
Abstract
A novel series of 5-nitro-1H-benzimidazole derivatives substituted at position 1 by heterocyclic rings was synthesized. Cytotoxicity and antiviral activity of the new compounds were tested. Compound 3 was more active than doxorubicin against A-549, HCT-116 and MCF-7. However, compound 3 showed no activity against human liver carcinoma Hep G-2 cell line. Compounds 9 and 17b (E) showed potency near to doxorubicin against the four cell lines. The acute toxicity of compound 9 on liver cancer induced in rats was determined in vivo. Interestingly, it showed restoration activity of liver function and pathology towards normal as compared to the cancer-bearing rats induced by DENA. Compounds 17a (Z), 17b (E) and 18a (Z) were the most promising compounds for their antiviral activity against rotavirus Wa strain.
Introduction
Liver cancer is one of the most common malignancies and the third most deadly cancers in the worldCitation1. The major risk factors in liver cancer include hepatitis viral infection, food additives, alcohol, aflatoxins, environmental and industrial toxic chemicals and air and water pollutantsCitation2,Citation3. Diethylnitrosamine (DENA) is a well-known hepatocarcinogenic agent present in tobacco smoke, water, cured and fried meals, cheddar cheese, agriculture chemicals and cosmetics and pharmaceutical productsCitation4. DENA is known to induce liver cancer in experimental animal models through inhibition of many enzymes involved in DENA repair mechanismCitation5. In rats, DENA is a potent hepatocarcinogen influencing the initiation stage of carcinogenesis during a period of enhanced cell proliferation accompanied by hepatocellular necrosis and induces DNA carcinogen adducts, DNA-strand breaks and in turn hepatocellular carcinomas without cirrhosis through the development of putative pre-neoplastic focal lesionsCitation6. Although there are many strategies for the treatment of liver cancerCitation7, their therapeutic outcome remains very poor. Therefore, prevention seems to be the best strategy for lowering the incidence of this disease. In this regard, many compounds have been tested and proved efficacy against experimentally induced hepatocarcinogenesis.
Benzimidazole heterocycle is an important multifunctional system for medicinal applications in various therapeutic fields. This is due to the presence of this heterocyclic moiety in many bioactive compounds and its interaction with many enzymes and different kinds of proteins. The broad range of activity of benzimidazole derivatives includes their activity as antihypertensive, anti-inflammatory, antibacterial, antifungal, anthelmintic, antiviral, antioxidant, antiulcer, antitumor and pyschoactivityCitation8–16. The 1-substituted-2-methyl-5-nitro benzimidazole derivatives with various heterocycles at position 1 were prepared by our laboratory group. It was found that thiadiazole ring linked at position 1 to benzimidazole ring through a methylene group, I, has cytotoxicity against breast cancer (MCF-7; )Citation17. The 1-substituted-2-methyl benzimidazole derivative II was reported to be cytotoxic against non-small lung cancer and breast cancerCitation18. It was evident that 1-(4-bromobenzyl) benzimidazole derivative III was the most potent androgen receptor antagonist for prostate cancerCitation19. This is in addition to our very recent published work in synthesis of novel series of cytotoxic benzimidazole derivativesCitation20–22. Moreover, many antiviral substituted benzimidazoles at position 1 were either clinically applied or evaluated using a wide range of virus strainsCitation23,Citation24. The nucleoside benzimidazole analogs with ribofuranosyl moiety at 1-position, 5,6-dichloro-1-(β-D-ribofuranosyl)benzimidazole (DRB IV) and its derivatives TCRB V, BDCRB VI and maribavir VII are well-known inhibitor of HCMV which inhibits viral RNA synthesis by blocking RNA polymerase II ()Citation25–28. The benzimidazole derivative VIII (JNJ 2408068) had an inhibitory activity against RSV at nanomolar concentration and 100 000 times better than that ribavirinCitation29. Japan Tobacco (JTK-109) IX, with cyclohexyl ring at position-1 of benzimidazole, is an important example of benzimidazole which was patented as HCV NS5B RNA-dependent RNA polymerase inhibitors ()Citation30,Citation31.
Figure 1. Benzimidazole structures I–XIV substituted at position 1 as lead potent active compounds for antitumor and antiviral activity.
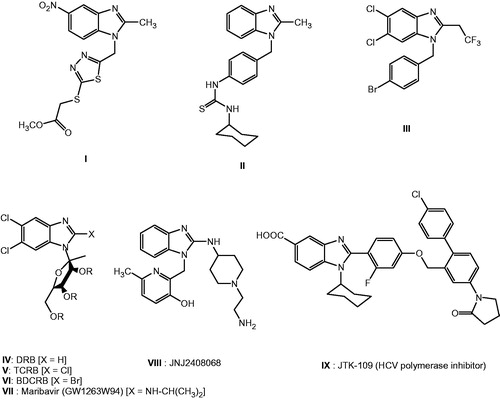
Depending upon the above-mentioned reasons, we have designed new kinds of 1-substituted benzimidazole derivatives through evaluating their cytotoxicity against various cell lines and evaluating their anticancer effect on diethylnitrosamine-induced hepatocellular carcinoma in rats, hoping to be used either alone or in combination with conventional chemotherapy in treatment of cancer. This study has also been achieved to obtain compounds with superior chemotherapeutic index in terms of increased bioavailability, higher cytotoxicity and lower side effects than the commercial available drugs.
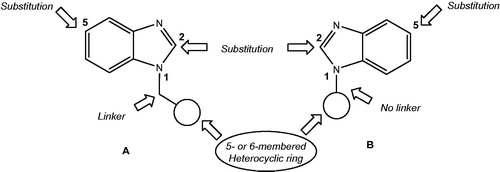
It is clear that the above-mentioned biologically active benzimidazoles, the heterocyclic substituents at position 1 are connected to the benzimidazole core either through linker or directly without linker. Therefore, by taking benzimidazoles I and JNJ 2408068 XIII as lead compounds, our strategy for the synthesis of new benzimidazole derivatives was to construct optimized cyclic or heterocyclic ring at position 1 of benzimidazole moiety that offers the potential to significantly advance out the antitumor activity (in vitro and in vivo) and antiviral activity. The general structures A (with heterocyclic rings connected to benzimidazole moiety at position 1 by methylene or ethylene linker) and B (with heterocyclic rings directly connected to benzimidazole moiety at position 1 without linker) were chosen to be used as the key for the synthesis of various benzimidazole derivatives ().
Figure 2. Structural requirements around benzimidazole nucleus for the study.
Experimental
Chemistry
Microanalyses, spectral data of the compounds were performed in the Microanalytical, National Research Centre, Cairo, Egypt. The IR spectra (4000–400 cm−1) were recorded using KBr pellets in a Jasco FT/IR 300E Fourier (Easton, MD) transform infrared spectrophotometer on a Perkin Elmer FT-IR 1650 (spectrophotometer, Waltham, MA). The NMR spectra were recorded using Joel EX-270 MHz and 500 MHz NMR spectrophotometers. Chemical shifts are reported in parts per million (ppm) from the tetramethylsilane resonance in the indicated solvent. Coupling constants are reported in Hertz (Hz); spectral splitting partners are designed as follow: singlet (s); doublet (d); triplet (t); multiplet (m). The mass spectra were carried out using Finnigan mat SSQ 7000 (Thermo. Inst. Sys. Inc., Waltham, MA) spectroscopy at 70 eV. Thin layer chromatography was performed on Merck aluminium sheet TLC Silica gel 60 F254. Column chromatography was carried out using Merck silica gel (230–400 mesh particle size). Detection of the zones was performed using ultraviolet irradiation at 254 nm.
Synthesis
Procedure for the preparation of 1-allyl-2-methyl-5-nitro-1H-benzo[d]imidazole (2)
Allyl bromide (0.98 ml, 28.0 mmol) was slowly added dropwise to a stirred solution of 5-nitro-2-methylbenzimidazole 1 (2 g, 11.3 mmol) and sodium hydride (0.6 g, 28.0 mmol) in dimethyl formamide (30 ml). The reaction was stirred at room temperature for about 8 h. The mixture was then poured onto water and the precipitate was collected by filtration. Yield: 80%. m.p.: 150–152 °C. IR (cm−1): 1618 (C=N), 1517, 1332 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.55 (s, 3H, CH3), 4.90–5.16 (m, 2H, CH2), 5.00 (d, J = 10.0 Hz, 1H, vinyl), 5.16 (d, J = 10.0 Hz, 1H, vinyl), 5.98–6.01 (m, 1H, vinyl CH2–CH=CH2), 7.68 (d, J = 9.1 Hz, 1H, H7), 8.09 (dd, J 6–7 = 9.1 Hz, J 6–4 = 2.0 Hz, 1H, H6), 8.39 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 14.24, 46.50, 107.45, 110.88, 114.84, 117.46, 117.91, 118.89, 133.30, 140.20, 141.99, 142.73, 143.02, 147.54, 157.03. MS: m/z 218, 24.7% (M+ + 1); m/z 217, 100% (M+); m/z 201, 10.9% (M+ – H – CH3); m/z 190, 2.3% (M+ – CH=CH2); m/z 177, 1.4% (M+ + H – CH2–CH=CH2); m/z 144, 16.4% (M+ – NO2 – CH=CH2). Anal. Calcd for C11H11N3O2 (217.22): C, 60.82; H, 5.10; N, 19.34. Found: C, 60.77; H, 5.15; N, 19.24.
Procedure for the preparation of 1-(2,3-dibromopropyl)-2-methyl-5-nitro-1H-benzo[d]imidazole (3)
Compound 2 (3 g, 13.8 mmol) was dissolved in 50 ml chloroform. The solution was cooled in ice bath with stirring. Then bromine (1.7 ml, 13.8 ml) was dissolved in 5 ml chloroform and added to the above mixture vigorously in three portions. The mixture was stirred in the ice bath for 2 h. The formed precipitate was filtered, washed with petroleum ether 60–80. Yield: 70%. m.p.: 172–174 °C. IR (cm−1): 1625 (C=N), 1532, 1344 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.50 (s, 3H, CH3), 4.12–4.14 (m, 2H, CH2), 4.80–5.01 (m, 3H, CHBr–CH2Br), 7.97 (d, J = 9.1 Hz, 1H, H7), 8.28 (d, J = 9.1 Hz, 1H, H6), 8.55 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 13.66, 36.28, 49.52, 50.96, 112.58, 116.17, 120.04, 132.79, 134.72, 137.60, 144.58, 157.34. MS: m/z 377, 16.4% (M+); m/z 201, 2.8% (M+ – H – 2Br – CH3); m/z 144, 100% (M+ – NO2 – CHBr-CH2Br). Anal. Calcd for C11H11Br2N3O2 (377.03): C, 35.04; H, 2.94; Br 42.39, N, 11.14. Found: C, 35.16; H, 2.77; Br 42.48, N, 11.20.
General procedure for the preparation of compounds (4–6)
A suspension of dibromo derivative 3 (3 g, 8 mmol) in ethanol was treated with potassium carbonate (16 mmol) for 2–3 h. The diamino derivative (8 mmol) was then added to the reaction mixture and refluxed for about 6–8 h. On cooling the reaction mixture, yellow to orange solid products separated out. They were collected by filtration, washed with water and dried.
4-[(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)methyl] imidazolidine-2-thione (4)
Yield: 70%. m.p.: 210–211 °C. IR (cm−1): 3344 (NH), 1616 (C=N), 1520, 1344 (NO2), 1322 (C=S). 1H NMR (DMSO-d6) δ (ppm): 2.55 (s, 3H, CH3), 3.05–3.15 (m, 1H, CH), 4.34–4.35 (m, 2H, CH2), 5.19–5.20 (m, 2H, CH2), 7.64 (d, J = 9.1 Hz, 1H, H7), 8.03 (d, J = 9.1 Hz, 1H, H6), 8.30 (s, 1H, H4). MS: m/z 218, 74.6% (M+ + H – NH–CS–NH) Anal. Calcd for C12H13N5O2S (291.07): C, 49.47; H, 4.50; N, 24.04; S, 11.01. Found: C, 49.61; H, 4.39; N, 24.16; S, 11.09.
2-Methyl-5-nitro-1-[(piperazin-2-yl)methyl]-1H-benzo[d] imidazole (5)
Yield: 64%. m.p.: 220–222 °C. IR (cm−1): 3244 (NH), 1629 (C=N), 1520, 1322 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.07–2.20 (m, 2H, CH2), 2.46 (s, 3H, CH3), 2.95–3.00 (m, 2H, CH2), 3.20–3.25 (m, 1H, CH), 4.34–4.35 (m, 2H, CH2), 5.23–5.24 (m, 2H, CH2), 7.54 (dd, J 6–7 = 9.1 Hz, J 6–4 = 2 Hz, 1H, H6), 7.98 (d, J = 9.1 Hz, 1H, H7), 8.11 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 14.38, 51.21, 109.45, 109.68, 115.22, 118.59, 121.48, 126.77, 139.77, 143.14, 141.94, 157.10. MS: m/z 274, 5.5% (M+ – 1); m/z 259, 11.0% (M+ – H – CH3). Anal. Calcd for C13H17N5O2 (275.31): C, 56.71; H, 6.22; N, 25.44. Found: C, 56.79; H, 6.32; N, 25.31.
1,2,3,4-Tetrahydro-2-[(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)methyl]quinoxaline (6)
Yield: 58%. m.p.: 194–196 °C. IR (cm−1): 3438 (NH), 1614 (C=N), 1519, 1337 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 3.30–3.37 (m, 1H, CH), 3.65–3.69 (m, 2H, CH2), 4.25–4.27 (m, 2H, CH2), 7.86–8.00 (m, 5H, aromatic protons + H6 benzimidazole), 8.19 (d, J = 9.0 Hz, 1H, H7), 8.25 (s, 1H, H4). Anal. Calcd for C17H17N5O2 (323.35): C, 63.15; H, 5.30; N, 21.66. Found: C, 63.24; H, 5.22; N, 21.75.
General procedure for the preparation of benzimidazole derivatives (7 and 8)
Compound 5 or 6 was added to sodium hydride in DMF (30 ml) and stirred for 15 min. Then acetyl chloride was added dropwise to the reaction mixture and stirred at room temperature for about 8 h. The mixture was poured into ice water and the precipitate was collected by filtration and crystallized from appropriate solvent.
2-Methyl-5-nitro-1-[(1,4-diacetylpiperazin-2-yl) methyl]-1H-benzo[d]imidazole (7)
Yield: 45%. m.p.: 183–184 °C. 1H NMR (DMSO-d6) δ (ppm): 2.07 (m, 2H, CH2), 2.24 (s, 6H, 2 COCH3), 2.31 (s, 3H, CH3), 2.95–2.98 (m, 2H, CH2), 3.22–3.25 (m, 1H, CH), 4.34–4.39 (m, 2H, CH2), 5.23–5.26 (m, 2H, CH2), 7.54 (dd, J 6–7 = 9.1 Hz, J 6–4 = 2.0 Hz, 1H, H6), 7.98 (d, J = 9.1 Hz, 1H, H7), 8.11 (s, 1H, H4). Anal. Calcd for C17H21N5O4 (359.16): C, 56.82; H, 5.89; N, 19.49. Found: C, 56.91; H, 5.77; N, 19.38.
1,2,3,4-Tetrahydro-2-[(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)methyl]1,4-diacetylquinoxaline (8)
Yield: 58%. m.p.: 176–178 °C. 1H NMR (DMSO-d6) δ (ppm): 2.37 (s, 6H, 2 COCH3), 2.46 (s, 3H, CH3), 3.34–3.37 (m, 1H, CH), 3.69–3.70 (m, 2H, CH2), 4.25–4.27 (m, 2H, CH2), 7.86–8.00 (m, 5H, aromatic protons + H6 benzimidazole), 8.19 (d, J = 9.1 Hz, 1H, H7), 8.24 (s, 1H, H4). Anal. Calcd for C21H21N5O2 (407.16): C, 61.91; H, 5.20; N, 17.19. Found: C, 61.83; H, 5.27; N, 17.25.
General procedure for the preparation of compounds 9–13
Methyl amine, glycine or dihydroxy derivative was added dropwise to a stirred solution of compound 3 (5 g, 13.3 mmol) and potassium carbonate (26.6 mmol) in acetone (30 ml). The mixture was stirred at 25 °C for about 10 h. Then the mixture was poured onto water. The formed precipitates were collected by filtration and crystallized from appropriate solvent.
N1,N2-Dimethyl-3-(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)propane-1,2-diamine (9)
Yield: 75%. m.p.: 165–167 °C. 1H NMR (DMSO-d6) δ (ppm): 2.20 (s, 6H, 2 NHCH3), 2.37 (s, 3H, CH3), 3.06–3.10 (m, 1H, CH), 3.95–3.99 (m, 2H, CH2), 4.82–4.85 (m, 2H, CH2), 7.56 (d, J = 9.1 Hz, 1H, H7), 7.96 (d, J = 9.1 Hz, 1H, H6), 8.30 (s, 1H, H4), 8.50 (broad band, 2 NH). 13C NMR (DMSO-d6) δ (ppm): 13.13, 99.35, 115.19, 117.82, 120.06, 124.02, 124.36, 125.01, 125.53, 126.51, 126.00, 139.72, 143.48, 146.44, 149.33, 152.76, 159.19. MS: m/z 277, 2.0% (M+); m/z 249, 5.0% (M+–2CH3+2H). Anal. Calcd for C13H19N5O2 (277.32): C, 56.30; H, 6.91; N, 25.25. Found: C, 56.22; H, 6.99; N, 25.39.
3-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl) propane-1,2-diaminoacetic acid (10)
Yield: 80%. m.p.: 150–152 °C. IR (cm−1): 3438, 3209 (NH + carboxylic OH), 1682 (carboxylic C=O), 1616 (C=N), 1516, 1340 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.37 (s, 3H, CH3), 2.43–2.45 (m, 2H, CH2), 2.59–2.32 (m, 2H, CH2), 3.05–3.08 (m, 1H, CH), 3.83–3.88 (m, 2H, CH2), 4.34–4.36 (m, 2H, CH2), 7.56 (d, J = 9.1 Hz, 1H, H7), 8.01 (d, J = 9.1 Hz, 1H, H6), 8.30 (s, 1H, H4), 8.45 (broad band, 2 NH), 10.48 (s, 2H, 2 COOH). 13C NMR (DMSO-d6) δ (ppm): 13.50, 43.22, 161.06, 166.90, 171.48. MS: m/z 364, 25.0% (M+ – 1); m/z 231, 24.5% (M+ – H – 2 CH2COOH – NH), 202, 100% (M+ – H – 2 CH2COOH – 2 NH – CH2), 173, 88.0% (M+ – 2 H – 2 CH2COOH – 2 NH – CH2 – CH – CH3). Anal. Calcd for C15H19N5O6 (365.34): C, 49.31; H, 5.24; N, 19.17. Found: C, 49.38; H, 5.17; N, 19.23.
1-[(1,4-Dioxan-2-yl)methyl]-2-methyl-5-nitro-1H-benzo[d] imidazole (11)
Yield: 83%. m.p.: 205–207 °C. IR (cm−1):1621 (C=N), 1519, 1334 (NO2).1H NMR (DMSO-d6) δ (ppm): 2.37 (s, 3H, CH3), 2.87–2.90 (m, 2H, CH2), 3.21–3.30 (m, 1H, CH), 3.76–3.85 (m, 2H, CH2), 4.36–4.38 (m, 2H, CH2), 4.82–4.87 (m, 2H, CH2), 7.62 (d, J = 9.1 Hz, 1H, H7), 8.03 (d, J = 9.1 Hz, 1H, H6), 8.30 (s, 1H, H4). MS: m/z 277, 0.6% (M+); m/z 191, 58.7% (M+ + H – 1,4-dioxane ring); m/z 178, 5.8% (M+ + 2H – 1,4-dioxane ring – CH2); m/z 163, 21.7% (M+ + 2H – 1,4-dioxane ring – CH2 – CH3). Anal. Calcd for C13H15N3O4 (277.28): C, 56.31; H, 5.45; N, 15.15. Found: C, 56.43; H, 5.39; N, 15.21.
1-[(2,3-Dihydrobenzo[b]1,4-dioxin-3-yl)methyl]-2-methyl-5-nitro-1H-benzo[d]imidazole (12)
Yield: 87%. m.p.: 208–210 °C. 1H NMR (DMSO-d6) δ (ppm): 2.34 (s, 3H, CH3), 3.30–3.36 (m, 1H, CH), 3.68–3.69 (m, 2H, CH2), 4.88–4.91 (m, 2H, CH2), 7.60–7.80 (m, 4H, aromatic), 7.94 (d, J = 9.1 Hz, 1H, H7), 7.97 (d, J = 9.1 Hz, 1H, H6), 8.07 (s, 1H, H4). Anal. Calcd for C17H15N3O4 (325.32): C, 62.76; H, 4.65; N, 12.92. Found: C, 62.85; H, 4.53; N, 12.83.
2-Methyl-1-[(5,7-dimethyl-1,4-dioxepan-2-yl) methyl]-5-nitro-1H-benzo[d]imidazole (13)
Yield: 77%. m.p.: 240–242 °C. IR (cm−1):1621 (C=N), 1520, 1337 (NO2). 1H NMR (DMSO-d6) δ (ppm): 1.09 (s, 6H, 2 CH3), 1.90 (s, 3H, CH2), 2.49 (s, 3H, CH3), 3.21–3.24 (m, 3H, 3 CH), 4.48–4.52 (m, 2H, CH2), 4.93–4.97 (m, 2H, CH2), 7.64 (d, J = 9.1 Hz, 1H, H7), 8.01 (d, J = 9.1 Hz, 1H, H6), 8.30 (s, 1H, H4). MS: m/z 319, 1.0% (M+). Anal. Calcd for C16H21N3O4 (319.36): C, 60.17; H, 6.63; N, 13.16. Found: C, 60.25; H, 6.54; N, 13.26.
Procedure for the preparation of 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenylethanone (14)
A solution of 5-nitro-2-methylbenzimidazole 1 (5 g, 28 mmol) and sodium hydride (0.6 g, 28 mmol) in dimethyl formamide (30 ml) was stirred at room temperature. Then phenacyl bromide (5.6 g, 28 mmol) was added dropwise to the reaction mixture. The reaction was allowed to stirring at room temperature for about 8 h. The mixture was then poured onto water and the precipitate was collected by filtration. Yield: 84%. m.p.: 146–147 °C. IR (cm−1): 1690 (C=O), 1617 (C=N), 1516, 1334 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.50 (s, 3H, CH3), 6.23 (s, 2H, CH2), 7.55–7.85 (m, 5H, aromatic), 8.00–8.25 (m, 3H, H7 + H6 + H4). MS: m/z 295, 22.5% (M+); m/z 279, 4.0% (M+ – H – CH3); m/z 190, 3.4% (M+ – C6H5 – CO). Anal. Calcd for C16H13N3O3 (295.10): C, 65.08; H, 4.44; N, 14.23. Found: C, C, 65.16; H, 4.32; N, 14.30.
General procedure for the preparation of 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-3-aryl-1-phenylprop-2-en-1-one (15–18)
A mixture of compound 14 (6.7 mmol), aromatic aldehydes (6.7 mmol) and 10% aqueous sodium hydroxide (10 ml) in ethanol (30 ml) was stirred at room temperature for about 3 h. The resulting solid was washed, dried and crystallized from ethanol. The two stereoisomers (E and Z) were separated into individual pure components 15a(Z)–18a(Z) as major components and 15b(E)–18b(E) as minor components using column chromatography on silica gel (200 × 30 mm, n-hexane, n-hexane/ethylacetate 10:1, 3:1, 2:1, 1:1).
(Z)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-p-tolylprop-2-en-1-one (15a)
Yield: 72%. m.p.: 150–152 °C. IR (cm−1): 1647 (C=O), 1602 (C=C), 1517, 1338 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.19 (s, 3H, CH3 p-tolyl), 2.33 (s, 3H, CH3 benzimidazole), 6.86 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.07 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.38 (d, J = 9.0 Hz, 1H, H7), 7.60 (t, J = 7.5 Hz, 2H, aromatic), 7.70 (t, J = 7.5 Hz, 1H, aromatic), 7.94 (s, 1H, CH=C), 7.95 (d, J = 7.5 Hz, 2H, aromatic), 8.04 (d, J = 9.0 Hz, 1H, H6), 8.51 (s, 1H, H4). MS: m/z 397, 4.0% (M+). Anal. Calcd for C24H19N3O3 (397.14): C, 72.53; H, 4.82; N, 10.57. Found: C, 72.61; H, 4.73; N, 10.44.
(E)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-p-tolylprop-2-en-1-one (15b)
Yield: 15%. m.p.: 135–136 °C. IR (cm−1): 1650 (C=O), 1518, 1330 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.20 (s, 3H, CH3 p-tolyl), 2.35 (s, 3H, CH3 benzimidazole), 6.87 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.07 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.61 (t, J = 7.5 Hz, 2H, aromatic), 7.71 (t, J = 7.5 Hz, 1H, aromatic), 7.84 (d, J = 9.0 Hz, 1H, H7), 7.92 (s, 1H, CH=C), 7.99 (d, J = 7.5 Hz, 2H, aromatic), 8.07 (d, J = 1.7 Hz, 1H, H4), 8.12 (d, J = 9.0 Hz, 1H, H6). MS: m/z 397, 75.0% (M+); m/z 382, 12.1%, (M+ – CH3); m/z 292, 14.1% (M+ – C6H5CO); m/z 246, 41.2% (M+ – C6H5CO– NO2). Anal. Calcd for C24H19N3O3 (397.14): C, 72.53; H, 4.82; N, 10.57. Found: C, 72.65; H, 4.71; N, 10.49.
(Z)-3-(2-Methoxyphenyl)-2-(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)-1-phenylprop-2-en-1-one (16a)
Yield: 68%. m.p.: 190–192 °C. IR (cm−1): 1636 (C=O), 1595 (C=C), 1522, 1343 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.37 (s, 3H, CH3), 3.69 (s, 3H, OCH3), 6.49 (d, J = 7.5 Hz, 1H, aromatic), 6.69 (m, 1H, aromatic), 7.05 (d, J = 7.5 Hz, 1H, aromatic), 7.30 (d, J = 9.0 Hz, 1H, H7), 7.33–7.4 (m, 1H, aromatic), 7.59–7.66 (m, 2H, aromatic), 7.72–7.75 (m, 1H, aromatic), 7.93 (d, J = 7.5 Hz, 2H, aromatic), 8.02 (d, J = 9.0 Hz, 1H, H6), 8.07 (s, 1H, CH=C), 8.47 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 13.67, 55.99, 110.58, 111.95, 114.75, 118.34, 119.81, 120.85, 129.40, 129.47, 133.09, 133.44, 136.60, 139.08, 139.43, 142.01, 143.16, 155.82, 157.84, 191.67. MS: m/z 413, 35.6% (M+). Anal. Calcd for C24H19N3O4 (413.43): C, 69.72; H, 4.63; N, 10.16. Found: C, 69.65; H, 4.70; N, 10.23.
(E)-3-(2-Methoxyphenyl)-2-(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)-1-phenylprop-2-en-1-one (16b)
Yield: 17%. m.p.: 115–116 °C. IR (cm−1): 1653 (C=O), 1597 (C=C), 1521, 1338 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.34 (s, 3H, CH3), 3.74 (s, 3H, OCH3), 6.56 (d, J = 7.5 Hz, 1H, aromatic), 6.66–6.75 (m, 1H, aromatic), 7.03 (d, J = 7.5 Hz, 1H, aromatic), 7.31–7.35 (m, 1H, aromatic), 7.60–7.65 (m, 2H, aromatic), 7.71–7.76 (m, 1H, aromatic), 7.82 (d, J = 7.5 Hz, 1H, aromatic), 7.94 (s, 1H, aromatic), 7.95 (s, 1H, CH=C), 7.98 (d, J = 9 Hz, 1H, H7), 8.04 (d, J = 9 Hz, 1H, H6), 8.07 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 14.44, 14.16, 56.48, 107.10, 111.03, 112.37, 115.22, 118.79, 120.59, 121.36, 128.66, 129.38, 133.47, 135.23, 137.14, 139.62, 140.40, 142.51, 143.70, 156.35, 157.90, 158.31, 192.07. MS: m/z 413, 9.4% (M+); m/z 382, 4.2%, (M+ – OCH3); m/z 226, 36.3% (M+ – C6H5CO). Anal. Calcd for C24H19N3O4 (413.43): C, 69.72; H, 4.63; N, 10.16. Found: C, 69.68; H, 4.71; N, 10.25.
(Z)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-3-(5-methylfuran-2-yl)-1-phenylprop-2-en-1-one (17a)
Yield: 70%. m.p.: 178–180 °C. IR (cm−1): 1614 (C=O), 1519, 1341 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.35 (s, 3H, CH3 furan), 2.47 (s, 3H, CH3 benzimidazole), 6.23 (d, J = 2.5 Hz, 1H, furan), 6.55 (d, J = 2.5 Hz, 1H, furan), 7.34 (d, J = 9.0 Hz, 1H, H7), 7.39–7.48 (m, 1H, aromatic), 7.59 (m, 2H, aromatic), 7.72 (s, 1H, CH=C), 7.87–7.90 (m, 2H, aromatic), 8.07 (d, J = 9.0 Hz, 1H, H6), 8.50 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 13.90, 14.02, 22.66, 51.08, 110.99, 111.19, 115.05, 118.65, 119.02, 124.14, 129.30, 129.57, 132.26, 137.55, 143.45, 146.31, 149.87, 159.41, 191.70. MS: m/z 387, 87.5% (M+). Anal. Calcd for C22H17N3O4 (387.12): C, 68.21; H, 4.42; N, 10.85. Found: C, 68.33; H, 4.35; N, 10.92.
(E)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-3-(5-methylfuran-2-yl)-1-phenylprop-2-en-1-one (17b)
Yield: 17%. m.p.: 137–139 °C. IR (cm−1): 1621 (C=O), 1520, 1337 (NO2). 1H NMR (DMSO-d6) δ (ppm): 1.92 (s, 3H, CH3 furan), 2.47 (s, 3H, CH3 benzimidazole), 6.24 (d, J = 2.5 Hz, 1H, furan), 6.55 (d, J = 2.5 Hz, 1H, furan), 7.58–7.61 (m, 2H, aromatic), 7.67–7.77 (m, 1H, aromatic), 7.70 (s, 1H, CH=C), 7.82 (d, J = 9.0 Hz, 1H, H7), 7.90–7.95 (m, 2H, aromatic), 8.09 (d, J = 9.0 Hz, 1H, H6), 8.11 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 13.86, 14.16, 51.09, 107.02, 111.20, 118.30, 119.25, 123.92, 124.01, 129.25, 129.67, 132.59, 132.92, 135.26, 137.60, 143.26, 146.37, 148.14, 158.26, 159.33, 191.67. MS: m/z 386, 9.4% (M+ – 1); m/z 292, 4.2%, (M+ + H – methylfuryl – CH3); m/z 262, 36.3% (M+ – C6H5 – NO2 – 2H); m/z 249, 36.3% (M+ – C6H5 – NO2 – CH3). Anal. Calcd for C22H17N3O4 (387.12): C, 68.21; H, 4.42; N, 10.85. Found: C, 68.37; H, 4.33; N, 10.94.
(Z)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-(thiophen-2-yl)prop-2-en-1-one (18a)
Yield: 68%. m.p.: 188–190 °C. IR (cm−1): 1643 (C=O), 1522, 1338 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 7.14 (m, 1H, H′3 thiophene), 7.61 (m, 2H, H′2 + H′4 thiophene), 7.70–7.78 (m, 1H, aromatic), 7.79–7.86 (m, 2H, aromatic), 7.86 (d, J = 9.0 Hz, 1H, H7), 7.97–7.99 (m, 2H, aromatic), 8.12 (d, J = 9.0 Hz, 1H, H6), 8.18 (s, 1H, H4), 8.37 (s, 1H, CH=C). MS: m/z 393, 5.0% (M+ + 2H + 2H). Anal. Calcd for C21H15N3O3S (389.43): C, 64.77; H, 3.88; N, 10.79l; S, 8.23. Found: C, 64.65; H, 3.96; N, 10.64; S, 8.31.
(E)-2-(2-Methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-(thiophen-2-yl)prop-2 -en-1-one (18b)
Yield: 17%. m.p.: 125–127 °C. IR (cm−1): 1648 (C=O), 1521, 1338 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 7.15–7.25 (m, 1H, H′3 thiophene), 7.60–7.72 (m, 2H, H′2 + H′4 thiophene), 7.69–7.75 (m, 1H, aromatic), 7.80–7.87 (m, 2H, aromatic), 7.87 (d, J = 9.0 Hz, 1H, H7), 7.96–7.99 (m, 2H, aromatic), 8.14 (d, J = 9.0 Hz, 1H, H6), 8.19 (s, 1H, H4), 8.39 (s, 1H, CH=C). 13C NMR (DMSO-d6) δ (ppm): 14.01, 106.97, 118.68, 119,55, 125.84, 128.70, 129.24, 129.77, 133.01, 134.42, 134.45, 136.37, 139.06, 140.27, 143.59, 148.49, 157.96, 191.59. MS: m/z 390, 0.3% (M+ + 1); m/z 260, 1.2%, (M+ – thiophene – NO2); m/z 226, 18.0% (M+ – C6H5 – thiophene – 3H). Anal. Calcd for C21H15N3O3S (389.43): C, 64.77; H, 3.88; N, 10.79; S, 8.23. Found: C, 64.62; H, 3.93; N, 10.69; S, 8.28.
General procedure for the preparation of 1-(2,3,6,7-tetrahydro-7-aryl-5-phenyl-1H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole derivatives (19–22); 5,6-dihydro-5-(2-methyl-5-nitro-1H-benzo[d] imidazol-1-yl)-6-aryl-4-phenylpyrimidine-2(1H)-thione derivatives (23–26)
Compounds 15, 16, 17 or 18 (4 mmol) were added to ethylenediamine or thiourea (4 mmol). Then, Sodium hydroxide (10 mmol) in ethanol (25 ml) was added. The reaction mixture was refluxed in for 8 h. The solution was poured onto ice water. The precipitate was filtered and crystallized from ethanol.
1-((Z)-2,3,6,7-Tetrahydro-5-phenyl-7-p-tolyl-1H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (19)
Yield: 60%. m.p.: 220–222 °C. IR (cm−1): 1615 (C=N), 1517, 1332 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.07 (s, 3H, CH3 p-tolyl), 2.46 (s, 3H, CH3 benzimidazole), 2.96 (s, 2H, CH2–NH), 4.32 (s, 2H, CH2-N), 5.23 (d, J = 1.5 Hz 1H, CH), 6.12 (d, J = 1.5 Hz, 1H, CH), 6.81 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.02 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.26 (m, 3H, aromatic), 7.54 (m, 3H, 2H aromatic + H7), 7.98 (d, J = 9.0 Hz, 1H, H6), 8.11 (s, 1H, H4). MS: m/z 440, 5.5% (M+ + H). Anal. Calcd for C26H25N5O2 (439.20): C, 71.05; H, 5.73; N, 15.93. Found: C, 71.13; H, 5.64; N, 15.86.
1-((Z)-3,4,5,6-Tetrahydro-5-(2-methoxyphenyl)-7-phenyl-2H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (20)
Yield: 71%. m.p.: 165–166 °C. IR (cm−1): 1598 (C=N), 1510, 1334 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.37 (s, 3H, CH3), 3.53 (s, 2H, CH2–NH), 3.69 (s, 3H, OCH3), 3.98 (s, 2H, CH2–N), 4.72 (d, J = 1.5 Hz 1H, CH), 5.64 (d, J = 1.5 Hz, 1H, CH), 6.48 (d, J = 7.5 Hz, 1H, aromatic), 6.67–6.69 (m, 1H, aromatic), 7.05 (d, J = 7.5 Hz, 1H, aromatic), 7.30–7.35 (m, 2H, aromatic), 7.61–7.65 (m, 2H, aromatic), 7.75–7.81 (m, 1H, aromatic), 7.94–7.99 (m, 2H, 1H aromatic + H7), 8.07 (d, J = 9.0 Hz, 1H, H6), 8.10 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 16.00, 30.90, 56.15, 63.33, 72.60, 112.91, 120.84, 128.12, 136.2. MS: m/z 413, 18.75% (M+ – H – NH(CH2)2N + O). Anal. Calcd for C26H25N5O3 (455.20): C, 68.56; H, 5.53; N, 15.37. Found: C, 68.63; H, 5.45; N, 15.46.
1-((Z)-3,4,5,6-Tetrahydro-5-(5-methylfuran-2-yl)-7-phenyl-2H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (21)
Yield: 50%. m.p.: 180–182 °C. IR (cm−1): 1624 (C=N), 1516, 1336 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.43 (s, 3H, CH3 furan), 2.50 (s, 3H, CH3 benzimidazole), 3.93 (s, 2H, CH2–NH), 4.30 (s, 2H, CH2–N), 4.70 (d, J = 1.5 Hz 1H, CH), 5.36 (d, J = 1.5 Hz, 1H, CH), 6.12 (d, J = 2.5 Hz, 1H, furan), 6.39 (d, J = 2.5 Hz, 1H, furan), 7.29–7.60 (m, 5H, aromatic), 7.75 (d, J = 9.0 Hz, 1H, H7), 7.97 (d, J = 9.0 Hz, 1H, H6), 8.29 (s, 1H, H4). MS: m/z 429, 0.06% (M+). Anal. Calcd for C24H23N5O3 (429.18): C, 67.12; H, 5.40; N, 16.31. Found: C, 67.23; H, 5.29; N, 16.40.
1-((Z)-2,3,6,7-Tetrahydro-5-phenyl-7-(thiophen-2-yl)-1H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (22)
Yield: 65%. m.p.: 188–190 °C. IR (cm−1): 1628 (C=O), 1516, 1334 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 2.96 (s, 2H, CH2–NH), 3.72 (s, 2H, CH2–N), 4.93 (d, J = 1.5 Hz 1H, CH), 5.98 (d, J = 1.5 Hz, 1H, CH),7.14–7.17 (m, 1H, H′3 thiophene), 7.60 (m, 2H, H′2 + H′4 thiophene), 7.69–7.72 (m, 1H, aromatic), 7.79–7.86 (m, 2H, aromatic), 7.87 (d, J = 9.0 Hz, 1H, H7), 7.97–8.00 (m, 2H, aromatic), 8.13 (d, J = 9.0 Hz, 1H, H6), 8.19 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 15.15, 30.90, 41.33, 41.42, 41.61, 63.26, 79.20, 124.18, 126.80, 128.55, 129.11, 129.24, 129.37, 136.32, 138.89, 142.30, 143.52, 144.45, 184.61. MS: m/z 431, 0.2% (M+); m/z 350, 0.4%, (M+ – thiophene + 2H); m/z 268, 2.0% (M+ – C6H5 – thiophene – 3H), m/z 177, 80% (M+ + H – 2,3,6,7-tetrahydro-5-phenyl-7-(thiophen-2-yl)-1H-1,4-diazepin-6-yl). Anal. Calcd for C23H21N5O2S (431.14): C, 64.02; H, 4.91; N, 16.23; S, 7.43. Found: C, 64.13; H, 4.82; N, 16.18; S, 7.35.
5,6-Dihydro-5-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-4-phenyl-6-p-tolylpyrimidine-2(1H)-thione (23)
Yield: 78%. m.p.: 210–212 °C. IR (cm−1): 1643 (C=N), 1521, 1338 (NO2), 1303 (C=S). 1H NMR (DMSO-d6) δ (ppm): 2.20 (s, 3H, CH3 p-tolyl), 2.35 (s, 3H, CH3 benzimidazole), 4.52 (d, J = 1.5 Hz 1H, CH), 5.90 (d, J = 1.5 Hz, 1H, CH), 6.87 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.05 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.59–7.65 (m, 3H, aromatic), 7.70–7.75 (m, 1H, aromatic), 7.85 (d, J = 9.0 Hz, 1H, H7), 7.97–7.99 (m, 1H, aromatic), 8.07 (s, 1H, H4), 8.10 (d, J = 9.0 Hz, 1H, H6). MS: m/z 422, 4.0% (M+ – SH). Anal. Calcd for C25H21N5O2S (455.14): C, 65.92; H, 4.65; N, 15.37; S, 7.04. Found: C, 65.85; H, 4.74; N, 15.27; S, 7.14.
5,6-Dihydro-6-(2-methoxyphenyl)-5-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-4-phenylpyrimidine-2(1H)-thione (24)
Yield: 91%. m.p.: 198–200 °C. IR (cm−1): 1598 (C=N), 1510, 1334 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.34 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 4.97 (d, J = 1.5 Hz 1H, CH), 5.86 (d, J = 1.5 Hz, 1H, CH), 6.56 (d, J = 7.5 Hz, 1H, aromatic), 6.67 (m, 1H, aromatic), 7.04 (d, J = 7.5 Hz, 1H, aromatic), 7.29–7.33 (m, 1H, aromatic), 7.60–7.65 (m, 2H, aromatic), 7.72–7.74 (m, 1H, aromatic), 7.79–7.83 (m, 1H, aromatic), 7.94 (d, J = 9.0 Hz, 1H, H7), 7.98–8.00 (m, 1H, aromatic), 8.04 (d, J = 9.0 Hz, 1H, H6), 8.07 (s, 1H, H4). MS: m/z 468, 0.01% (M+ – 3H). Anal. Calcd for C25H21N5O3S (471.14): C, 63.68; H, 4.49; N, 14.85; S, 6.80. Found: C, 63.76; H, 4.57; N, 14.75; S, 6.72.
5,6-Dihydro-5-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-6- (5-methylfuran-2-yl)-4-phenylpyrimidine-2(1H)-thione (25)
Yield: 80%. m.p.: 180–182 °C. 1H NMR (DMSO-d6) δ (ppm): 1.92 (s, 3H, CH3 furan), 2.35 (s, 3H, CH3 benzimidazole), 4.34 (d, J = 1.5 Hz 1H, CH), 5.09 (d, J = 1.5 Hz, 1H, CH), 6.23 (d, J = 2.5 Hz, 1H, furan), 6.54 (d, J = 2.5 Hz, 1H, furan), 7.58–7.80–7.82 (m, 4H, aromatic), 7.91–7.93 (m, 2H, 1H aromatic + H7), 8.10–8.12 (m, 2H, H6 + H4). 13C NMR (DMSO-d6) δ (ppm): 15.37, 22.71, 31.24, 62.93, 117.70, 129.20, 129.99, 142.68. MS: m/z 444, 3.0% (M+ – 1); m/z 418, 0.5%, (M+– 2 CH3 + 3H). Anal. Calcd for C23H19N5O3S (445.50): C, 62.01; H, 4.30; N, 15.72; S, 7.20. Found: C, 62.13; H, 4.43; N, 15.82; S, 7.13.
5,6-Dihydro-5-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-4-phenyl-6-(thiophen-2-yl)pyrimidine-2(1H)-thione (26)
Yield: 68%. m.p.: 205–207 °C. IR (cm−1): 1622 (C=N), 1520, 1338 (NO2), 1266 (C=S). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 4.72 (d, J = 1.5 Hz 1H, CH), 5.45 (d, J = 1.5 Hz, 1H, CH), 7.14–7.22 (m, 1H, H′3 thiophene), 7.60–7.64 (m, 2H, H′2 + H′4 thiophene), 7.70–7.74 (m, 1H, aromatic), 7.79–7.83 (m, 2H, aromatic), 7.87 (d, J = 9.0 Hz, 1H, H7), 7.96–8.00 (m, 2H, aromatic), 8.15 (d, J = 9.0 Hz, 1H, H6), 8.19 (s, 1H, H4). 13C NMR (DMSO-d6) δ (ppm): 31.23, 40.67, 128.70, 131.35, 148.43, 155.89, 155.89, 206.92. MS: m/z 447, 0.03% (M+); m/z 434, 0.1%, (M+ – CH3 + 2H). Anal. Calcd for C22H17N5O2S2 (447.53): C, 59.04; H, 3.83; N, 15.65; S, 14.33. Found: C, 59.12; H, 3.73; N, 15.76; S, 14.24.
General procedure for the preparation of 1-(4,5-dihydro-5-aryl-3-phenyl-1H-pyrazol-4-yl)-2- methyl-5-nitro-1H-benzo[d]imidazole derivatives (27–30)
Compounds 15, 16, 17 or 18 (21 mmol) were stirred with hydrazine hydrate (99.9% sol., 26 mmol) in ethanol (20 ml) for 3 h. The reaction was stirred at room temperature and monitored by TLC. The solvent was evaporated under reduced pressure and the solids obtained were crystallized from diethyl ether.
1-(4,5-Dihydro-3-phenyl-5-p-tolyl-1H-pyrazol-4-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (27)
Yield: 66%. m.p.: 212–214 °C. IR (cm−1): 1683 (C=N), 1516, 1340 (NO2), 1299 (C=S). 1H NMR (DMSO-d6) δ (ppm): 2.07 (s, 3H, CH3 p-tolyl), 2.46 (s, 3H, CH3 benzimidazole), 5.23 (d, J = 1.5 Hz 1H, CH), 6.49 (d, J = 1.5 Hz, 1H, CH), 6.83 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.02 (d, J = 8.0 Hz, 2H, p-tolyl AB-system), 7.20–7.27 (m, 2H, aromatic), 7.54–7.57 (m, 3H, aromatic),), 8.00 (dd, J 6–7 = 9.2 Hz, J 6–4 = 2.2 Hz, 1H, H6), 8.11 (d, J = 2.3 Hz, 1H, H4), 8.69 (d, J = 9.2 Hz, 1H, H7). MS: m/z 234, 100%, (M+ – H – 2-methyl-5-nitro-1H-benzo[d]imidazole part); m/z 177, 32%, (M+ + H – 4,5-dihydro-3-phenyl-5-p-tolyl-1H-pyrazol-4-yl part). Anal. Calcd for C24H21N5O2 (411.17): C, 70.06; H, 5.14; N, 17.02; S, 7.78. Found: C, 70.13; H, 5.20; N, 17.09; S, 7.64.
1-(4,5-Dihydro-5-(2-methoxyphenyl)-3-phenyl-1H-pyrazol-4-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (28)
Yield: 56%. m.p.: 195–197 °C. IR (cm−1): 1600 (C=N), 1520, 1338 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.35 (s, 3H, CH3), 3.68 (s, 3H, OCH3), 4.65 (d, J = 1.5 Hz 1H, CH), 6.17 (d, J = 1.5 Hz, 1H, CH), 6.66–6.72 (m, 2H, aromatic), 7.03–7.07 (m, 1H, aromatic), 7.31–7.36 (m, 1H, aromatic), 7.60–7.65 (m, 3H, aromatic), 7.70–7.76 (m, 1H, aromatic), 7.80–7.84 (m, 1H, aromatic), 7.94 (d, J = 9.0 Hz, 1H, H6), 7.97 (d, J = 9.0 Hz, 1H, H7), 8.10 (s, 1H, H4). MS: m/z 411, 0.05% (M+ – CH3 – H). Anal. Calcd for C24H21N5O3 (427.46): C, 67.44; H, 4.95; N, 16.38. Found: C, 67.56; H, 4.84; N, 16.27.
1-(4,5-Dihydro-5-(5-methylfuran-2-yl)-3-phenyl-1H-pyrazol-4-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (29)
Yield: 70%. m.p.: 182–184 °C. IR (cm−1): 1598 (C=N), 1522, 1339 (NO2). 1H NMR (DMSO-d6) δ (ppm): 1.92 (s, 3H, CH3 furan), 2.37 (s, 3H, CH3 benzimidazole), 4.62 (d, J = 1.5 Hz 1H, CH), 5.76 (d, J = 1.5 Hz, 1H, CH), 6.23 (d, J = 2.5 Hz, 1H, furan), 6.55 (d, J = 2.5 Hz, 1H, furan), 7.56–7.68 (m, 4H, aromatic), 7.82–7.85 (m, 1H, aromatic), 7.91 (d, J = 9.0 Hz, 1H, H7), 8.10–8.12 (m, 2H, H6 + H4). 13C NMR (DMSO-d6) δ (ppm): 63.73, 72.89, 79.71, 159.95. MS: m/z 318, 16.47% (M+ – H – 5-methylfuryl). Anal. Calcd for C22H19N5O3 (401.15): C, 65.83; H, 4.77; N, 17.45. Found: C, 65.75; H, 4.65; N, 17.34.
1-(4,5-Dihydro-3-phenyl-5-(thiophen-2-yl)-1H-pyrazol-4-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole (30)
Yield: 68%. M.p.: 203–205 °C. IR (cm−1): 1618 (C=N), 1520, 1339 (NO2). 1H NMR (DMSO-d6) δ (ppm): 2.38 (s, 3H, CH3), 4.85 (d, J = 1.5 Hz 1H, CH), 5.60 (d, J = 1.5 Hz, 1H, CH), 7.14–7.18 (m, 1H, H′3 thiophene), 7.61–7.76 (m, 2H, H′2 + H′4 thiophene), 7.71–7.76 (m, 1H, aromatic), 7.79–7.84 (m, 2H, aromatic), 7.87 (d, J = 9.0 Hz, 1H, H7), 7.98–8.13 (m, 2H, aromatic), 8.14 (d, J = 9.0 Hz, 1H, H6), 8.19 (s, 1H, H4). MS: m/z 297, 0.55% (M+ + H – Ph – CH3 – NH). Anal. Calcd for C21H17N5O2S (403.11): C, 62.52; H, 4.25; N, 17.36; S, 7.95. Found: C, 62.65; H, 4.13; N, 17.45; S, 7.88.
Antitumor activity
Materials and methods of in vitro studies
The MTT assay developed by MosmannCitation32 was modified by MiuraCitation33 and used to determine the in vitro inhibitory effects of test compounds on cell growth. Briefly, medium containing 10 × 103 cells (Hep-G2, HCT-116, MCF-7 or A-549 cells) in fresh complete growth medium was seeded into each well of a 96-well microplate, with the compound solution added simultaneously to triplicate wells, before the final volume was made up to 100 µl. The plate was incubated at 37 °C for 72 h in a humidified atmosphere of 5% CO2 using a water jacketed carbon dioxide incubator (Sheldon, TC2323, Cornelius, OR). Media was aspirated, fresh medium (without serum) was added and cells were incubated either alone (negative control) or with different concentrations of sample to give a final concentration of (100, 50, 25, 12.5, 6.25, 3.125, 1.56 and 0.78 µg/ml). Cells were suspended in RPMI 1640 medium for (Hep-G2, MCF-7 and HCT-116), and DMEM for (A549), 1% antibiotic–antimycotic mixture (10 000 U/ml potassium penicillin, 10 000 µg/ml streptomycin sulfate and 25 µg/ml amphotericin B) and 1% l-glutamine in 96-well flat bottom microplate at 37 °C under 5% CO2. After 48 h of incubation, medium was aspirated, 200 μl of 10% sodium dodecyl sulphate (SDS) in de-ionized water was added to each well and further incubated overnight at 37 °C under 5% CO2. A 200 μl of 10% SDS in de-ionized water was added to each well to stop the reaction and to solubilize any MTT-formazan that had formed and then incubated overnight at 37 °C. Doxorubicin, which is known natural cytotoxic agent, was used as positive control in 100 µg/ml and gave 100% lethality under the same conditionsCitation34. A 100 µl of 0.02N HCl/50% N,N-dimethyl formamide/20% SDS was added to solubilize any MTT-formazan that had formed. The optical density of each well was measured at 575 nm (OD575) using a microplate multi-well reader (Bio-Rad Laboratories Inc., model 3350, Hercules, CA) and the inhibition of cell growth (%) was calculated as (1 – T/C) × 100, where C is the mean OD575 of the control group and T is that of the treated group. The IC50 was determined from the dose-response curve. A statistical significance was tested between samples and negative control (cells with vehicle) using independent t-test by SPSS 11 program (Chicago, IL).
Materials and methods of in vivo study
Animals
The animal care and handling was done according to the guidelines set by the World Health Organization, Geneva, Switzerland, and according to approval from the ethics committee for animals care at the National Research Centre, Egypt (ethic No. 10-230). Adult male Sprague–Dawley rats (180 ± 20 g, body weight), were purchased from the animal house of National Research Centre, Egypt. They were kept for a week under environmentally controlled conditions (constant temperature 25–27 °C, with 12 h light/dark cycle) for 1 week prior to starting the experiments. The mice were kept as 10 animals per cage, and they were provided with tap water and commercial diets
Materials
Diethylnitrosamine (DENA) and carbon tetrachloride (CCl4) were purchased from Sigma Chemical Co. (St. Louis, MO). DENA was dissolved in saline and injected in a single dose (200 mg/kg, i.p.) to initiate hepatic carcinogenesis, while CCl4 was used in a single dose (2 ml/kg) by gavage as 1:1 dilution in corn oil to stimulate liver cell proliferation and regenerationCitation35. Aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP) and total bilirubin (Bili) kits were obtained from Biomerieux (France).
Experimental design
In vivo assay of acute toxicity of the compounds
The acute toxicity of the synthesized compounds was determined in vivo according to Prieur et al.Citation36 and GhoshCitation37. Briefly, animals were divided into subgroups (10 rats each) administrated i.v. in a single dose with gradually doses ranged from 0 to 2500 µg/kg. Control animals received the vehicle alone (DMSO). Mortality of the animals was observed up to one month post-treatment. LD50 (the median lethal doses) of each compound was determined as (the dose resulted in 50% mortality of the animals) was calculated using a computer program for Probit analysis. The compounds which exhibited acute cytotoxicity will be further examined for its protective effect on DENA-induced liver cancer in rats.
In vivo antitumor activity of the compounds on liver cancer induced by DENA
Adult male Sprague–Dawley rats were divided into three groups with eight animals in each group. Group 1 (untreated control group): animals were fed on a standard diet and given water throughout the course of the experiment. Group 2 (DENA treated group): Rats were injected with a single dose of DENA (200 mg/kg, i.p.) and 2 weeks later received a single dose of CCl4 (2 ml/kg) by oral gavage at 1:1 dilution in corn oil for 32 weeks. Group 3 (DENA and compound 9 group): Rats-bearing cancer from Group 2 were treated with compound 9 by intravenous administration at dose of 1/10 of the LD50 value and continued for 10 consecutive days and the experiment was ended after 30 days from last injection.
At the end of the treatment protocol, animals were anesthetized with ether and blood samples were drawn from the orbital venous plexus. Serum was separated by centrifugation for 5 min at 1500 × g and stored at −20 °C until analysis. This was then used to determine liver function by assaying ALT, AST and ALP activities and total bilirubin spectrophotometrically according to the manufacturer's instructions, using reagent kits obtained from Roche Diagnostics (Mannheim, Germany).
Histopathological examination
For histopathological examination, animals were sacrificed by decapitation and their livers were rapidly excised, weighed, washed with saline, portions of liver were fixed in 10% formalin in saline, then embedded in paraffin wax, serially sectioned and stained according to Conn et al.Citation38 using a standard method of hematoxylin and eosin (H&E).
Statistical analysis
The results are reported as mean ± standard error (SE) of eight rats. Statistical differences were analyzed according to followed by one way ANOVA test followed by student's t-test wherein the differences were considered to be significant at p < 0.05.
Antiviral activity
Cytotoxicity test
It was done according to Simoes et al.Citation39 as well as Walum et al.Citation40. Briefly, all samples (100 mg) were dissolved in 500 μl of DMSO. Decontamination of samples was done by adding 12 μl of 100× of antibiotic–antimycotic mixture to 500 μl of each sample. Then, bi-fold dilutions were done to 100 μl of original dissolved samples and 100 μl of each dilutions were inoculated in Hep-2 and MA104 cell lines (obtained from the holding company for Biological Products & Vaccines VACSERA, Egypt) previously cultured in 96 multi-well plates (Greiner-Bio One, Germany) to estimate the non-toxic dose of the tested samples. Cytotoxicity assay was done using cell morphology evaluation by inverted light microscope and cell viability test applying trypan blue dye exclusion method.
Cell morphology evaluation by inverted light microscopy
Hep-2 and MA104 cell cultures (2 × 105 cells/ml) were prepared in 96-well tissue culture plates (Greiner-Bio One, Germany). After 24 h incubation at 37 °C in a humidified 5% (v/v) CO2 atmosphere cell monolayers were confluent, the medium was removed from each well and replenished with 100 μl of bi-fold dilutions of different samples tested prepared in DMEM (GIBCO BRL). For cell controls, 100 μl of DMEM without samples was added. All cultures were incubated at 37 °C in a humidified 5% (v/v) CO2 atmosphere for 72 h. Cell morphology was observed daily for microscopically detectable morphological alterations, such as loss of confluence, cell rounding and shrinking and cytoplasm granulation and vacuolization. Morphological changes were scored (Simoes et al.)Citation39.
Cell viability assay
It was done by trypan blue dye exclusion method (Walum et al.)Citation40. Hep-2 and MA104 cell cultures (2 × 105 cells/ml) were grown in 12-well tissue culture plates Greiner-Bio One, Germany). After 24 h incubation, the same assay described above for tested samples cytotoxicity was followed by applying 100 μl of tested samples dilutions (bi-fold dilutions) per well. After 72 h, the medium was removed, cells were trypsinized and an equal volume of 0.4% (w/v). Trypan blue dye aqueous solution was added to cell suspension. Viable cells were counted under the phase contrast microscope.
Determination of adenovirus type 7 and rotavirus Wa strain titers using plaque assay
Non-toxic dilutions were mixed (100 μl) with 100 μl of different doses of adenovirus type 40 (1 × 105, 1 × 106, 1 × 107) and the same doses of rotavirus Wa strain. The infectivities of the rotavirus stocks were activated with 10 μg/ml trypsin for 1/2 h at 37 °C. The mixture was incubated for 1/2 h in 37 °C. The inoculation of (100 μl) 10-fold dilutions of treated and untreated Adenovirus type 7 and rotavirus Wa strain was carried out into Hep 2 and MA 104 cell lines for Adenovirus type 7 and rotavirus Wa strain, respectively, in 12 multi-well plates. After 1 h of incubation for adsorption at 37 °C in a 5% CO2-water vapor atmosphere without constant rocking. The plates were rocked intermittently to keep the cells from drying. After adsorption, 1 ml of 2× media [Dulbecco's Modified Eagle Medium (DMEM), Gibco-BRL] plus 1 ml 1% agarose was added to each well, 0.5 μg/ml was added to the media-agarose mixture in the case of rotavirus Wa strain and the plates were incubated at 37 °C in a 5% CO2-water vapor atmosphere. After the appropriate incubation period, the cells were stained with 0.4% crystal violet after formalin fixation, and the number of plaques was counted. The viral titers were then calculated, and expressed as plaque-forming units per milliliter (pfu/ml).
Results and discussion
Chemistry
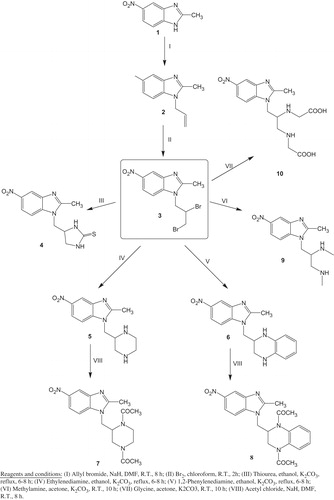
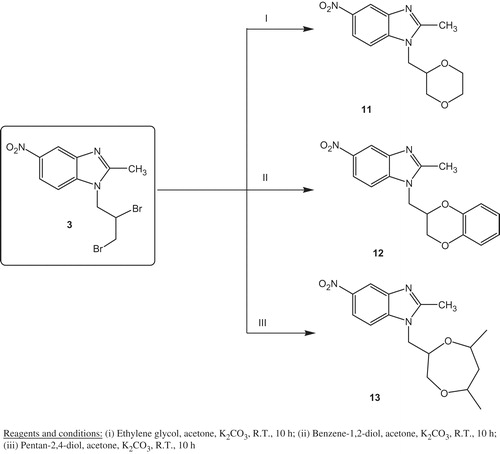
2-Methyl-5-nitro-1H-benzimidazole 1, previously described by our groupCitation17,Citation18,Citation20,Citation41, was used as a starting material to prepare benzimidazole derivatives. During transformation of compound 1 into 2, alkylation occurred using allyl bromide with stirring at room temperature in DMF and sodium hydride. Addition of bromine at the double bond of the allyl group was accomplished in chloroform to produce compound 3. Compound 3 was reacted with thiourea, ethylene diamine and o-phenylene diamine in ethanol and triethylamine to form the benzimidazole derivatives 4, 5 and 6, respectively, with different heterocyclic rings (imidazolidine-2-thione ring in 4, piperazine ring in 5, and 1,2,3,4-tetrahydroquinoxaline ring in 6) at position-1 of the benzimidazole nucleus (Scheme 1)Citation42. Diacetylation of 5 and 6 was performed using acetyl chloride to form the diacetyl benzimidazole derivatives 7 and 8, respectively. Next, Compound 3 reacted with two equivalents of methyl amine or glycine in the presence of potassium carbonate in acetone to achieve the diamino derivatives 9 and 10 respectively (Scheme 1). Scheme 2 represents the formation of three novel benzimidazole derivatives having heterocyclic rings containing oxygen via the reaction of 3 with the dihydroxy compounds. Compound 3 reacted with ethylene glycol, benzene-1, 2-diol, or pentan-2,4-diol in the presence of potassium carbonate in acetone to form the benzimidazole derivatives 11, 12 and 13, respectively, with different oxygen containing heterocyclic rings (1,4-dioxane ring in 11, 2,3-dihydro-benzo[1,4]dioxine ring in 12, and 5,7-dimethyl-[1,4]dioxepane ring in 13) at position-1 of the benzimidazole nucleus in good to excellent yields (Scheme 2).
Scheme 1. Synthesis of novel benzimidazole derivative 3–10 connected at position 1 via methylene linker.
Scheme 2. Synthesis of benzimidazole derivatives 11–13 with heterocyclic rings connected at position 1 via methylene linker (using different diols).
Indeed, 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenylethanone 14, which is considered as an important starting material for synthesis of various benzimidazole derivatives with antiviral and anticancer activity, was prepared by the reaction of 2-methyl-5-nitro-1H-benzimidazole 1 with phenacyl bromide with stirring at room temperature in DMF and sodium hydride for 8 h. Condensation reaction of 14 with various functionalized aromatic and heterocyclic aldehydes was performed under stirring at room temperature in 10% NaOH and ethanol as a solvent to form the benzimidazole derivatives, 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-3-aryl-1-phenylprop-2-en-1-one, 15–18, with α,β-unsaturated ketones (Scheme 3)Citation43. Compounds 15–18 reacted with ethylene diamineCitation44, thioureaCitation44 and hydrazine hydrateCitation45 in ethanol to form the benzimidazole derivatives, 1-(2,3,6,7-tetrahydro-7-aryl-5-phenyl-1H-1,4-diazepin-6-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole derivatives 19–22, 5,6-dihydro-5-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-6-aryl-4-phenylpyrimidine-2(1H)-thione 23–26 derivatives and 1-(4,5-dihydro-5-aryl-3-phenyl-1H-pyrazol-4-yl)-2-methyl-5-nitro-1H-benzo[d]imidazole derivatives 27–30 with seven, six and five-membered heterocyclic rings, respectively, at the position 1 of the benzimidazole nucleus in good to excellent yields (Scheme 3). Interestingly, the two stereoisomers (E and Z) were separated successfully into individual pure components [15a(Z)–18a(Z) as major components and 15b(E)–18b(E) as minor components] by column chromatography on silica gel and characterized by combination of 1H, 13C NMR, IR and Mass Spectrometry. The structures of all the new products were established also by 1H, 13C NMR, IR and Mass Spectrometry. The 1H, 13C NMR spectra as well as the X-ray structure analysis allowed the complete assignment of all signals of the stereoisomers. As the chemical shift is an electronic environment dependent, a similar degree of conjugation between the aromatic ring and the olefinic α,β-unsaturated conjugated carbonyl system provides a closed chemical shift values for the (E) and (Z) isomers. However, the significant differences between the isomers appear in the vinyl proton and the aromatic region in the 1H NMR. The olefinic proton (CH=C) of the (Z) isomers was appeared at more downfield shift than in the case of the olefinic proton (CH=C) of the (E) isomer. This is attributed to the direction of the carbonyl group which is in the same side of the olefinic double bond in the case of the (Z) isomer (Scheme 3). Therefore, the carbonyl group causes de-shielding of the olefinic proton in (Z) isomers. This behavior was observed in the most cases of the (E) and (Z) isomers. For example, in 3-(2-Methoxyphenyl)-2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenylprop-2-en-1-one 16a (Z) the olefinic proton was appeared at δ 8.07 ppm whereas in 16b (E) was appeared at δ 7.95 ppm. This downfield shift in the case of the olefinic proton of 16a (Z) is due to the existence of the carbonyl group and the olefinic double bond at the same side. The 13C NMR signals leads to the observation that the chemical shifts of the carbon nuclei change by about 1–3 ppm between the two (E) and (Z) isomers. The 13C NMR spectra of both (E) and (Z) isomers display CH3 carbon of benzimidazole at position 2 at 13–15 ppm, olefinic carbon–carbon double bond at 130–157 ppm and carbonyl carbon at 190–193 ppm. Interestingly, structure of 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-p-tolylprop-2-en-1-one; 15a(Z); was established by X-ray crystal structure analysis (). The crystallographic data, collection, refinement and the geometric parameters (bond length, bond angle and torsion angle) of 15a(Z) are represented in the Supplementary Material. It is clear from X-ray structure that the (Z) isomer reduces the possible steric interaction between tolyl unit and the phenyl ring adjacent to the carbonyl group. This could much favor the direction of the reaction toward the preference of formation of the favorable (Z) isomer as a more stable conformer. This reason also explains the high yields of the (Z) isomers as the major product compared to the (E) isomers.
Figure 3. X-ray structure of 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-p-tolylprop-2-en-1-one; 15a(Z).
![Figure 3. X-ray structure of 2-(2-methyl-5-nitro-1H-benzo[d]imidazol-1-yl)-1-phenyl-3-p-tolylprop-2-en-1-one; 15a(Z).](/cms/asset/648ecf7b-28bc-4005-914f-ac7c5510c789/ienz_a_979344_f0003_b.jpg)
Scheme 3. Synthesis of benzimidazole derivatives 14–30 with heterocyclic rings directly connected at position 1.
Antitumor activity
In vitro cytotoxicity studies
The in vitro cytotoxic screening of the synthesized compounds against four different cell lines was reported in . From these results, the following important points can be detected. In the case of in vitro cytotoxic activity against human lung adenocarcinoma cells (A-549), three of the tested compounds 3, 9 and 17b (E) (IC50: 28.91, 134.90, 123.70 µM, respectively) showed anticancer activity comparable to doxorubicin (84.10 µM). On the other hand, the other compounds show no appreciable cytotoxic activity against A-549 cell line. For the in vitro cytotoxic activity against human colorectal carcinoma cells (HCT-116), the same three compounds 3, 9 and 17 b (E) (IC50: 97.60, 175.90, 135.90 µM, respectively) from all the tested compounds were found to possess anticancer activity as compared to doxorubicin (111.70 µM). While in the case of the in vitro cytotoxic activity against human hepatocellular carcinoma (Hep G-2), three of the tested compounds, 9, 16a (Z) and 17b (E) exhibited less cytotoxic activity (IC50: 141.70, 135.00, 89.40 µM, respectively) than the standard (IC50: 63.60 µM; ). In the case of in vitro cytotoxic activity against human breast cancer cells (MCF-7), four of the tested compounds showed significant activity which are 3, 9, 13 and 17b (E) (IC50: 142.45, 329.40, 196.00, 161.40 µM, respectively). Compound 3 and 17b (E) were the most active against MCF-7 than doxorubicin (IC50: 163.80 µM). However, the four compounds exhibited less cytotoxic activity than the standard ().
Table 1. % Growth inhibitory activity at 300 µM of the synthesized compounds, indicating IC50 (µg/ml) between brackets. .
.
In vivo toxicity and antitumor study
In vivo assay of acute toxicity
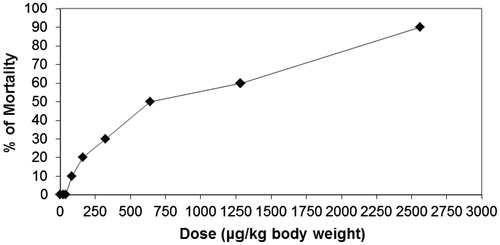
The result revealed that although the prepared compounds which gave positive in vitro were tested for the acute toxicity in healthy animals, compound 9 was the only compound that exhibited acute toxicity in healthy animals. The concentrations required by compound 9 for 50% mortality of the animals was found to be 640 µg/kg body weight as shown in .
Figure 4. In vivo assay of acute toxicity LD50 of the synthesized compound 9 µg/kg body weight.
Hepatoprotective effect of the synthesized compound 9 against liver cancer induced by DENA
As shown in , DENA-treated rats showed significant (p < 0.05) increase in serum AST, ALT and ALP activities, along with significant (p < 0.05) increase in total bilirubin level compared to control. The treatment with compound 9 at a dose of 1/10 of the LD50 values in the rats resulted in normalization in AST, ALT and ALP activities as well as the total bilirubin level compared to DENA-treated (). This means that compound 9 is a potent anticancer agent and has effect on liver cancer induced in rats.
Table 2. Liver function test of normal, DENA- and compound 9-treated DENA rats.
In this study, serum obtained from tumor bearing rats showed significant increase in activities of AST, ALT, ALP along with significant increase in bilirubin level compared to control animals. The elevation of these enzyme activities was indicative of the toxic effect of DENA on the liver tissue. It is known that N-nitroso compounds act as strong carcinogens in various mammals including primatesCitation46. DENA has been shown to be metabolized by cytochrome P-450 IIE1 (CYP 2E1) to its active ethyl radical metabolite, which could interact with DNA causing mutation and carcinogenesisCitation47. Administration of compound 9 to DENA treated rats showed restoration activity of AST, ALT, ALP and bilirubin level towards normal. Such reverse in serum enzyme activities could be attributed to the ability of 9 to inhibit CYP 2E1 activity, presumably by serving as a competitive inhibitor, leading to a decrease in the formation and/or bioactivation of these nitrosamines.
Histopathological examination
The microscopic examinations of liver sections from different groups were summarized in . In this study, histological examination of rat liver sections was consistent with the results obtained from biochemical studies. Liver of control animals as presented in () revealed normal architecture characterized by polyhedral shaped hepatocytes and cytoplasm granulated with small uniform nuclei. Hepatocytes were arranged in well-organized hepatic cords and separated by narrow blood sinusoids.
Figure 5. Liver section of untreated normal control rats [A] showed unremarkable pathological changes. Liver section from rats treated with DENA showed: hyperchromatism, hyperplasia, proliferating hepatocytes [B]; loss architecture, both hepatic and portal with significant tumor thrombi within portal vessels, slightly larger tumor cells having more irregular nuclei [C]; malignant nuclei [D]; megalocytosis, hyperchromatic nuclei, nuclear vacuolization and prominence [E]; dissolution of hepatic cords appeared as empty vacuoles aligned by strands of necrotic hepatocytes [F]; disarrangement of normal hepatic cells with intense centrilobular necrosis [G]. Liver of the DENA-rats treated with compound 9 showed normal hepatic lobule architecture [H] (H&E, magnification 400×).
![Figure 5. Liver section of untreated normal control rats [A] showed unremarkable pathological changes. Liver section from rats treated with DENA showed: hyperchromatism, hyperplasia, proliferating hepatocytes [B]; loss architecture, both hepatic and portal with significant tumor thrombi within portal vessels, slightly larger tumor cells having more irregular nuclei [C]; malignant nuclei [D]; megalocytosis, hyperchromatic nuclei, nuclear vacuolization and prominence [E]; dissolution of hepatic cords appeared as empty vacuoles aligned by strands of necrotic hepatocytes [F]; disarrangement of normal hepatic cells with intense centrilobular necrosis [G]. Liver of the DENA-rats treated with compound 9 showed normal hepatic lobule architecture [H] (H&E, magnification 400×).](/cms/asset/b854eb39-21cf-4eeb-91e6-d02032e58d6d/ienz_a_979344_f0005_b.jpg)
Liver of rats treated with DENA alone showed distortion in the tissue organization with hyperchromatism, hyperplasia, proliferating hepatocytes (), both hepatic and portal with significant tumor thrombi within portal vessels, tumor cells are slightly larger have more irregular nuclei and numerous mitotic figures () with malignant nuclei (). Some section showed megalocytosis, hyperchromatic nuclei as well as nuclear vacuolization and nuclear prominence () dissolution of hepatic cords which appeared as empty vacuoles aligned by strands of necrotic hepatocytes (). Also, disarrangement of normal hepatic cells with intense centrilobular necrosis were observed induced cancer liver (). Theses marked changes in the hepatic architecture could be explained based on that DENA treatment manifested its toxic effects through the generation of ROS. The resulting effect was the production of elevated amounts of malondialdehyde and conjugated dienes, which caused deleterious effects on the membranous components of hepatocytes.
Liver of the DENA-rats treated with compound 9 showed improvement in the hepatic pattern with more or less normal hepatic lobule architecture (). The histological results confirmed the biochemical results which improved that compound 9 is a potent anticancer agent.
Taken together, from the biochemical and histological data, our results demonstrate that the synthesized compound 9 may be potent anticancer agents for inclusion in modern clinical trials.
Antiviral activity
represented the non-toxic doses of tested materials on MA104 and Hep2 cell lines. Similar sensitivity of the two types of cell lines to toxic materials in tested samples was observed. However, slight high resistance to toxic materials was observed with Hep-2 cell line. Considerable antiviral activity against rotavirus Wa strain which percentage of reduction of infectious rotavirus particles was 66.7, 70 and 63.3% for materials 18a (Z), 17b (E) and 17a (Z), respectively (). Less percentage of reduction was observed with adenovirus type 7 which was 56.7 and 50% for materials 17b (E) and 17a (Z), respectively (). The higher resistance to effect of tested materials for adenovirus than rotavirus may return to the nature of the genome of the two viruses. Adenovirus is a DNA virus while rotavirus is RNA virus. The activity of the rest of the materials is weak which ranged from 10% to 20% on rotavirus Wa strain and 0% to 20% on adenovirus type 7 (data not shown).
Table 3. Non-toxic doses of tested materials on MA104 and Hep2 cell lines.
Table 4. Anti-rotavirus Wa strain activity of non-toxic doses from tested materials.
Table 5. Anti-adenovirus type 7 strain activity of non-toxic doses from tested materials.
Structure–activity relationship
The best substitution at position 1 of benzimidazole was found to be the 2,3-dibromopropyl substituent in compound 3 to obtain the best cytotoxicity. This may be due to the flexible aliphatic chain substituted with the voluminous electron donating two bromine atoms making the best fitting with the receptor. Also, the propyl-1,2-diamine was a favored substitution at position 1 of benzimidazole in compound 9 as it also possesses both flexibility as well as the electronegative two amino groups. It was noticed that 9 was more active than 10 having the diglycinyl groups, as the presence of the two carboxylic groups demolishes the activity. Substitution at position 1 with the 2-propen-1-one chain having 2-methylfuryl ring in the E configuration was favored due to steric factors as well as the possible hydrophobic interaction of the 2-methylfuryl ring with the active site. Similarly compound 17b was found to be the most active among the series against rotavirus Wa strain, which can be attributed to the same previously mentioned effects. Compounds 17a and 18a were active against the virus but to a lesser extent than 17b. The activity of both compounds may be also due to the hydrophobic interaction of the 2-methyl furyl and of the thienyl ring with the receptor. The antitumor activity was abolished by the presence of heterocyclic rings directly linked or linked through a methylene group to position 1.
Conclusion
A series of novel benzimidazole derivatives substituted at position 1 by heterocyclic ring systems were synthesized and evaluated for their cytotoxicity against A-549, HCT-116, Hep-G2 and MCF-7 cell lines (in vitro and in vivo) and antiviral activity (against rotavirus Wa strain and adenovirus type 7). The in vitro cytotoxic screening of the new compounds against four different cell lines was achieved. Compound 3 was more active than doxorubicin against A-549, HCT-116 and MCF-7. Compound 9 and 17b (E) showed potency near to doxorubicin against the four cell lines. The acute toxicity of compound 9 on liver cancer induced in rats was determined in vivo. Interestingly, it showed restoration activity of liver function and pathology towards normal as compared to the cancer-bearing rats induced by DENA. In addition, compounds 18a (Z), 17b (E) and 17a (Z) were the most promising antiviral compounds against rotavirus Wa strain, while 17b (E) and 17a (Z) showed considerable antiviral activity against adenovirus type 7.
Supplementary material available online.
Supplemental Material.pdf
Download PDF (2.2 MB)Declaration of interest
The authors report no conflicts of interest.
References
- Qian Y, Ling CQ. Preventive effect of Ganfujian granule on experimental hepatocarcinoma in rats. World J Gastroenterol 2004;10:755–7
- Farazi PA, Depinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674–87
- Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin 2009;59:225–49
- Reh BD, Fajen JM. Worker exposures to nitrosamines in a rubber vehicle sealing plant. Am Ind Hyg Assoc J 1996;57:918–23
- Bansal AK, Bansal M, Soni G, Bhatnagar D. Protective role of Vitamin E pre-treatment on N-nitrosodiethylamine induced oxidative stress in rat liver. Chem Biol Interact 2005;156:101–11
- Sayed-Ahmed MM, Aleisa AM, Al-Rejaie SS, et al. Thymoquinone attenuates diethylnitrosamine induction of hepatic carcinogenesis through antioxidant signaling. Oxid Med Cell Longev 2010;3:254–61
- Hsu PC, Buxton JA, Tu AW, et al. Publicly funded pegylated interferon-alpha treatment in British Columbia: disparities in treatment patterns for people with hepatitis C. Can J Gastroenterol 2008;22:359–64
- Denny WA, Rewcastle GW, Baguly BC. Potential antitumor agents. 59. Structure-activity relationships for 2-phenylbenzimidazole-4-carboxamides, a new class of “minimal” DNA-intercalating agents which may not act via topoisomerase II. J Med Chem 1990;33:814–9
- McKellar QA, Scott EW. The benzimidazole anthelmintic agents – a review. J Vet Pharmacol Ther 1990;13:223–47
- Spasov AA, Yozhitsa IN, Bugaeva LI, Anisimova VA. Benzimidazole derivatives: spectrum of pharmacological activity and toxicological properties (a review). Pharm Chem J 1999;33:232–43
- Rossignol JF, Maisonneuve H. Benzimidazoles in the treatment of trichuriasis: a review. Ann Trop Med Parasitol 1984;78:135–44
- Boiani M, Gonzalez M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini Rev Med Chem 2005;5:409–24
- Desai GK, Desai RK. Green route for the heterocyclization of 2-mercaptobenzimidazole into beta-lactum segment derivatives containing -CONH- bridge with benzimidazole: screening in vitro antimicrobial activity with various microorganisms. Bioorg Med Chem Soc 2006;14:8271–9
- Patil A, Ganguly S, Surana S. A systematic review of benzimidazole derivatives as an antiulcer agent. Rasayan J Chem 2008;1:447–60
- Dubey AK, Sanyal PK. Benzimidazoles in a Wormy World. Online Vet J 2010;5:63
- Narasimhan B, Sharma D, Kumar P. Benzimidazole: a medicinally important heterocyclic moiety. Med Chem Res 2012;21:269–83
- Ramla MM, Omar MA, El-Khamry AM, El-Diwani HI. Synthesis and antitumor activity of 1-substituted-2-methyl-5-nitrobenzimidazoles. Bioorg Med Chem 2006;14:7324–32
- El-NaemSh I, El-Nazhawy AO, El-Diwani HI, Abdel Hamid AO. Synthesis of 5-substituted 2-methylbenzimidazoles with anticancer activity. Arch Pharm 2003;336:7–17
- Ng RA, Guan J, Alford VC, et al. 2-(2,2,2-Trifluoroethyl)-5,6-dichlorobenzimidazole derivatives as potent androgen receptor antagonists. Bioorg Med Chem Lett 2007;17:955–8
- Omar MA, Shaker YM, Galal SA, et al. Synthesis and docking studies of novel antitumor benzimidazoles. Bioorg Med Chem 2012;20:6989–7001
- Temirak A, Shaker YM, Ragab FAF, et al. Part I. Synthesis, biological evaluation and docking studies of novel 2-furylbenzimidazoles as anti-angiogenic agents. Eur J Med Chem 2014;1–13. (in press)
- Temirak A, Shaker YM, Ragab FAF, et al. Part II. Synthesis, biological evaluation and docking studies of new 2-furylbenzimidazoles as anti-angiogenic agents. Arch Pharm Chem Life Sci 2014;347:291–304
- Tsay SC, Hwu JR, Singha R, et al. Coumarins hinged directly on benzimidazoles and their ribofuranosides to inhibit hepatitis C virus. Eur J Med Chem 2013;63:290–8
- Li YF, Wang GF, Luo Y, et al. Identification of 1-isopropylsulfonyl-2-amine benzimidazoles as a new class of inhibitors of hepatitis B virus. Eur J Med Chem 2007;42:1358–64
- Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J Biol Chem 1989;264:2250–7
- Townsend LB, Devivar RV, Turk SR, et al. Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(beta-D-ribofuranosyl)benzimidazoles. J Med Chem 1995;38:4098–105
- Biron KK. Antiviral drugs for cytomegalovirus diseases. Antiviral Res 2006;71:154–63
- Williams JD, Chen JJ, Drach JC, Townsend LB. Synthesis and antiviral activity of 3-formyl- and 3-cyano-2,5,6-trichloroindole nucleoside derivatives. J Med Chem 2004;47:5766–72
- Andries K, Moeremans M, Gevers T, et al. Substituted benzimidazoles with nanomolar activity against respiratory syncytial virus. Antiviral Res 2003;60:209–19
- Hirashima S, Suzuki T, Ishida T, et al. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J Med Chem 2006;49:4721–36
- Hirashima S, Oka T, Ikegashira K, et al. Further studies on hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors toward improved replicon cell activities: benzimidazole and structurally related compounds bearing the 2-morpholinophenyl moiety. Bioorg Med Chem Lett 2007;17:3181–6
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Miura S, Yoshimura Y, Endo M, et al. Antitumor activity of a novel orally effective nucleoside, 1-(2-deoxy-2-fluoro-4-thio-β-d-arabinofuranosyl)cytosine. Cancer Lett 1998;129:103–10
- Thabrew MI, Hughes RD, McFarlane IG. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol 1997;49:1132–5
- Al-Rejaie SS, Aleisa AM, Al-Yahya AA, et al. Progression of diethylnitrosamine-induced hepatic carcinogenesis in carnitine-depleted rats. World J Gastroenterol 2009;15:1373–80
- Prieur DJ, Young DM, Davis RD, et al. Procedures for preclinical toxicologic evaluation of cancer chemotherapeutic agents: protocols of the laboratory of toxicology. Cancer Chemother Rep 1973;4:1–28
- Ghosh MN. Fundamentals of experimental pharmacology. Calcutta, India: Scientific Book Agency; 1984:153–8
- Conn HJ, Darrow MA, Emmel VM, Staining procedure used by the biological stain commission. 2nd ed. Baltimore, MD: Williams & Wilkins Co.; 1960:289
- Simões CMO, Amoros M, Girre L. Mechanism of antiviral activity of triterpenoid saponins. Phytother Res 1999;13:323–8
- Walum E, Stenberg K, Jenssen D. Understanding cell toxicology: principles and practice Ellis Horwood. J Appl Toxicol 1991;11:389–90
- Ramla MM, Omar MA, Tokuda H, El-Diwani HI. Synthesis and inhibitory activity of new benzimidazole derivatives against Burkitt's lymphoma promotion. Bioorg Med Chem 2007;15:6489–96
- Holla BC, Akberali PM, Shivananda MK. Studies on nitrophenylfuran derivatives – part XII. Synthesis, characterization, antibacterial and antiviral activities of some nitrophenylfurfurylidene-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines. IL Farmaco 2001;56:919–27
- Turan-Zitouni G, Ozdemir A, Guven K. Synthesis of some 1-[(N,N-disubstituted thiocarbamoylthio)acetyl]-3-(2-thienyl)-5-aryl-2-pyrazoline derivatives and investigation of their antibacterial and antifungal activities. Arch Pharm Chem Life Sci 2005;338:96–104
- Turan-Zitouni G, Chevallet P, Kilic FS, Erol K. Synthesis of some thiazolyl-pyrazoline derivatives and preliminary investigation of their hypotensive activity. Eur J Med Chem 2000;35:635–41
- Zampieri D, Mamolo MG, Laurini E, et al. Antifungal and antimycobacterial activity of 1-(3,5-diaryl-4,5-dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives. Bioorg Med Chem 2008;16:4516–22
- Swenberg JA, Hoel DG, Magee PN. Mechanistic and statistical insight into the large carcinogenesis bioassays on N-nitrosodiethylamine and N-nitrosodimethylamine. Cancer Res 1991;51:6409–14
- Anis KV, Rajeshkumar NV, Kuttan R. Inhibition of chemical carcinogenesis by berberine in rats and mice. J Pharm Pharmacol 2001;53:763–8