Abstract
In order to study the structure–activity relationship of Flavokawain B Mannich-based derivatives as acetylcholinesterase (AChE) inhibitors in our recent investigation, 20 new nitrogen-containing chalcone derivatives (4 a–8d) were designed, synthesized, and evaluated for AChE inhibitory activity in vitro. The results suggested that amino alkyl side chain of chalcone dramatically influenced the inhibitory activity against AChE. Among them, compound 6c revealed the strongest AChE inhibitory activity (IC50 value: 0.85 μmol/L) and the highest selectivity against AChE over BuChE (ratio: 35.79). Enzyme kinetic study showed that the inhibition mechanism of compound 6c against AChE was a mixed-type inhibition. The molecular docking assay showed that this compound can both bind with the catalytic site and the peripheral site of AChE.
Introduction
Alzheimer's disease (AD), one of the most common diseases in the elderly population, is a chronic and progressive neurodegenerative disorder characterized by memory loss, language impairment, and intellectual ability degressionCitation1–3. Although the precise aetiology of AD is not elucidated enough, the decrease of acetylcholine level in brain was thought as the widely approved reason and AChE inhibitors were primary drugs for the therapy of AD which can improve central cholinergic functionCitation4,Citation5.
In recent years, natural products were considered to be new promising sources for the treatment of AD. Many natural products or their derivatives were discovered or synthesized which revealed AChE inhibitory activityCitation6–11. Flavokawain B, a natural product with chalcone scaffold, was selected to be as material to synthesize a series of Flavokawain B Mannich-base derivatives which revealed a moderate AChE inhibitory activity in our previous studyCitation12. Among them, the derivatives containing dimethylamine, diethylamine, piperidine, or pyrrolidine groups showed better inhibitory activity than others. In order to study the structure–activity relationship (SAR) of Flavokawain B derivatives as AChE inhibitors, we decided to modify the structures. By studying the characteristics of AChE inhibitors in clinical application or in the development, we supposed that tertiary amine groups were the possible key pharmacophores for AChE inhibitory activityCitation13–15.
Based on our previous investigations, a new series of chalcone derivatives with different tertiary amine groups were synthesized and evaluated for their biological activity. Additionally, we measured their Logarithm 1-octanol/water partition coefficients (log P values), which can be used to evaluate the ability to penetrate the blood brain barrier (BBB). Then kinetic experiments were performed to clarify their binding mode to AChE and molecular docking studies were carried out to explore their binding mode with AChE and BuChE.
Materials and methods
Chemistry
All chemicals and reagents were of analytical reagent grade and used without further purification. The melting points were measured on a WRS-lA melting point detector. 1H NMR spectra were recorded on a Bruker 400 MHz instrument (Bruker Nano, Inc., Billerica, MA) in CDCl3 with TMS as the internal reference. Mass spectra were obtained from Finnigan LCQ Advantage MAX by electrospray ionization (ESI-MS) (ThermoFinnigan, San Jose, CA). Infrared spectrum was obtained from Shimadzu Infinity-1 infrared spectrometer (Shimadzu Corporation, Kanagawa, Japan). The purity of compounds was checked by Shimadzu LC-20 A high-performance liquid chromatography (Shimadzu Corporation, Kanagawa, Japan).
Synthesis of 3′-hydroxychalcone (2)
Benzaldehyde (2.6 mL, 26 mmol), EtOH (20 mL), 20% NaOH (10 mL) were added into a solution containing 3-hydroxyacetophenone (2.72 g, 20 mmol). After stirring at room temperature for 24 h, 15% HCl was added to adjust the pH of solution to 3, followed by the appearance of the precipitate. The reaction mixture was filtrated and a light yellow solid product was gained with a yield of 87.8%. Mp: 175–177 °C.
General procedure for the synthesis of 3a–3e
A mixture of compound 2 (2.24 g, 10 mmol), K2CO3 (4.13 g, 30 mmol), and α,ω-dibromoalkane (60 mmol) were dissolved in DMF (20 mL) and stirred at 80 °C until compound 2 disappeared by monitoring with TLC. Then the mixture was poured into ice water (120 mL), extracted with ethyl acetate (2 × 100 mL), and washed with saturated NaCl solution (2 × 100 mL). The organic phase was dried with anhydrous Na2SO4 and concentrated in vacuum. Then the residue was purified by a silica-gel column chromatography to afford the final product.
1 (E)-1 -(3-(2-bromoethoxy)phenyl)-3-phenylprop-2-en-1-one (3a)
Compound 3a was synthesized from compound 2 (2.24 g, 10 mmol) with K2CO3 (4.13 g, 30 mmol) in DMF (20 mL) and 1,2-dibromoethane (5.2 mL, 60 mmol), followed by purification using silica-gel column chromatography with ethyl acetate/petroleum ether (1:12, v/v) as an eluent. Yield: 53.8%, light yellow solid; m.p. 122–124 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 3.69 (2H, t, J = 6.0 Hz, BrCH2), 4.37 (2H, t, J = 6.0 Hz, OCH2), 7.12–7.16 (1H, m, 4′-H), 7.40–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.55–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: IR (KBr) ν/cm−1: 3028, 2952, 2866, 2763, 1659, 1601, 1576, 1341, 1221, 1182, 833, 769. MS m/z (ESI): 331 [M + H]+.
(E)-1-(3-(3-bromopropoxy)phenyl)-3-phenylprop-2-en-1-one (3b)
Compound 3b was synthesized from compound 2 (2.24 g, 10 mmol) with K2CO3 (4.13 g, 30 mmol) in DMF (20 mL) and 1,3-dibromopropane (6.2 mL, 60 mmol), followed by purification using silica-gel column chromatography with ethyl acetate/petroleum ether (1:14, v/v) as an eluent. Yield: 61.3%, light yellow solid. m.p. 114–116 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 2.34–2.53 (2H, m, CH2CH2Br), 3.66 (2H, t, J = 6.0 Hz, CH2CH2Br), 4.21 (2H, t, J = 6.0, OCH2CH2), 7.12–7.15 (1H, m, 4′-H), 7.40–7.44 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.83 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3030, 2955, 2877, 2761, 1658, 1609, 1587, 1506, 1338, 1229, 1175, 834, 771. MS m/z (ESI): 345 [M + H]+.
(E)-1-(3-(4-bromobutoxy)phenyl)-3-phenylprop-2-en-1-one (3c)
Compound 3c was synthesized from compound 2 (2.24 g, 10 mmol) with K2CO3 (4.13 g, 30 mmol) in DMF (20 mL) and 1,4-dibromobutane (7.2 mL, 60 mmol), followed by purification using silica-gel column chromatography with ethyl acetate/petroleum ether (1:16, v/v) as an eluent. Yield: 65.4%, white solid. m.p. 108–110 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.92–2.03 (2H, m, OCH2CH2), 2.09–2.15 (2H, m, CH2CH2Br), 3.51 (2H, t, J = 6.0 Hz, CH2CH2Br), 4.09 (2H, t, J = 6.0, OCH2CH2), 7.11–7.15 (1H, m, 4′-H), 7.40–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.57–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2941, 2863, 2756, 1655, 1603, 1588, 1339, 1231, 1175, 835, 769. MS m/z (ESI): 359 [M + H]+.
(E)-1-(3-(5-bromopentyloxy)phenyl)-3-phenylprop-2-en-1-one (3d)
Compound 3d was synthesized from compound 2 (2.24 g, 10 mmol) with K2CO3 (4.13 g, 30 mmol) in DMF (20 mL) and 1,5-dibromopentane (8.2 mL, 60 mmol), followed by purification using silica-gel column chromatography with ethyl acetate/petroleum ether (1:18, v/v) as an eluent. Yield: 70.6%, white solid. m.p. 91–93 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.63–1.71 (2H, m, CH2CH2CH2), 1.80–1.87 (2H, m, CH2CH2Br), 1.91–1.98 (2H, m, OCH2CH2), 3.47 (2H, t, J = 6.0 Hz, CH2CH2Br), 4.08 (2H, t, J = 6.0, OCH2CH2), 7.11–7.16 (1H, m, 4′-H), 7.39–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2941, 2863, 2758, 1655, 1603, 1588, 1339, 1231, 1175, 835, 768. MS m/z (ESI): 373 [M + H]+.
(E)-1-(3-(6-bromohexyloxy)phenyl)-3-phenylprop-2-en-1-one (3e)
Compound 3e was synthesized from compound 2 (2.24 g, 10 mmol) with K2CO3 (4.13 g, 30 mmol) in DMF (20 mL) and 1,6-dibromohexane (9.2 mL, 60 mmol), followed by purification using silica-gel column chromatography with ethyl acetate/petroleum ether (1:20, v/v) as an eluent. Yield: 78.7%, white solid. m.p. 88–90 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.57–1.56 (4H, m, CH2CH2CH2CH2 and CH2CH2CH2CH2), 1.78–1.92 (4H, m, CH2CH2Br and OCH2CH2), 3.43 (2H, t, J = 6.0 Hz, CH2CH2Br), 4.06 (2H, t, J = 6.0, OCH2CH2), 7.11–7.15 (1H, m, 4′-H), 7.40–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.56–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3030, 2955, 2877, 2759, 1657, 1609, 1587, 1506, 1341, 1230, 1174, 834, 771. MS m/z (ESI): 387 [M + H]+.
General procedure for the synthesis of 4a–8d
A mixture of compound 3a–3e (1 mmol), secondary amines (dimethylamine, diethylamine, piperidine, and pyrrolidine), K2CO3 (0.412 g, 3 mmol), NaI (0.008 g, 0.05 mmol), acetone (15 mL) were refluxed. The solvent was removed, then the residue was dissolved with ethyl acetate (30 mL), followed by washing with saturated NaCl solution (2 × 20 mL). The organic phase was dried with anhydrous Na2SO4 and evaporated in vacuum, giving a crude product which was purified on silica gel with different ratio of methanol/dichloromethane as an eluent to afford the compounds 4a–8d.
(E)-1-(3-(2-(dimethylamino)ethoxy)phenyl)-3-phenylprop-2-en-1-one (4a)
The crude product was gained by the reaction of compound 3a (0.331 g, 1 mmol) with dimethylamine (0.34 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:40, v/v) as an eluent to give light yellow solid with a yield of 62.9%. m.p. 123–125 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 2.87 (6H, s, 2 × NCH3), 3.40 (2H, t, J = 6.0 Hz, OCH2CH2), 4.55 (2H, t, J = 6.0 Hz, OCH2CH2), 7.16–7.19 (1H, m, 4′-H), 7.42–7.47 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.57–7.69 (3H, m, 2′-H and 5′-H and 6′-H), 7.83 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3030, 2949, 2875, 2782, 1665, 1606, 1593, 1510, 1339, 1221, 1168, 836, 767. MS m/z (ESI): 296 [M + H]+.
(E)-1-(3-(2-(diethylamino)ethoxy)phenyl)-3-phenylprop-2-en-1-one (4b)
The crude product was gained by the reaction of compound 3a (0.331 g, 1 mmol) with diethylamine (0.31 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:45, v/v) as an eluent to give light yellow solid with a yield of 72.8%. m.p. 105–107 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.49 (6H, t, J = 8.0 Hz, 2 × NCH2CH3), 3.29 (4H, s, J = 6.0 Hz, 2 × NCH2CH3), 3.50 (2H, t, J = 6.0 Hz, OCH2CH2), 4.63 (2H, t, J = 6.0 Hz, OCH2CH2), 7.14–7.18 (1H, m, 4′-H), 7.42–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.49 (1H, d, J = 16.0 Hz, α-H), 7.55–7.68 (3H, m, 2′-H and 5′-H and 6′-H), 7.83 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2965, 2869, 2785, 1663. 1606, 1589, 1509, 1337, 1220, 1172, 834, 767. MS m/z (ESI): 324 [M + H]+.
(E)-3-phenyl-1-(3-(2-(piperidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (4c)
The crude product was gained by the reaction of compound 3a (0.331 g, 1 mmol) with piperidine (0.3 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:45, v/v) as an eluent to give light yellow solid with a yield of 77.4%. m.p. 98–100 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.48–1.56 (2H, m, CH2CH2CH2), 1.72–1.78 (4H, m, piperidine-H), 2.73 (4H, t, J = 6.0 Hz, 2 × NCH2CH2), 2.98 (2H, t, J = 6.0 Hz, NCH2CH2), 4.32 (2H, t, J = 6.0 Hz, OCH2CH2), 7.13–7.16 (1H, m, 4′-H), 7.40–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.61–7.69 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3029, 2961, 2875, 2771, 1659, 1608, 1591, 1511, 1339, 1221, 835, 771. MS m/z (ESI): 336 [M + H]+.
(E)-3-phenyl-1-(3-(2-(pyrrolidin-1-yl)ethoxy)phenyl)prop-2-en-1-one (4d)
The crude product was gained by the reaction of compound 3a (0.331 g, 1 mmol) with pyrrolidine (0.25 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:40, v/v) as an eluent to give light yellow oil with a yield of 66.7%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.78–1.88 (2H, m, pyrrolidine-H), 2.66–2.74 (4H, m, 2 × NCH2CH2), 2.97 (2H, t, J = 6.0 Hz, 2 × NCH2CH2), 4.21 (2H, t, J = 6.0 Hz, OCH2CH2), 7.14–7.17 (1H, m, 4′-H), 7.39–7.44 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.59–7.68 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2948, 2859, 2785, 1657, 1605, 1589, 1511, 1340, 1220, 1175, 833, 768. MS m/z (ESI): 322 [M + H]+.
(E)-1-(3-(3-(dimethylamino)propoxy)phenyl)-3-phenylprop-2-en-1-one (5a)
The crude product was gained by the reaction of compound 3b (0.344 g, 1 mmol) with dimethylamine (0.34 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:45, v/v) as an eluent to give a light yellow oil with a yield of 75.3%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.99–2.09 (2H, m, OCH2CH2), 2.31 (6H, s, 2 × NCH3), 2.52 (2H, t, J = 6.0 Hz, NCH2CH2), 4.11 (2H, t, J = 6.0 Hz, OCH2CH2), 7.12–7.15 (1H, m, 4′-H), 7.39–7.44 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3030, 2955, 2877, 2771, 1658, 1609, 1587, 1506, 1348, 1229, 1175, 834, 772. MS m/z (ESI): 310 [M + H]+.
(E)-1-(3-(3-(diethylamino)propoxy)phenyl)-3-phenylprop-2-en-1-one (5b)
The crude product was gained by the reaction of compound 3b (0.344 g, 1 mmol) with diethylamine (0.31 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:50, v/v) as an eluent to give light yellow oil with a yield of 72.7%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.12 (6H, t, J = 6.0 Hz, 2 × NCH2CH3), 1.99–2.03 (2H, m, OCH2CH2), 2.56–2.64 (6H, m, 3 × NCH2), 4.18 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.15 (1H, m, 4′-H), 7.40–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.61–7.68 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2955, 2869, 2771, 1653, 1605, 1577, 1509, 1337, 1221, 1173, 831, 769. MS m/z (ESI): 338 [M + H]+.
(E)-3-phenyl-1-(3-(3-(piperidin-1-yl)propoxy)phenyl)prop-2-en-1-one (5c)
The crude product was gained by the reaction of compound 3b (0.344 g, 1 mmol) with piperidine (0.3 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:50, v/v) as an eluent to give light yellow oil with a yield of 82.3%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.51–1.56 (2H, m, CH2CH2CH2), 1.79–1.85 (4H, m, piperidine-H), 2.16–2.21 (2H, m, OCH2CH2), 2.57–2.61 (6H, m, 3 × NCH2), 4.10 (2H, t, J = 6.0 Hz, OCH2CH2), 7.11–7.14 (1H, m, 4′-H), 7.39–7.44 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.59–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.83 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm − Citation1: 3032, 2944, 2861, 2776, 1659, 1610, 1592, 1503, 1338, 1222, 1172, 832, 768. MS m/z (ESI): 350 [M + H]+.
(E)-3-phenyl-1-(3-(3-(pyrrolidin-1-yl)propoxy)phenyl)prop-2-en-1-one (5d)
The crude product was gained by the reaction of compound 3b (0.344 g, 1 mmol) with pyrrolidine (0.25 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:45, v/v) as an eluent to give light yellow oil with a yield of 70.4%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.51–1.56 (2H, m, CH2CH2CH2), 1.82–1.88 (4H, m, pyrrolidine-H), 2.02–2.07 (2H, m, OCH2CH2), 2.61–2.72 (6H, m, 3 × NCH2), 4.13 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.14 (1H, m, 4′-H), 7.38–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.61–7.68 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2955, 2868, 2767, 1657, 1609, 1589, 1508, 1345, 1226, 1174, 830, 769. MS m/z (ESI): 336 [M + H]+.
(E)-1-(3-(4-(dimethylamino)butoxy)phenyl)-3-phenylprop-2-en-1-one (6a)
The crude product was gained by the reaction of compound 3c (0.358 g, 1 mmol) with dimethylamine (0.34 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:50, v/v) as an eluent to give light yellow solid with a yield of 78.5%. m.p. 85–87 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.75–1.88 (4H, m, OCH2CH2 and NCH2CH2), 2.36 (6H, s, 2 × NCH3), 2.50 (2H, t, J = 6.0 Hz, NCH2CH2), 4.07 (2H, t, J = 6.0 Hz, OCH2CH2), 7.11–7.15 (1H, m, 4′-H), 7.39–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2941, 2863, 2756, 1655, 1603, 1588, 1339, 1231, 1175, 835, 768. MS m/z (ESI): 324 [M + H]+.
(E)-1-(3-(4-(diethylamino)butoxy)phenyl)-3-phenylprop-2-en-1-one (6b)
The crude product was gained by the reaction of compound 3c (0.358 g, 1 mmol) with diethylamine (0.31 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow oil with a yield of 68.4%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.21 (6H, t, J = 6.0 Hz, 2 × NCH2CH3), 1.78–1.90 (4H, m, OCH2CH2 and NCH2CH2), 2.36 (6H, s, 2 × NCH3), 2.72–2.84 (6H, m, 3 × NCH2CH2), 4.07 (2H, t, J = 6.0 Hz, OCH2CH2), 7.11–7.15 (1H, m, 4′-H), 7.39–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.59–7.68 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2948, 2858, 2751, 1658, 1606, 1586, 1341, 1230, 1175, 836, 769. MS m/z (ESI): 352 [M+H]+.
(E)-3-phenyl-1-(3-(4-(piperidin-1-yl)butoxy)phenyl)prop-2-en-1-one (6c)
The crude product was gained by the reaction of compound 3c (0.358 g, 1 mmol) with piperidine (0.3 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow solid with a yield of 76.5%. m.p. 73–75 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.46–1.51 (2H, m, CH2CH2CH2), 1.65–1.71 (4H, m, piperidine-H), 1.74–1.88 (4H, m, OCH2CH2 and NCH2CH2), 2.47–2.56 (6H, m, 3 × NCH2CH2), 4.06 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.13 (1H, m, 4′-H), 7.38–7.45 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2952, 2861, 2755, 1657, 1608, 1589, 1339, 1232, 1174, 838, 771. MS m/z (ESI): 364 [M + H]+.
(E)-3-phenyl-1-(3-(4-(pyrrolidin-1-yl)butoxy)phenyl)prop-2-en-1-one (6d)
The crude product was gained by the reaction of compound 3c (0.358 g, 1 mmol) with pyrrolidine (0.25 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:50, v/v) as an eluent to give light yellow oil with a yield of 64.1%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.85–1.98 (8H, m, pyrrolidine-H and OCH2CH2 and NCH2CH2), 2.75–2.92 (6H, m, 3 × NCH2CH2), 4.07 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.13 (1H, m, 4′-H), 7.39–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.59–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3032, 2952, 2863, 2755, 1656, 1608, 1583, 1341, 1233, 1175, 837, 768. MS m/z (ESI): 350 [M + H]+.
(E)-1-(3-(5-(dimethylamino)pentyloxy)phenyl)-3-phenylprop-2-en-1-one (7a)
The crude product was gained by the reaction of compound 3d (0.372 g, 1 mmol) with dimethylamine (0.34 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow oil with a yield of 74.2%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.60–1.66 (2H, m, CH2CH2CH2), 1.87–1.92 (2H, m, NCH2CH2), 1.95–2.01 (2H, m, OCH2CH2), 2.86 (6H, s, 2 × NCH3), 3.11 (2H, t, J = 6.0 Hz, NCH2CH2), 4.08 (2H, t, J = 6.0 Hz, OCH2CH2), 7.09–7.13 (1H, m, 4′-H), 7.38–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm− Citation1: 3031, 2954, 2867, 2779, 1656, 1605, 1589, 1505, 1340, 1225, 1175, 835, 767. MS m/z (ESI): 338 [M + H]+.
(E)-1-(3-(5-(diethylamino)pentyloxy)phenyl)-3-phenylprop-2-en-1-one (7b)
The crude product was gained by the reaction of compound 3d (0.372 g, 1 mmol) with diethylamine (0.31 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:60, v/v) as an eluent to give light yellow oil with a yield of 62.7%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.44 (6H, t, J = 6.8 Hz, 2 × NCH2CH3), 1.58–1.66 (2H, m, CH2CH2CH2), 1.86–1.99 (4H, m, NCH2CH2 and OCH2CH2), 3.07 (2H, t, J = 6.0 Hz, NCH2CH2), 3.20 (4H, m, 2 × NCH2CH3), 4.08 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.14 (1H, m, 4′-H), 7.40–7.47 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3032, 2958, 2875, 2776, 1656, 1605, 1587, 1508, 1342, 1223, 1175, 835, 768. MS m/z (ESI): 366 [M + H]+.
(E)-3-phenyl-1-(3-(5-(piperidin-1-yl)pentyloxy)phenyl)prop-2-en-1-one (7c)
The crude product was gained by the reaction of compound 3d (0.372 g, 1 mmol) with piperidine (0.30 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:60, v/v) as an eluent to give light yellow oil with a yield of 75.3%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.43–1.54 (4H, m, piperidine-H and CH2CH2CH2), 1.58–1.61 (6H, m, NCH2CH2 and piperidine-H), 1.79–1.88 (2H, m, OCH2CH2), 2.38–2.51 (6H, m, 3 × NCH2CH2), 4.05 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.13 (1H, m, 4′-H), 7.40–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3029, 2956, 2873, 2771, 1657, 1608, 1585, 1508, 1343, 1222, 1175, 834, 767. MS m/z (ESI): 378 [M + H]+.
(E)-3-phenyl-1-(3-(5-(pyrrolidin-1-yl)pentyloxy)phenyl)prop-2-en-1-one (7d)
The crude product was gained by the reaction of compound 3d (0.372 g, 1 mmol) with pyrrolidine (0.25 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow oil with a yield of 68.1%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.51–1.59 (2H, m, CH2CH2CH2), 1.69–1.82 (6H, m, NCH2CH2 and pyrrolidine-H), 1.91–1.96 (2H, m, OCH2CH2), 2.68–2.79 (6H, m, 3 × NCH2CH2), 4.04 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.13 (1H, m, 4′-H), 7.40–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2959, 2878, 2777, 1656, 1607, 1589, 1507, 1341, 1223, 1174, 833, 768. MS m/z (ESI): 364 [M + H]+.
(E)-1-(3-(6-(dimethylamino)hexyloxy)phenyl)-3-phenylprop-2-en-1-one (8a)
The crude product was gained by the reaction of compound 3e (0.386 g, 1 mmol) with diethylamine (0.31 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow oil with a yield of 65.6%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.48–1.61 (4H, m, CH2CH2CH2CH2 and CH2CH2CH2CH2), 1.80–1.96 (4H, m, NCH2CH2 and OCH2CH2), 2.86 (6H, s, 3 × NCH3), 3.09 (2H, t, J = 6.0 Hz, NCH2CH2), 4.05 (2H, t, J = 6.0, OCH2CH2), 7.09–7.13 (1H, m, 4′-H), 7.38–7.47 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.58–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3029, 2961, 2877, 2773, 1657, 1606, 1585, 1507, 1343, 1223, 1174, 833, 771. MS m/z (ESI): 352 [M + H]+.
(E)-1-(3-(6-(diethylamino)hexyloxy)phenyl)-3-phenylprop-2-en-1-one (8b)
The crude product was gained by the reaction of compound 3e (0.386 g, 1 mmol) with dimethylamine (0.34 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:60, v/v) as an eluent to give light yellow oil with a yield of 68.3%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.44 (6H, t, J = 6.8 Hz, 2 × NCH2CH3), 1.49–1.60 (4H, m, CH2CH2CH2CH2 and CH2CH2CH2CH2), 1.81–1.95 (4H, m, NCH2CH2 and OCH2CH2), 3.05 (2H, t, J = 6.0 Hz, NCH2CH2), 3.19 (4H, m, 2 × NCH2CH3), 4.04 (2H, t, J = 6.0, OCH2CH2), 7.09–7.14 (1H, m, 4′-H), 7.38–7.46 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.58–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3031, 2960, 2875, 2778, 1657, 1605, 1589, 1508, 1342, 1225, 1172, 834, 768. MS m/z (ESI): 380 [M + H]+.
(E)-3-phenyl-1-(3-(6-(piperidin-1-yl)hexyloxy)phenyl)prop-2-en-1-one (8c)
The crude product was gained by the reaction of compound 3e (0.386 g, 1 mmol) with piperidine (0.3 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:60, v/v) as an eluent to give light yellow oil with a yield of 70.9%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.37–1.45 (2H, m, piperidine-H), 1.49–1.57 (4H, m, 4H, m, CH2CH2CH2CH2 and CH2CH2CH2CH2), 1.71–1.88 (8H, piperidine-H and NCH2CH2 and OCH2CH2), 2.57–2.73 (m, 6H, 3 × NCH2CH2), 4.05 (2H, t, J = 6.0 Hz, OCH2CH2), 7.09–7.13 (1H, m, 4′-H), 7.39–7.47 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.51 (1H, d, J = 16.0 Hz, α-H), 7.60–7.67 (3H, m, 2′-H and 5′-H and 6′-H), 7.81 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3033, 2954, 2877, 2773, 1657, 1607, 1589, 1508, 1343, 1223, 1173, 833, 767. MS m/z (ESI): 392 [M + H]+.
(E)-3-phenyl-1-(3-(6-(pyrrolidin-1-yl)hexyloxy)phenyl)prop-2-en-1-one (8d)
The crude product was gained by the reaction of compound 3e (0.386 g, 1 mmol) with pyrrolidine (0.25 mL, 3 mmol). Then it was purified using silica-gel column chromatography with methanol/dichloromethane (1:55, v/v) as an eluent to give light yellow oil with a yield of 58.5%. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.38–1.53 (4H, m, pyrrolidine-H), 1.58–1.65 (2H, m, NCH2CH2), 1.79–1.88 (6H, CH2CH2CH2CH2 and CH2CH2CH2CH2 and OCH2CH2), 2.50–2.61 (6H, 3 × NCH2CH2), 4.04 (2H, t, J = 6.0 Hz, OCH2CH2), 7.10–7.14 (1H, m, 4′-H), 7.39–7.47 (5H, m, 2-H and 3-H and 4-H and 5-H and 6-H), 7.52 (1H, d, J = 16.0 Hz, α-H), 7.60–7.66 (3H, m, 2′-H and 5′-H and 6′-H), 7.82 (1H, d, J = 16.0 Hz, β-H). IR (KBr) ν/cm−1: 3029, 29638, 2875, 2778, 1655, 1606, 1587, 1506, 1341, 1222, 1175, 835, 768. MS m/z (ESI): 378 [M + H]+.
Log P measurement
Octanol–water partition coefficients of compounds 4a–8d were measured by the shake flask method described previously with slight modificationCitation16. The aqueous phase was replaced by PBS (pH = 7.4). Both the octanol and the aqueous phase were saturated with each other before use. Different from the described method above, in the present experiment, ultrasonic method was applied to accelerate this progression. The assay mixture containing test compounds was shaken at 37 °C over night and then centrifuged at 2000 rpm for 20 min, followed by the analysis with HPLC. A C18 column (250 nm × 4.6 mm, 5 μm) was used with the mobile phase of methanol–0.1% triethylamine (TEA) (85:15, v/v) at a flow rate of 1.0 mL min−1 and the detection wavelength of 319 nm at 32 °C. Experiments were conducted in triplicate and log P values were calculated.
AChE and butyrylcholinesterase (BuChE) inhibition assay
AChE/BuChE activity assays were conducted by the Ellman method with slight modificationCitation17. Each compound was dissolved in Tween 80 and diluted with water to various concentrations immediately before use. The reaction mixture containing 40 μL AChE/BuChE, 100 μL acetylthiocholine iodide/S-butyrylthiocholine iodide, 2.76 mL Na2HPO4/NaH2PO4 buffer (pH 8.0, 0.1 mol/L), and 100 μL different concentrations of tested compounds were incubated at 30 °C for 25 min. Then the reaction was terminated via adding 100 μL 20% sodium dodecylsulfate (SDS) and 100 μL 10 mmol/L 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB) was added to generate the yellow anion 5-thio-2-nitro-benzoic acid. The absorbance of each assay mixture was measured at 412 nm by UV spectroscopy. The IC50 values were calculated by the Bliss method and expressed as mean ± SD of the replicates.
Kinetic studies
Kinetic characterization of AChE was conducted by a reported method with some modificationsCitation18. Compound 6c was added into the assay solution and preincubated with the enzyme at 30 °C, followed by the addition of 100 µL acetylthiocholine iodide including five concentrations. The assay solution contained 100 µL compound 6c, 100 µL DTNB, 2.76 mL 0.1 mol/L Na2HPO4/NaH2PO4 buffer (pH 8.0). Kinetic characterization of the hydrolysis of acetylthiocholine iodide catalyzed by AChE was conducted spectrometrically at 412 nm. Additionally, the parallel control experiment was made without compound 6c in the mixture.
Molecular docking
Molecular modelling was performed by Molecular Operating Environment (MOE) software package (Chemical Computing Group, Montreal, Canada). The X-ray crystallographic structures of AChE (PDB code: 1EVE) and BuChE (PDB code: 1P0I) were gained from protein data bank. The 3D structure of the strongest AChE inhibitor 6c was established by virtue of the builder interface of MOE program (Chemical Computing Group, London, UK), and docked into the active site of the protein after energy being minimized. The Dock scoring in MOE software (Chemical Computing Group, Montreal, Canada) was done by ASE scoring function.
Results and discussion
The synthetic routes to get the target compounds 4a–8d are outlined in Scheme 1. Compound 2 was gained from the reaction of 3-hydroxyacetophenone with benzaldehyde in the presence of NaOH/EtOH. Then, compound 2 was treated with dibromoalkanes and K2CO3 in N,N-dimethylformamide (DMF) at 80 °C to generate compounds 3a–3e. Finally, compounds 4a–8d were achieved by refluxing the mixture including compounds 3a–3e and secondary amines (dimethylamine, diethylamine, piperidine and pyrrolidine) in acetone in the presence of K2CO3 and NaI. The structures of the compounds were characterized by proton nuclear magnetic resonance spectroscopy (1H NMR), infrared spectrum (IR) and mass spectrometry (MS). In addition, the purities of all synthesized compounds were confirmed to be higher than 97% by HPLC.
Scheme 1. Reagents and conditions: (a) benzaldehyde, NaOH, EtOH, rt; (b) Br(CH2)nBr, K2CO3, DMF, 80 °C; (c) secondary amine, K2CO3, NaI, acetone, reflux.
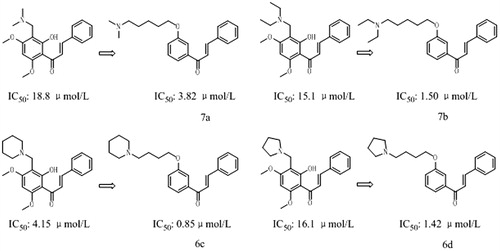
Inhibiting effects of new synthesized compounds in the present study against AChE and BuChE were evaluated by the modified Ellman method, using Rivastigmine as the positive control. The half maximal inhibitory concentration (IC50 values) for AChE and BuChE as well as the selectivity for AChE are summarized in . The results indicated that all compounds exhibited higher inhibitory activities against AChE than the precursor compound 2 (IC50 > 500 µmol/L). Among them, compounds 7a, 7b, 6c and 6d with IC50 values of 3.82, 1.50, 0.85, and 1.42 µmol/L, respectively, showed more potent than Rivastigmine (IC50 = 10.54 µmol/L). Interestingly, compound 6c displayed the strongest AChE inhibitory activity (IC50 = 0.85 μmol/L) and highest selectivity (ratio: 35.79).
Table 1. Inhibition of AChE and BuChE (IC50 values and selectivity) and log P values of nitrogen-containing chalcone derivatives.
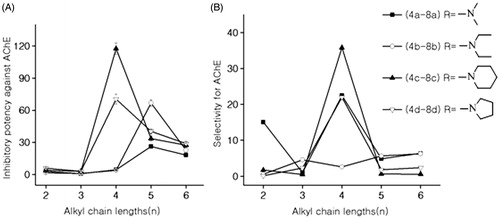
Based on the enzyme inhibition data (), it seemed that the alteration of the spacer length between amino-alkyl side chain and chalcone scaffold could dramatically affect the AChE inhibitory potency. Generally, the inhibitory potency (=1/IC50*100) of chalcone derivatives presented a parabola profile with the growth of carbon spacer length. For dimethylmine or diethylamine containing chalcone derivatives, the compounds with a five-carbons spacer (compounds 7a and 7b) revealed the highest inhibitory potency among the homologues. However, for piperidine or pyrrolidine containing chalcone derivatives, the compounds with a four-carbons spacer (compounds 6c and 6d) showed the highest inhibition activity among the homologues against AChE yet. In addition, the compounds with a three-carbons spacer showed the poorest inhibition activity among the homologues.
Figure 1. (A) Effects of alkyl chain lengths on anti-AChE activities; (B) effects of alkyl chain lengths on selectivity for AChE.
As shown in , it appeared that some of the new synthesized chalcone derivatives (compounds 6c, 6d, 7a and 7b) exhibited 5–10 folds inhibitory activities against AChE than relative Flavokawain B Mannich base derivatives which were reported in our previous investigation. For the modification or optimization of flavokawain B Mannich base derivatives, structure simplification was employed, which was a widely used method in the drug discovery and developmentCitation19–21. This method is benefit for the screening for pharmacophore or important structure domain in natural products, enhancing bioactivity or attenuating side effects. For instance, Procaine, simplified from natural compound Cocaine, had stronger local anaesthesia effect than Cocaine, but had no addiction. Rivastigmine, a simplified compound from natural alkaloid Eserine, had more potent AChE inhibition than Eserine as well as satisfactory stability. Thereby, simplified nitrogen chalcone derivatives from Flavokawain B were more convenient to investigate the structure–activity relationship (Appendix Figure A1) According to the results of the present study, it indicated that not only the variations of amino-alkyl groups but also the spacer length between amino-alkyl side chain and chalcone scaffold were important for AChE inhibitory activity of chalcone derivatives.
Figure 2. The comparison of the inhibitory activity between the corresponding Flavokawain B derivatives and chalcone derivatives against AChE.
Compound 6c, possessing the highest activity against AChE among the new synthesized chalcone derivatives in the present investigation, was selected for kinetic studies. The linear Lineweaver–Burk equation of the Michaelis–Menten was applied to evaluate the inhibition profile. The graphical analysis of the steady-state inhibition data of compound 6c is shown in Appendix Figure A2. According to the analysis, Km but not Vmax increased with the increasing concentration of compound 6c, which presented a mixed-type inhibition. The competitive inhibition constant (Ki) and the non-competitive constant (Ki’) are 0.69 μmol/L and 1.19 μmol/L, respectively (Appendix: Table A1).
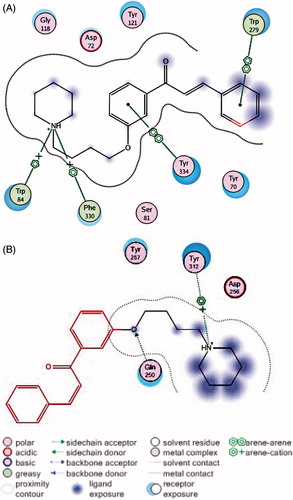
To explore the possible interacting mode of the chalcone derivatives with AChE, molecular docking was performed for compound 6c with software MOE2008 (Chemical Computing Group, London, UK). As shown in Appendix Figure A3, this compound exhibited multiple points binding modes with AChE (Appendix Figure A3). In the top of the gorge, the aromatic moiety adopted an appropriate orientation for its binding to PAS, via the π–π stacking interaction with Trp279. The conformation of the side chain conformed to the shape of the mid-gorge and interacted with Tyr334. In the bottom of the gorge, the charged nitrogen of piperidine ring was observed to bind to CAS via a cation–π interaction between Trp84 and Phe330. That is, compound 6c binds simultaneously to catalytic active sites and peripheral anionic site of AChE, while it binds with BuChE only with a cation–π interaction between Tyr312 and the nitrogen of piperidine (Appendix Figure A3). These results may partial explain its potent inhibition for AChE and its high selective inhibition for AChE over BuChE.
As a potential compound for treatment of AD, log P was thought as an important physical chemistry parameter to valuate or predict the ability to cross blood–brain barrier (BBB). It was reported that the log P with the optimum central nervous system (CNS) penetration was around 2 ± 0.7Citation22. As shown in , log P values of new synthesized compounds ranged from 1.54 to 1.98, which indicated that all the compounds are possible sufficiently lipophilic to pass the BBB in vivo.
Conclusions
The present studies indicated that the length of carbon chain-linked chalcone scaffold and the amino alkyl group had markedly influence in AChE inhibitory activity. Among them, compound 6c revealed the strongest AChE inhibitory activity (IC50 value: 0.85 μmol/L) and highest selectivity (ratio: 35.79). Enzyme kinetic study suggested that the inhibition mechanism of compound 6c was a mixed-type inhibition. Besides, molecular docking further explained its potent inhibition of AChE and high selectivity for AChE over BuChE. Over all, compound 6c might serve as a potential agent for the treatment of AD.
Declaration of interest
The authors report that that they have no conflicts of interest. The present investigation was supported by the grant of “The Natural Science Foundation of China (No. 21342015)”, “The Project of Science and Technology of Hu’nan Province (No. 2012SK3183)”and “The Research Funds for younger investigators of Hu’nan University (No. HNU2013066)”.
References
- Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology 2014;76:27–50
- Pivtoraiko VN, Abrahamson EE, Leurgans SE, et al. Cortical pyroglutamate amyloid-β levels and cognitive decline in Alzheimer's disease. Neurobiol Aging 2015;36:12–19
- García-Alberca JM. Cognitive intervention therapy as treatment for behavior disorders in Alzheimer disease: evidence on efficacy and neurobiological correlations. Neurología (English Edition) 2015;30:8–15
- Molinuevo JL, Gauthier S. Benefits of combined cholinesterase inhibitor and memantine treatment in moderate–severe Alzheimer’s disease. Alzheimers Dement 2013;9:326–31
- Small G, Bullock R. Defining optimal treatment with cholinesterase inhibitors in Alzheimer’s disease. Alzheimers Dement 2011;7:177–84
- Silva T, Reis J, Teixeira J, et al. Alzheimer's disease, enzyme targets and drug discovery struggles: from natural products to drug prototypes. Ageing Res Rev 2014;15:116–45
- Mukherjee PK, Kumar V, Mal M, et al. Acetylcholinesterase inhibitors from plants. Phytomedicine 2007;14:289–300
- Li RS, Wang XB, Hu XJ, et al. Design, synthesis and evaluation of flavonoid derivatives as potential multifunctional acetylcholinesterase inhibitors against Alzheimer’s disease. Bioorg Med Chem Lett 2013;23:2636–41
- Asadipour A, Alipour M, Jafari M, et al. Novel coumarin-3-carboxamides bearing N-benzylpiperidine moiety as potent acetylcholinesterase inhibitors. Eur J Med Chem 2013;70:623–30
- Liew SY, Khaw KY, Murugaiyah V, et al. Natural indole butyrylcholinesterase inhibitors from Nauclea officinalis. Phytomedicine 2015;22:45–8
- Liu HR, Liu XJ, Fan HQ, et al. Design, synthesis and pharmacological evaluation of chalcone derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 2014;22:6124–33
- Liu HR, Huang XQ, Lou DH, et al. Synthesis and acetylcholinesterase inhibitory activity of Mannich base derivatives flavokawain B. Bioorg Med Chem Lett 2014;24:4749–53
- Konrath EL, Passos CS, Klein-Júnior LC, et al. Alkaloids as a source of potential anticholinesterase inhibitors for the treatment of Alzheimer’s disease. J Pharm Pharmacol 2013;65:1701–25
- Darras FH, Wehle S, Huang G, et al. Amine substitution of quinazolinones leads to selective nanomolar AChE inhibitors with ‘inverted’ binding mode. Bioorg Med Chem 2014;22:4867–81
- Darvesh S, Macdonald LR, Martin E. Selectivity of phenothiazine cholinesterase inhibitors for neurotransmitter systems. Bioorg Med Chem Lett 2013;23:3822–5
- Sharma J, Singla AK, Dhawan S. Zinc–naproxen complex: synthesis, physicochemical and biological evaluation. Int J Pharm 2003;260:217–27
- Ellman GL, Courtney KD, Andres VJ, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95
- Skrzypek A, Matysiak J, Niewiadomy A, et al. Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur J Med Chem 2013;62:311–19
- Magedov IV, Kireev AS, Jenkins AR, et al. Structural simplification of bioactive natural products with multicomponent synthesis. 4: 4H-pyrano-[2,3-b]naphthoquinones with anticancer activity. Bioorg Med Chem Lett 2012;22:5195–8
- Magedov IV, Manpadi M, Rozhkova E, et al. Structural simplification of bioactive natural products with multicomponent synthesis: dihydropyridopyrazole analogues of podophyllotoxin. Bioorg Med Chem Lett 2007;17:1381–5
- Yang R, Zhao R, Chen D, et al. Discovering selective agonists of endothelial target for acetylcholine (ETA) via diversity-guided pharmacophore simplification and simulation. Bioorg Med Chem Lett 2004;14:3017–25
- Glave WR, Hansch CJ. Relationship between lipophilic character and anesthetic activity. J Pharm SCI-US 1972;61:589–91
Appendix
Figures A1–A3 and Table A1.
Figure A1. Some examples of the derivatives from natural products with structural simplification.
Figure A2. (A) Lineaweaver–Burk plot for the inhibition of AChE by compound 6c; (B) the replots of the intercept versus concentration of compound 6c; (C) the replots of the slope versus concentration of compound 6c.
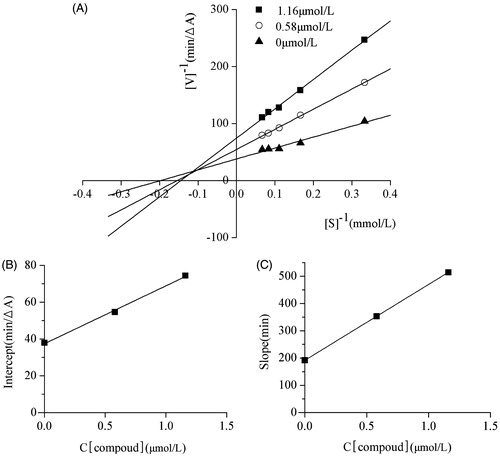
Figure A3. Molecular modelling of compound 6c with AChE (A) and BuChE (B) generated with MOE2008.