
Abstract
Enterovirus 71 (EV71) is a highly infectious pathogen primarily responsible for Hand, Foot, and Mouth Disease, particularly among children. Currently, no approved antiviral drug has been developed against this disease. The EV71 3C protease is deemed an attractive drug target due to its crucial role in viral polyprotein processing. Rupintrivir, a peptide-based inhibitor originally developed to target the human rhinovirus 3C protease, was found to inhibit the EV71 3C protease. In this communication, we report the inhibitory activities of 30 Rupintrivir analogs against the EV71 3C protease. The most potent inhibitor, containing a P2 ring-constrained phenylalanine analog (compound 9), was found to be two-fold more potent than Rupintrivir (IC50 value 3.4 ± 0.4 versus 7.3 ± 0.8 μM). Our findings suggest that employing geometrically constrained residues in peptide-based protease inhibitors can potentially enhance their inhibitory activities.
Introduction
Enterovirus 71 (EV71) is a single-stranded, positive-sense RNA virus belonging to the genus Enterovirus, family PicornaviridaeCitation1. This highly infectious pathogen is spread through the oral-fecal route and is primarily responsible for Hand, Foot, and Mouth Disease (HFMD) in humans, particularly young children, with outbreaks occurring in the Asia-Pacific region including Australia, China, Malaysia, Singapore, Taiwan, Thailand and VietnamCitation2. Infection symptoms include fever, vomiting, vesicular rashes on the hands, feet, buttocks, and oropharyngeal ulcersCitation3. Although the disease is generally self-limiting, a small percentage of patients develop severe neurological complications including encephalitis, aseptic meningitis, and poliomyelitis-like flaccid paralysisCitation2,Citation4. The virus is highly contagious; a 2008 outbreak in China resulted in 488 955 human infections with 126 fatalitiesCitation5 and, more recently, a 2011 outbreak resulted in more than 1.6 million human infections causing more than five hundred deathsCitation6. These grim statistics highlight an urgent need for the development of antivirals against EV71 as there is a current lack of effective drugs for treating this diseaseCitation2,Citation6.
An appealing drug target is the viral 3C cysteine protease which plays a crucial role in cleaving virus-encoded polyproteins into mature viral proteins specifically at the polyprotein’s Gln–Gly peptide bond junctionCitation6,Citation7. Indeed, peptidomimetics mimicking the protease’s recognition substrate ALFQ-X, with ‘X’ representing a reactive electrophile like an aldehyde or an acrylate Michael acceptor, have been shown to inhibit the EV71 3C proteaseCitation8–10. Rupintrivir (AG-7088; ), a peptide-based drug candidate with an electrophilic ethyl propenoate Michael acceptor at its C-terminus, was originally developed as a human rhinovirus virus (HRV) 3C protease inhibitor by Agouron PharmaceuticalsCitation12–15. Recently, Rupintrivir showed promising results when used for treating EV71-infected miceCitation16. The rationale for this was because HRV, like EV71, belongs to the same Picornaviridae family of viruses, suggesting that a viral protease inhibitor developed for HRV might also be effective on EV71Citation12. When bound to the viral 3C protease active site, the ethyl propenoate moiety reacts with the protease’s nucleophilic Cys147 –SH via a Michael addition to form an S–C covalent bond, irreversibly inhibiting the protease. Indeed, this mechanism of action was further supported by X-ray crystallography studies using co-crystals of Rupintrivir bound to the EV71 3C proteaseCitation9,Citation10. However, it must be emphasized that Rupintrivir was originally optimized to target the HRV 3C protease and not EV71. A primary sequence comparison between the two viral proteases revealed a 43% sequence identity over 183 residues, suggesting that there might be room for further structural optimization for the EV71 3C protease. In this structure–activity relationship (SAR) study, we report the synthesis and inhibitory activities (IC50) of 30 Rupintrivir analogs against the EV71 3C protease.
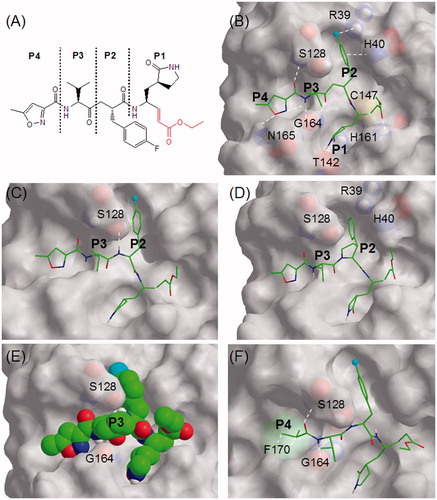
Figure 1. (A) Schematic diagram of Rupintrivir showing P1, P2, P3, and P4 regions using conventional terminologyCitation11. The ethyl propenoate moiety is depicted in red; (B) the stick model of Rupintrivir (green) in the EV71 3C protease catalytic site (3SJO.pdb). Protease residues involved in ligand interaction are numbered and shown as CPK representations, the cyan sphere represents fluorine and white hashes represent possible interactions; (C) Cpd. 2 showing the extra H-bond between the P2 NH and S128; (D) Cpd. 9 with its P2 phenylpyrrolidine moiety oriented in the S2 subsite; (E) the CPK model of Cpd. 16 showing its P3 cyclohexyl sandwiched between S128 and G164; (F) Cpd. 21 showing hydrophobic interactions between its P4 N-cap and F170 phenyl group (green CPK representation) in the S4 subsite. Figures C–F were obtained from molecular modeling.
Methods
The general synthetic procedure for compounds 2–31 is outlined in Scheme 1. Detailed synthetic protocols and spectral data can be found in the Supplementary notes. Inhibitory activities of the synthesized peptidomimetics were generated using a published EV71 3C protease inhibition assayCitation17 (see Supplementary information for details) and are summarized in . Molecular modeling and energy minimization were performed using Macromodel 9.9 (Schrödinger, New York, NY) (see Supplementary information for details).
Scheme 1. General synthetic procedure for compounds 2–31. Reagents and conditions: (a) swell 2-chlorotrityl chloride resin in DMF, 25 °C, 30 min; (b) appropriate Fmoc-protected amino acid: Fmoc-R1-OH, DIPEA, CH2Cl2, 25 °C, 30 min; (c) 20% piperidine in DMF (v/v), 25 °C, 30 min; (d) appropriate Fmoc-protected amino acid: Fmoc-R2-OH, HBTU, HOBT, DIPEA, DMF, 25 °C, 1 h; (e) appropriate carboxylic acid: R3COOH, HBTU, HOBt, DIPEA, DMF, 25 °C, 1 h; (f) 95% TFA in CH2Cl2 (v/v), 25 °C, 30 min; (g) (S,E)-ethyl 4-amino-5-(S-2-oxopyrrolidin-3-yl)pent-2-enoate, DIC, HOBT, DMF, 25 °C, 16 h.
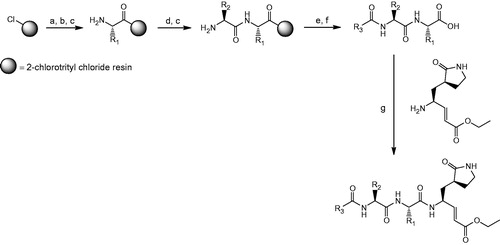
Table 1. Structures and inhibitory data of Rupintrivir analogs against EV71 3C protease.
Results and discussion
Rupintrivir (compound 1; ), used as a reference in this study, possessed an IC50 value of 7.3 ± 0.8 μM against the EV71 3C protease (), close to the 2.3 ± 0.5 μM previously reportedCitation10. A structure of Rupintrivir bound to the EV71 3C protease active site obtained using X-ray crystallography (; 3SJO.pdb)Citation9 revealed several noteworthy features: (a) the overall protease active site was relatively shallow with the exception of the S1 and S2 subsites; (b) the catalytic Cys147 formed a S–C covalent bond to the inhibitor’s ethyl propenoate moiety; (c) the S1 subsite appeared to accommodate a 5-membered lactam well with H-bonding interactions to Thr142 and His161 of the protease; (d) the relatively narrow S2 subsite could accommodate the P2 phenyl and is involved in π–π interactions with His40 while the aromatic para-F was likely involved in electrostatic interactions with the cationic Arg39; (e) the S3 subsite was located above the inhibitor backbone, sandwiched between protease residues Ser128 and Gly164 with the P3 sidechain being solvent-exposed; (f) the S4 subsite spanned a relatively large but shallow area, suggesting that the potential for further P4 optimization might be somewhat limited.
Our SAR study started with the P2 residue. The first analog, compound 2 (), was identical to Rupintrivir except that the backbone methylene between P2 and P3 was substituted with NH (). Surprisingly, this did not significantly affect its IC50 value compared with Rupintrivir (IC50 value 6.2 ± 0.6 versus 7.3 ± 0.8 μM), suggesting that the additional hydrogen bond between the P2 and P3 amide NH to the Ser128 side-chain hydroxyl did not play a significant role in binding (). We next moved the aromatic para-F to the ortho- and meta- positions (compounds 3 and 4, respectively) and observed approximate two-fold reduction in inhibitory activities (). This suggested that the protease preferred a para-F and that the electrostatic-like interaction between the para-F to Arg39 played an important role in inhibitor binding. We further tested this hypothesis by removing the fluorine (compound 5). As expected, a two-fold reduction in inhibitory activity was observed compared with compound 2 (IC50 value 13.0 ± 1.9 versus 6.2 ± 0.6 μM, respectively; ). Substituting the para-F with para-trifluromethyl and para-carboxyl moieties (compounds 6 and 7, respectively) yielded interesting results; the para-CF3 (compound 6) substitution resulted in similar activity to para-F (compound 2) with IC50 values of approximately 6 μM while the para-COOH (compound 7) substitution abrogated inhibitory activity (IC50 value >100 μM), suggesting that the carboxyl group was too large to fit into the S2 subsite. To further explore the S2 subsite specificity, we replaced the P2 phenyl with a 5-membered thiophene (compound 8), a known phenyl group bioisostereCitation18. Intriguingly, this improved inhibition almost two-fold compared to the P2 phenylalanine of compound 5 (IC50 value 7.5 ± 0.9 versus 13.0 ± 1.9 μM), suggesting that thiophene was a suitable bioisostere for phenylalanine. In our final P2 analog, we substituted the P2 phenylalanine with a 3-phenylpyrrolidine moiety which could be considered as a geometrically constrained phenylalanine analog (compound 9; ). Interestingly, assay results revealed it to be approximately four-fold more potent than compound 5 with a P2 phenylalanine (IC50 value 3.4 ± 0.4 versus 13.0 ± 1.9 μM) and two-fold more potent than the reference compound Rupintrivir (IC50 value 3.4 ± 0.4 versus 7.3 ± 0.8 μM). Molecular modelingCitation19–21 suggested that the 5-membered pyrrolidine moiety could fit into the active site with the phenyl ring optimally oriented in the S2 subsite (). In addition, the highly constrained ring structure could have played a significant role in reducing the entropic binding penalty, resulting in a two-fold increase in inhibitory activity over Rupintrivir. This finding could potentially open avenues towards the design of new peptidomimetic inhibitors containing geometrically constrained phenylalanine surrogates.
We next shifted our focus to the P3 residue. Close inspection of the X-ray crystal (3SJO.pdb) of Rupintrivir’s P3 valine residue revealed that its N-terminal NH and C-terminal CO were involved in H-bonding to Gly164 while the iso-propyl side-chain was flanked by Ser128 and Gly 164 and was mainly solvent exposed (). Thus, any P3 variation was not expected to affect IC50 values significantly. Surprisingly, our analog with a P3 glycine (compound 10; ) caused a five-fold decrease in inhibitory activity compared with compound 2 (32.8 ± 4.1 versus 6.2 ± 0.6 μM). We postulated this to be due to the higher entropy penalty paid on ligand-protease binding since glycine, lacking a side-chain, was significantly more flexible than valine in compound 2 (). Replacing the P3 valine with alanine (compound 11) altered the IC50 value slightly (7.7 ± 0.5 versus 6.2 ± 0.6 μM), while an amino iso-butyric acid residue replacement (compound 12) resulted in a two-fold decrease in inhibitory activity over compound 2 (13.4 ± 1.1 versus 6.2 ± 0.6 μM). The latter result suggested that the P3 subsite was too small to fit an extra α-methyl group. Replacing the P3 valine with more bulky residues (compounds 13–19; ) proved detrimental to inhibitory activities (). For example, substituting the P3 valine with cyclohexylglycine (compound 16) resulted in a three-fold decrease in inhibitory activity over compound 2 (18.3 ± 1.7 versus 6.2 ± 0.6 μM; ). Molecular modeling suggested it to be due to unfavorable steric hindrance between the cyclohexyl ring and flanking residues Ser128 and Gly164 ().
Our final series of analogs involved varying the P4 N-cap. In Rupintrivir, this was represented by a 5-methylisoxazole-3-carbonyl moiety (compound 1; ). Closer inspection of revealed its N to be involved in intramolecular H-bonding to the P3 valine NH while its aromatic O and carbonyl O were H-bonded to Asn165 and Ser128, respectively (). This intricate H-bonding network, although originally optimized for the HRV 3C protease, seemed coincidentally well suited for the EV71 3C protease as both viral proteases shared a relatively high (43%) sequence identity. Indeed, our first P4 analog, with an acetyl N-cap (compound 20) exhibited a 1.5-fold decrease in inhibitory activity compared with compound 2 (9.5 ± 0.7 versus 6.2 ± 0.6 μM; ) and we attributed this to the loss of intermolecular H-bonding interactions with Asn165 which were present for the 5-methylisoxazole-3-carbonyl N-cap (). Interestingly, replacing the acetyl with an iso-propyl moiety (compound 21) did not alter inhibitory activity significantly when compared with compound 2 (IC50 value 7.2 ± 0.6 versus 6.2 ± 0.6 μM; ). This was postulated to be due to hydrophobic interactions between the iso-propyl and the phenyl group of Phe170 located in the S4 subsite (). Replacing the N-cap with phenyl and the larger naphthalene moieties (compounds 22–25) resulted in less potent inhibitors (IC50 values ranging from 12.9 to 33.3 μM; ), suggesting that larger ring systems were unsuitable for the protease’s S4 subsite. We postulated this to be due to steric hindrance caused by Asn165 located at the edge of the S4 subsite. On this premise, we focused our attention to 5-membered isoxazole analogs (compounds 26–31; ). The 5-membered 3-furoate moiety of compound 26 exhibited a 1.5-fold decrease in inhibitory activity compared with compound 2 (9.3 ± 0.7 versus 6.2 ± 0.6 μM, respectively) and was suspected to be due to the loss of the intramolecular H-bonding interaction between the P4 aromatic N and the backbone amide NH of the P3 valine (). Compound 27 with a 2-furoate moiety suffered a more than 2-fold decrease in inhibitory activity compared to compound 2 (IC50 value 16.1 ± 1.9 versus 6.2 ± 0.6 μM, respectively; ) and this was attributed to the loss of H-bonding to Asn165 located at the edge of the S4 subsite. Intriguingly, removing the 5-methyl group from the P4 isoxazole (compound 28; ) did not significantly affect its IC50 value when compared with compound 2 (7.0 ± 0.9 versus 6.2 ± 0.6 μM, respectively; ), suggesting that the isoxazole’s methyl group did not play a significant role in hydrophobic interactions in the S4 subsite. Indeed, replacing the methyl group with longer and bulkier alkyl groups did not alter IC50s significantly (compounds 29–31; ), suggesting that the potential for further P4 optimization might be limited.
Conclusions
EV71 is a highly infectious pathogen primarily responsible for HFMD, particularly in children. To date, there are no effective antiviral drugs to treat this diseaseCitation2,Citation6. The EV71 3C protease is deemed an attractive drug target due to its role in viral polyprotein processing. Rupintrivir, a peptide-based HRV 3C inhibitor, has been shown to inhibit EV71 3C proteaseCitation8–10. As Rupintrivir was originally designed to inhibit the HRV 3C protease, we postulated that there could be room for further optimization against the EV71 protease. Our SAR study using Rupintrivir analogs identified compound 9 to be the most potent inhibitor. Compound 9 contained a P2 ring-constrained phenylalanine analog that was two-fold more potent than Rupintrivir in our protease inhibition assay (IC50 value 3.4 ± 0.4 versus 7.3 ± 0.8 μM, respectively). This suggested that employing geometrically constrained residues was a plausible strategy to improve the inhibitory activities of peptide-based protease inhibitors and should provide valuable insights into the design of future peptide-based antivirals against HFMD.
Supplementary material available online
Supplementary data
Supplemental Material.pdf
Download PDF (170.4 KB)Declaration of interest
The authors report that they have no conflicts of interest. We thank A*STAR Biomedical Research Council for financial support.
References
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 2005;3:765–76
- Wu KX, Ng MM-L, Chu JJH. Developments towards antiviral therapies against enterovirus 71. Drug Discov Today 2010;15:1041–51
- McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 2002;26:91–107
- Rhoades RE, Tabor-Godwin JM, Tsueng G, Feuer R. Enterovirus infections of the central nervous system. Virology 2011;411:288–305
- Yang F, Ren L, Xiong Z, et al. Enterovirus 71 outbreak in the People's Republic of China in 2008. J Clin Microbiol 2009;47:2351–2
- Shang L, Xu M, Yin Z. Antiviral drug discovery for the treatment of enterovirus 71 infections. Antiviral Res 2013;97:183–94
- Norder H, De Palma AM, Selisko B, et al. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antiviral Res 2011;89:204–18
- Kuo CJ, Shie JJ, Fang JM, et al. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem 2008;16:7388–98
- Lu G, Qi J, Chen Z, et al. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J Virol 2011;85:10319–31
- Wang J, Fan T, Yao X, et al. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol 2011;85:10021–30
- Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 1967;27:157–62
- Binford SL, Maldonado F, Brothers MA, et al. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother 2005;49:619–26
- Matthews DA, Dragovich PS, Webber SE, et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci USA 1999;96:11000–7
- Patick AK, Binford SL, Brothers MA, et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother 1999;43:2444–50
- Patick AK. Rhinovirus chemotherapy. Antiviral Res 2006;71:391–6
- Zhang X, Song Z, Qin B, et al. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Res 2013;97:264–9
- Wang QM, Johnson RB, Cox GA, et al. A continuous colorimetric assay for rhinovirus-14 3C protease using peptide p-nitroanilides as substrates. Anal Biochem 1997;252:238–45
- Poulie CBM, Bunch L. Heterocycles as nonclassical bioisosteres of α-amino acids. ChemMedChem 2013;8:205–15
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 2001;105:6474–87
- Polak E, Ribière G. Note on the convergence of methods of conjugate directions. Recherche opérationelle, Série Rouge 1969;16:35–43
- Hasel W, Hendrickson TF, Still WC. A rapid approximation to the solvent accessible surface areas of atoms. Tetrahedron Comput Methodol 1988;1:103–16