
Abstract
The protozoan parasite Trypanosoma cruzi is the agent responsible for trypanosomiasis (Chagas disease) in humans and other animals. It has been recently reported that this pathogen encodes for an α-class carbonic anhydrase (CA, EC 4.2.1.1), denominated TcCA, which was shown to be crucial for its life cycle. Inhibition studies of a class of 4-oxoquinazoline containing a benzensulfonamide moiety and their 4-thioxo bioisosteres against the protozoan enzyme TcCA are described here. Most of 4-oxoquinazoline sulfonamides showed nanomolar TcCA inhibition activity with KIs in the same order of magnitude of acetazolamide (AAZ), whereas their thioxo bioisosters showed moderate anti-Trypanosoma CA potency with KIs in the micromolar range. The discovery of compounds incorporating a 4-oxoquinazoline ring as a low-nanomolar TcCA inhibitor is quite promising and it may be useful for developing anti-Trypanosoma agents with a novel mechanism of action compared to the clinically used drugs (such as benznidazole, nifurtimox) for which significant resistance and serious adverse effects due to their high-toxicity appeared.
Introduction
American trypanosomiasis or Chagas disease affects millions of people mostly in South and Central Americas, but nowadays it spreads also in Europe and North AmericaCitation1, being caused by the protozoan pathogen Trypanosoma cruzi. The only two clinically used drugs for its treatment are nitroazoles, such as benznidazole and nifurtimox. Their mechanism of action most probably involves the formation of free radicals from the nitro groups of the drugs towards which the parasite results to be particularly sensitive and consequently reduction of detoxification capabilities of T. cruziCitation2. However, since these two drugs are not always effective to abolish the chronic infectionCitation3, new anti-infective drugs started to be explored ultimatelyCitation1.
Our groups recently reported that an α-CA with very high-catalytic activity is present in this pathogenic protozoanCitation2. This enzyme, denominated TcCA, was cloned and characterizedCitation2, and further investigations showed that this enzyme may be inhibited by sulfonamides and thiolsCitation2,Citation4, two of the main classes of CA inhibitors (CAIs)Citation5–10 as well as a wide range of inorganic anionsCitation6. Interestingly, most aromatic/heterocyclic sulfonamides as well as some 5-mercapto-1,3,4-thiadiazoles investigated earlier were not highly effective as TcCA inhibitors in vitroCitation2,Citation11. However, some of these thiols, incorporating heterocyclic scaffolds, were among the most potent in vitro TcCA inhibitors (KIs of 21.1–79.0 nM) and some of them also showed anti-trypanosomal activity in vivo, inhibiting the epimastigotes growth of two T. cruzi strainsCitation2.
The inhibition studies of TcCA by inorganic and complex anions and other molecules interacting with zinc proteins, such as sulfamide, sulfamic acid, phenylboronic/arsonic acids, have also been reportedCitation6. It showed that the protozoan enzyme TcCA was inhibited in the low-micromolar range by iodide, cyanate, thiocyanate, hydrogensulfide and trithiocarbonate as well as by diethyldithiocarbamate (KI = 5 μM). Therefore, protozoan CA inhibition may be a valid strategy to control infection such as Chagas disease mainly caused by a pathogen agent like the protozoa T. cruzi.
In this study, we reported the inhibition studies of new classes of quinazoline derivatives which were designed and then synthesised as potential selective TcCA inhibitors.
Materials and methods
Chemistry
Melting points (°C) were determined in open capillaries on a Gallenkamp melting point apparatus (Sanyo Gallenkamp, Southborough, UK) and were uncorrected. Precoated silica gel plates (silica gel 0.25 mm, 60G F254; Merck, Germany) were used for thin layer chromatography, and the dichloromethane/methanol (9.5:0.5) mixture was used as a developing solvent system. Iodine was used for staining together with ultraviolet light. Infrared spectra were recorded in KBr discs using IR-470 Shimadzu spectrometer (Shimadzu, Tokyo, Japan). 1H-NMR spectra were recorded on Bruker AC-400 Ultra Shield NMR spectrometer (Bruker, Flawil, Switzerland, δ ppm) at 300 MHz for 1H and 75 MHz for 13C, using TMS as internal standard, and peak multiplicities are designed as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Electron impact mass spectra were recorded on a Shimadzu GC-MS-QP 5000 instrument (Shimadzu, Tokyo, Japan). Elemental analyses were performed, on Carlo Erba 1108 Elemental Analyzer (Heraeus, Hanau, Germany), at the Micro-analytical Unit, Faculty of Science, Cairo University, Cairo, Egypt, and the results found were within ±0.4% of the theoretical values.
The synthesis and the full characterization of the compounds investigated here (8a–h and 9a–e) as CA inhibitors are reported elsewhereCitation12.
CA inhibition studies
An applied photophysics stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activityCitation13. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.4) and 20 mM NaBF4 (for maintaining constant ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled–deionized water, and dilutions up to 0.01 nM were done thereafter with distilled–deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver-Burk plots, as reported earlierCitation14–16 represent the mean from at least three different determinations. All CAs were recombinant proteins obtained as reported earlier by these groupsCitation2,Citation14–16.
Results and discussion
Chemistry
The present findings extend our previous studies which reported the synthesis of a new series of 2,3,5,6- and/or 8-substituted-4-oxoquinazoline derivatives and their bioisosters 4-thioxoquinazolineCitation12, presenting here the inhibition studies of these compounds against the α-CA from T. cruzi (TcCA). The approach to prepare the target potentially bioactive quinazoline derivatives 8a–h and thioxoquinazolin-4-one analogues 9a–e was via a generalized route described in The starting materials, thioxoquinazoline derivatives 3a–h and their 2,4-dithioxo isosteres 4a–e, were prepared according to reported literature procedureCitation17,Citation18. The reaction of the anthranilic acid derivatives appropriate 3,5 or 6 substituted with the selected isothiocyanates in boiling ethanol containing catalytic amount of triethylamine afforded the key intermediates 3,5,6 and/or 8 substituted-2-thio-4-oxoquinazoline and 2-thio-4-thioxoquinazoline derivatives 3a–h and 4a–e, which were reacted with 2-chloro-N-(4-sulfamoyl-phenyl)-acetamide to give the new sulfonamide derivatives containing quinazoline ring 8a–h and their isosteres 9a–e, respectively (). These isothiocyanates were chosen in such a way as to contain moieties which were shown earlier to lead to interesting CA inhibitory compounds in the quinazoline-containing sulfonamide derivativesCitation19,Citation20.
Scheme 1. Synthesis of the 2,3,5,6 and/or 8-substituted-4-oxoquinazoline derivatives 8a–h and their bioisosters 4-thioxoquinazoline derivatives 9a–e.
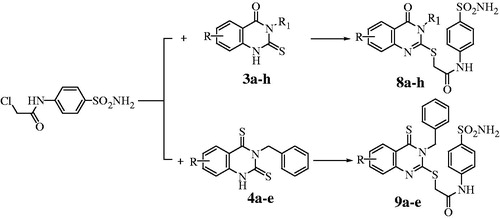
In order to obtain compounds with higher biological activityCitation21–29, it has been thought that the oxoquinazoline derivatives could be converted to their thioxo isosteres. Therefore, the conversion of the thioxoquinazolin-4-one 3a–e derivatives to their corresponding di-thioxo 4a–e isosteres was obtained by the treatment of compounds 3a–e with phosphorus pentasulfide in dry pyridine. 2-Chloro-N-(4-sulfamoylphenyl)-acetamide was prepared by acylation of sulfanilamide at room temperature under basic condition.
The new compounds and their intermediates were characterized by elemental analyses, IR, 1H- and 13C-NMR as well as by mass spectroscopy (see Experimental Protocols for detailsCitation12).
CA inhibition
The inhibition studies of a new quinazolin-4-one sulfonamide series (8a–h) and their 4-thioxo bioisosteres (9a–e), as well as acetazolamide (AAZ, 5-acetamido-1,3,4-thiadiazole-2-sulfonamide, a clinically used drug) against the human (h) CA isoforms hCA I, II, and the protozoan enzyme TcCA are reported in . Data for the selectivity ratios of the dominant and physiologically relevant human carbonic anhydrase isoforms (hCA I and II) over the TcCA enzyme with these compounds are also included in , as they may be the main off-targets in case they are very efficient TcCA inhibitors. The following structure–activity relationship (SAR) for TcCA inhibition with the new group of quinazoline-sulfonamide derivatives 8a–h and 9a–e reported here can be observed from .
(i) The cytosolic slow isoform hCA I was generally poorly inhibited by the new quinazoline derivatives and their bioisosteres reported here with inhibition constants in the range of 674–5432 nM. However, some derivatives belonging to the 4-oxoquinazoline series such as 8a–c showed to be high/medium potent inhibitors against this human isoform, with KIs of 86.5–119 nM ().
(ii) Against the physiologically dominant human isoform hCA II, the derivatives 8a–c were very potent inhibitors, with KIs in the range of 1.3–1.7 nM. Indeed, they showed to have better inhibition activity than the clinically used acetazolamide AAZ (KI of 12 nM). The derivative 8g showed KI against this cytosolic human isoform of the same order of magnitude as acetazolamide (12.1 nM). Most of the 4-thioxoquinazoline derivatives, such as 9a–b, 9d and 9e, were medium potency hCA II inhibitors (KIs in the range of 44.0–91.9 nM). The remaining compounds reported here such as 8d–8f, 8h and 9c showed ineffective hCA II inhibitory activity, with KIs in the range of 114.2–233.8 nM ().
(iii) As reported in , acetazolamide (AAZ) acts as a medium-potency CA inhibitor against the protozoan enzyme TcCA (KI of 61.6 nM)Citation2. In the same way, the new sulphonamides belonging to the 4-oxoquinazoline series 8 reported here generally acted as medium potent inhibitors of TcCA, with KIs of the same order of magnitude as acetazolamide in the range of 43.3–68.4 nM (except for 8f with KI equal to 272.7 nM and thus 4.4 times higher than AAZ, and 8c with KI of 6.6 nM about 10 times lower than AAZ). Only one 4-thioxoquinazoline derivative 9d with KI of 56.7 nM showed similar inhibition potency of acetazolamide.
(iv) Although most of the 4-oxoquinazoline sulphonamides reported here inhibited medium efficiently TcCA, no significant change of KI values has been observed. Therefore, SAR is almost impossible to delineate as all substitution patterns lead to highly effective inhibitors of this protozoan CA, TcCA.
(v) However, it is also interesting to notice that the best TcCA inhibitor was derivative 8c (KI of 6.6 nM), which was also a very strong inhibitor of the cytosolic isoform hCA II with KI of 1.7 nM and a medium potency inhibitor of the other cytosolic human isoform CA I with KI of 86.5 nM.
(vi) Generally, the derivatives 8a–h belonging to the 4-oxoquinazoline series are more potent TcCA inhibitors than their bioisosters 4-thioxoquinazoline derivatives 9a–9e (except for compounds 8f and 9d which showed KI of 272.7 and 56.7 nM, respectively). Indeed, the least effective TcCA inhibitors were mostly the 4-thioxoquinazoline derivatives, such as 9a–c and 9e, which showed KIs in the range of 349.2–763.5 nM being thus low-potency inhibitors of this protozoan enzyme together with the only 4-oxoquinazoline derivatives 8f with KI of 272.7 nM.
(vii) Furthermore, the 8-methyl-3-benzyl (8d) and the 6-nitro-3-allyl 4-oxoquinazoline (8g) derivatives with KI of 34.9 and 43.3 nM, respectively, were slightly more active compounds compared to the standard drug AAZ. However, keeping the nitro side chain in the same 6th position and just changing the substitution group in 3rd position from allyl to benzyl (such as in the 8h derivative) bring a small increase of KI from 43.3 to 68.4 nM (). However, due to the presence of the nitro group, these two derivatives most probably possess the same mechanism of action as TcCA inhibitors of the only two clinically available drugs, benznidazole and nifurtimox. They are indeed nitroazoles whose mechanism of action is based on the production of free radicals involving the NO2 groups of the drugs, to which T. cruzi is particularly sensitive given its reduced detoxification capabilitiesCitation2.
(viii) The remaining oxoquinazoline derivatives with either 3-allyl or 3-benzyl side chain mostly possessed very similar inhibition activity against the protozoan enzyme TcCA. However, changing position of the methoxy group from 6 to 8 in the quinazoline ring lead an enhancement of the inhibition constant by a factor of 4.5 as observed for derivative 8f (KI 272.7 nM).
(ix) Some of the TcCA inhibitors reported here also showed a high-selective inhibition of the protozoan CA over the human isoforms. Indeed, compound 8d which shows to be one of the most potent TcCA inhibitor with KI of 34.9 nM represents also the most selective inhibitor against the protozoan enzyme over the human CA isoforms reported here with selectivity ratios as high as 34.9 against hCA I and 5.98 against hCA II (). Other derivatives of the same quinazolin-4-one series possessing the inhibition activity against TcCA of the same order of magnitude as AAZ showed good selectivity ratios for inhibiting the bacterial over the human enzymes, such as 8e and 8h ().
Table 1. Inhibition data of quinazoline-sulfonamides 8a–h, 9a–e and acetazolamide AAZ (as standard inhibitor) against human (h) isoforms hCA I, II (cytosolic) and the protozoan enzyme TcCA by a stopped-flow CO2 hydrase assay (13). The selectivity ratios are also reported.
Although, most of the compounds reported here acted as medium selective inhibitors for the protozoan isoform over the human ones, some of them presented better selectivity inhibition ratios against hCA I and hCA II than the drug clinically used AAZ which shows a selectivity ratios for inhibiting TcCA against hCA I and II equal to 4.06 and 0.19, respectively. However, it is interesting to notice how acetazolamide acts as an efficient TcCA inhibitor even showing low-selectivity ratios against hCA I and II.
Conclusions
We evaluated a new series of quinazolin-4-one and quinazolin-4-thione derivatives containing a benzenesulfonamide moiety for the inhibition of TcCA, an α-class CA recently cloned from Trypanosoma cruzi, the causative agent of Chagas disease, which was recently shown to be crucial for its life cycle. The inhibition studies of the protozoan enzyme TcCA with the reported compounds revealed that the 4-oxoquinazoline derivatives were generally more efficient than their 4-thioxoquinazoline bioisosters 4. However, most of the novel tested 4-oxoquinazoline sulfonamides, showing moderate/high inhibition against TcCA, were as potent as acetazolamide AAZ (as standard inhibitor) and one of them high-effectively inhibited TcCA at a subnanomolar concentration. In order to determine the selectivity ratios for inhibiting the protozoan over the human enzymes, the CA inhibitory activity of these compounds was also determined against the human cytosolic isoforms hCA I and II. According to the results obtained in the present study, many of the potent TcCA inhibitors reported here also possessed moderate selectivity ratios for inhibiting the protozoan over the human enzymes. The discovery of the 6-methyl-3-allyl 4-oxoquinazoline derivative as a low-nanomolar TcCA inhibitor may be useful to detect lead molecules for developing anti-Trypanosoma agents with a different mechanism of action compared to clinically used drugs (benznidazole, nifurtimox) for which significant resistance appeared. Indeed, the quinazoline-sulfonamides may represent a new class of compounds with potent inhibitory activity against protozoan enzymes, adding thus a relevant biological profile to the sulfonamides with CA inhibitory actionCitation30–33.
Declaration of interest
Most of the authors declare no financial interest. CTS reports conflict of interest as author of many patents on CA inhibitors. This research was funded by National Plan of Science, Technology and Innovation (Grant No. 11-MED1874-02), King Saud University, Riyadh (to AF), Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCT/CNPq) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Rio de Janeiro (FAPERJ).
References
- Coura JR, Borges-Pereira J. Chagas disease. What is known and what should be improved: a systemic review. J Rev Soc Bras Med Trop 2012;45:286–96
- Pan P, Vermelho AB, Capaci Rodrigues G, et al. Cloning, characterization, and sulfonamide and thiol inhibition studies of an α-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J Med Chem 2013;56:1761–71
- Salomon CJ. First century of changes' disease: an overview on novel approaches to nifurtimox and benzonidazole delivery systems. J Pharm Sci 2012;101:888–94
- Güzel-Akdemir Ö, Akdemir A, Pan P, et al. A class of sulfonamides with strong inhibitory action against the α-carbonic anhydrase from Trypanosoma cruzi. J Med Chem 2013;56:5773–81
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Pan P, Vermelho AB, Scozzafava A, et al. Anion inhibition studies of the a-carbonic anhydrase from the protozoan pathogen Trypanosoma cruzi, the causative agent of Chagas disease. Bioorg Med Chem 2013;21:4472–6
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun 2012;48:1868–70
- Di Fiore A, Maresca A, Alterio V, et al. Carbonic anhydrase inhibitors: X-ray crystallographic studies for the binding of N-substituted benzenesulfonamides to human isoform II. Chem Commun 2011;47:11636–8
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discovery 2011;10:767–77
- Capasso C, Supuran CT. Antiinfective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704
- Alafeefy AM, Ceruso M, Al-Tamimi AS, et al. Quinazoline-sulfonamides with potent inhibitory activity against the α-carbonic anhydrase from Vibrio cholera. Bioorg Med Chem (in press)
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors. Part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents with potential use as antiglaucoma drugs. Eur J Med Chem 1998;33:247–54
- Wilkinson BL, Bornaghi LF, Houston TA, et al. Carbonic anhydrase inhibitors: inhibition of isozymes I, II and IX with triazole-linked O-glycosides of benzene sulphonamides. J Med Chem 2007;50:1651–7
- Supuran CT. Inhibition of bacterial carbonic anhydrases and zinc proteases: from orphan targets to innovative new antibiotic drugs. Curr Med Chem 2012;19:831–44
- Al-Omar MA, Abdel-Hamide SG, Hamad AA, El-Subbagh IE. Synthesis and biological screening of some new substituted-3H-quinazolin-4-one analogs as antimicrobial agents. Saudi Pharm J 2004;12:63–71
- Del Giudice MR, Borioni A, Mustazza C, Gatta F. New [g]-fused [1,2,4]triazolo[1,5-c]pyrimidines: synthesis of pyrido[3,2-e] and [4,3-e][1,2,4]triazolo[1,5-c]pyrimidine, pyrimido[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine and [1,2,4]triazolo[1,5-c]pteridine derivatives. J Heterocyclic Chem 1994;31:1503–7
- Alafeefy AM, Alqasoumi SI, Ashour AE, Alshebly MM. Quinazoline-sulfonamides as potential antitumor agents: synthesis and biological testing. J Enzyme Inhib Med Chem 2013;28:375–83
- Samel AB, Pai NR. Synthesis and antimicrobial activity of some novel hydrazinyl quinazoline amine derivatives. Res J Pharm Technol 2011;4:263–7
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012; 27:138–47
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta-carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J Enzyme Inhib Med Chem 2013;28:388–91
- Maresca A, Scozzafava A, Vullo D, Supuran CT. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:384–7
- Supuran CT, Maresca A, Gregáň F, Remko M. Three new aromatic sulfonamide inhibitors of carbonic anhydrases I, II, IV and XII. J Enzyme Inhib Med Chem 2013;28:289–93
- Bonneau A, Maresca A, Winum JY, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401
- Allouche F, Chabchoub F, Carta F, Supuran CT. Synthesis of aminocyanopyrazoles via a multi-component reaction and anti-carbonic anhydrase inhibitory activity of their sulfamide derivatives against cytosolic and transmembrane isoforms. J Enzyme Inhib Med Chem 2013;28:343–9
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Del Prete S, De Luca V, Scozzafava A, et al. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium Vibrio cholerae. J Enzyme Inhib Med Chem 2014;29:23–7
- Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29:292–6
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87
- Durdagi S, Scozzafava G, Vullo D, et al. Inhibition of mammalian carbonic anhydrases I-XIV with grayanotoxin III: solution and in silico studies. J Enzyme Inhib Med Chem 2014;29:469–75