAs. The following structure-activity relationship (SAR) has been delineated:
The slow cytosolic isoform hCA I was moderately inhibited by sulfonamides 3a–3d, 4a–4d, 5a–5d and 7a, 7b, with inhibition constants in the range of 86.4–753 nM, and weakly inhibited by derivatives 10a–10d (KIs in the range of 6.54–32.8 µM). It is interesting to note that the Schiff’s bases 3a–3d showed a rather similar behavior as hCA I inhibitors, with a relatively small variation of the inhibition constants irrespective of the nature of moiety Ar. The strongest inhibitor in the subseries was the phenyl derivative 3a, but its substitution with 4-Cl, 4-MeO groups, or its replacement by a thienyl scaffold, led to a slight diminution of the inhibitor potency in compounds 3b–3d. For piperidines 4a–4d, the range of inhibition was already higher compared to derivatives 3, with the thienyl derivative 4d being the most active in the subseries (KI of 91.3 nM) and the 4-methoxyphenyl one 4c the least active (KI of 247 nM, ). Sulfones 5 also showed a significant variation of the inhibitory power with the nature of the Ar group, with the best inhibitor being the phenyl derivative 5a (KI of 130 nM) and the least effective one the thienyl derivative 5d (KI of 753 nM). For compounds 7, the methyl derivative 7a showed effective hCA I inhibitory properties (this was the best inhibitor of this isoform among the reported derivatives, being almost 3-fold more effective than the clinically used sulfonamide (AAZ) whereas the bulkier Ph derivative 7b was much less effective as inhibitor of this isoform. Even a stronger loss of activity was then observed for sulfonamides 10, which probably due to their more rigid scaffold (the thiadiazole ring is directly connected to the benzene one in these compounds) can be less well-accommodated within the restricted space of the hCA I active siteCitation28.
All new compounds reported here were excellent inhibitors of the physiologically dominant (in humans) isoform hCA II. Indeed, only 3a showed an inhibition constant of 17.1 nM (comparable to that of acetazolamide, 12.1 nM) whereas all the remaining derivatives were sub-nanomolar or low nanomolar hCA II inhibitors, with KIs in the range of 0.55–8.6 nM. The SAR is thus almost impossible to delineate as all these substitution patterns seem to be highly favorable for obtaining tight-binding hCA II inhibitors. Among the best such compounds were the Schiff’s base 3d (incorporating the thienyl moiety), the piperidine 4b (incorporating a 4-chlorophenyl moiety), the sulfones 5b and 5c, incorporating 4-Cl- and 4-MeO-phenyl moieties, the bulky phenyl-substituted 7b as well as the 1,3,4-thiadiazoles 10c and 10d. All these compounds showed inhibition constants <1 nM, being among the most effective hCA II inhibitors reported up until now.
Against hCA IX the derivatives 3–10 showed with KIs in the range of 4.5–553 nM. The most ineffective inhibitors of this isoform were 3d and 4d (with KIs of 354–553 nM), which both incorporate 2-thienyl fragment at the end of the bulky tail derivatizing the sulfanilamide scaffold. It is interesting to note that other chloro derivatives 3 or piperidines 4, incorporating a different substitution patterns (e.g. in 3a–3c or 4a–4c) were quite effective (3a and 3c) or medium potency (3b, 4a–4c) hCA IX inhibitors (the last group of compounds showed with KIs in the range of 16.8–63.0 nM, ). Interestingly, irrespective of the substitution pattern, all the sulfones 5 and the [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl-substituted derivatives 7 were medium potency hCA IX inhibitors, with KIs in the range of 42.7–85.1 nM. The same is true for 10d, incorporating two sulfamoyl functionalities but on a quite bulky scaffold (KI of 50.1 nM), whereas the remaining 2-(cyanophenyl-methylene)-1,3,4-thiadiazol-3(2H)-yl derivatives 10a–10c, which incorporate more compact functionalities compared to 10d, were quite efficient hCA IX inhibitors, with KIs in the range of 4.5–8.9 nM. These compounds are more effective compared to acetazolamide AAZ as hCA IX inhibitors (together with 3a and 3c). Thus, SAR for hCA IX inhibition with compounds 3–10 is much more complicated compared to what we observed for the inhibition of the cytosolic isoforms hCA I and II discussed earlier.
The second transmembrane isoforms, hCA XII; was also inhibited by sulfonamides 3–10 investigated here, with KIs in the range of 0.85–376 nM (). As for hCA IX discussed earlier, the least effective hCA XII inhibitors were the same two compounds (3d and 4d) incorporating the 2-thienyl tail (KIs of 207–376 nM). Medium potency inhibition, with KIs in the range of 11.7–41.2 nM was observed for aroylhydrazones 3a–3c, piperidines 4a and 4c as well as sulfones 5a–5c. The remaining derivatives, i.e. 4b, 5d, 7a, b and 10a–10d, were highly effective hCA XII inhibitors with KIs in the range of 0.85–8.5 nM (). The best and only subnanomolar hCA XII inhibitor was 7b (KI of 0.85 nM). Many of the investigated compounds were more effective or similar to acetazolamide for the inhibition of this isoform.
The inhibition profile of the four subclasses of derivatives investigated here, which all carry the sulfanilamide head group, but highly diverse tail moieties, was very different and specific for each of them. For example derivatives 7 were highly effective as hCA II and XII inhibitors, medium potency hCA IX inhibitors and rather weak hCA I inhibitors. Although no highly hCA IX/XII – selective compounds were detected in this study, the inhibition profiles of these derivatives are of great interest considering the many applications that CAIs possess in various pharmacological fields, for obtaining diureticsCitation32, antiepilepticsCitation33, antiobesityCitation34 and antiglaucomaCitation35,Citation36 agents.
Conclusions
We investigated a series of recently reported benzenesulfonamides, incorporating aroylhydrazone, [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenyl-methylene)-1,3,4-thiadiazol-3(2H)-yl moieties as inhibitors of four α-CAs, the isoforms hCA I, II (cytosolic, offtarget enzymes) and hCA IX and XII (transmembrane, tumor-associated isoforms). Low nanomolar activity was observed against hCA II (KIs of 0.56–17.1 nM) with these sulfonamides, whereas the slow cytosolic isoform hCA I was less inhibited by these compounds (KIs of 86.4 nM–32.8 µM). Most of the sulfonamides investigated here also significantly inhibited CA IX, with KIs in the range of 4.5–47.0 nM, although some of the derivatives incorporating bulkier bicyclic moieties as well as thienyl fragments, showed a weaker activity against this isoform (KIs in the range 50.1–553 nM). All the investigated compounds also inhibited CA XII with KIs in the range 0.85–376 nM. The best inhibitors were those incorporating bulky [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl moieties and 1,3,4-thiadiazol-3(2H)-yl groups. Although no hCA IX/XII – selective compounds were detected in this study, the inhibition profiles of these derivatives are of great interest considering the many applications of CAIs for obtaining diuretics, antiepileptics, antiobesity and antiglaucoma agents.
Declaration of interest
This research was financed by a 7th FP EU grant, METOXIA (to C.T.S.) and by the National Plan of Science, Technology and Innovation (Grant No. 10-MED1188-02), King Saud University, Riyadh and by the Graduate Studies and Scientific Research Agency, Salman bin Abdulaziz University, Grant No. 2.H.33, Alkharj, Saudi Arabia (to A.M.A).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34. doi: 10.3389/fphar.2011.00034
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Supuran CT. Carbonic anhydrase inhibition with natural products: novel chemotypes and inhibition mechanisms. Mol Divers 2011;15:305–16
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- Kolayli S, Karahalil F, Sahin H, et al. Characterization and inhibition studies of an alpha-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J Enzyme Inhib Med Chem 2011;26:895–900
- Alp C, Ozsoy S, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;27:818–24
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Kazancıoğlu EA, Güney M, Sentürk M, Supuran CT. Simple methanesulfonates are hydrolyzed by the sulfatase carbonic anhydrase activity. J Enzyme Inhib Med Chem 2012;27:880–5
- Cavdar H, Ekinci D, Talaz O, et al. α-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–54
- Ekinci D, Kurbanoglu NI, Salamcı E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8
- Ekinci D, Al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7
- Singh S, Supuran CT. QSARs on human carbonic anhydrase VA and VB inhibitors of some new not yet synthesized, substituted aromatic/heterocyclic sulphonamides as anti-obesity agent. J Enzyme Inhib Med Chem 2012;27:666–72
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47
- Sahin H, Can Z, Yildiz O, et al. Inhibition of carbonic anhydrase isozymes I and II with natural products extracted from plants, mushrooms and honey. J Enzyme Inhib Med Chem 2012;27:395–402
- Sentürk M, Ekinci D, Göksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2012;27:365–9
- Bootorabi F, Jänis J, Hytönen VP, et al. Acetaldehyde-derived modifications on cytosolic human carbonic anhydrases. J Enzyme Inhib Med Chem 2011;26:862–70
- Chohan ZH, Shad HA, Supuran CT. Synthesis, characterization and biological studies of sulfonamide Schiff’s bases and some of their metal derivatives. J Enzyme Inhib Med Chem 2012;27:58–68
- Ozensoy O, Arslan M, Supuran CT. Carbonic anhydrase inhibitors: purification and inhibition studies of pigeon (Columba livia var. domestica) red blood cell carbonic anhydrase with sulfonamides. J Enzyme Inhib Med Chem 2011;26:749–53
- Sahin H, Aliyazicioglu R, Yildiz O, et al. Honey, pollen, and propolis extracts show potent inhibitory activity against the zinc metalloenzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2011;26:440–4
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Alafeefy AM, Abdel-Aziz HA, Vullo D, et al. Inhibition of carbonic anhydrases from the extremophilic bacteria Sulfurihydrogenibium yellostonense (SspCA) and S. azorense (SazCA) with a new series of sulfonamides incorporating aroylhydrazone-, [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenylmethylene)-1,3,4-thiadiazol-3(2H)-yl moieties. Bioorg Med Chem 2014;22:141–7
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20
- Dubois L, Peeters SG, van Kuijk SJ, et al. Targeting carbonic anhydrase IX by nitroimidazole based sulfamides enhances the therapeutic effect of tumor irradiation: a new concept of dual targeting drugs. Radiother Oncol 2013;108:523–8
- Said HM, Hagemann C, Carta F, et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg Med Chem 2013;21:3949–57
- Lounnas N, Rosilio C, Nebout M, et al. Pharmacological inhibition of carbonic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas. Cancer Lett 2013;333:76–88
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91
- Supuran CT, Di Fiore A, De Simone G. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2008;13:383–92
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16
- Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin Ther Pat 2012;22:79–88