
Abstract
A series of 2-substituted 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxides were synthesized and evaluated for their affinity to the glycine binding site of the N-methyl-d-aspartate (NMDA) receptor. The binding affinity was determined by the displacement of radioligand [3H]MDL-105,519 from rat cortical membrane preparations. The most attractive structures in the search for prospective NMDA receptor ligands were identified to be 2-arylcarbonylmethyl substituted 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxides. It has been demonstrated for the first time that the replacement of NH group in the ligand by sp3 CH2 is tolerated. This finding may pave the way for previously unexplored approaches for designing new ligands of the NMDA receptor.
Introduction
The N-methyl-d-aspartate (NMDA) receptor is a member of the glutamate receptor superfamily and plays an important role in neurotransmission within the central nervous system (CNS), as well as in the synaptic plasticity that underlies learning processes and memoryCitation1,Citation2. Dysfunction of this receptor is thought to be involved in various disorders including epilepsy, ischemic brain damage and neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseasesCitation3,Citation4. Overstimulation of the NMDA receptor caused by excessive glutamate in the synaptic region is believed to contribute to the symptomatology of these disorders. The NMDA receptor, being organized as a ligand-gated ion channel, is activated by glutamate in the presence of a co-agonist – glycine or d-serine. The presence of a co-agonist, which binds to a specific strychnine–insensitive glycine binding site (glycine B) of the NMDA receptor, is necessary for ion channel activityCitation5. Therefore, the glycine binding site was perceived as a unique pharmacological target and several highly potent glycine binding site antagonists have been discoveredCitation6–8. As, unsurprisingly, glycine B antagonist pharmacophoreCitation9 does not look like a brain penetrant, this was one of the reasons for low efficacy in vivo. The NMDA receptors are present not only in the CNS but also in the periphery and there is growing interest in the potential use of glycine antagonist as treatment for different neuropathic pain conditions. An example of peripheral activity of a brain impermeant glycine selective antagonist is MRZ 2/596 (V)Citation4, demonstrating efficacy in several pain modelsCitation10. Successful optimisation of peripherally restricted NMDAR glycine binding site antagonists, based on the quinoxaline-2,3-dione structure VI, has already been reportedCitation11. As it targets peripheral NMDA receptors, the glycine antagonist pharmacophore model is much more promising for the optimisation of bioavailability of active compounds.
To search for new starting points in the design of effective glycine B antagonists based on structure IICitation12, we focused on information from the early studies of 5,7-dichlorokynurenic acid (I, DCKA)Citation13. It has been shown that 4-O-substitution dramatically reduced the activity of the compounds. The carbonyl group in structure I ensured the interaction with the receptor via water molecule hydrogen bondingCitation14. Alternatively, the activity was improved by the introduction of acidic functional group. This occurred in our series of naphthalenes II, where the most active compounds contained a carboxyl group in the substituent R ().
Figure 1. Structure of DCKA, naphthoic acid II, target compounds III and IV, MRZ-2/596 and quinoxaline VI.
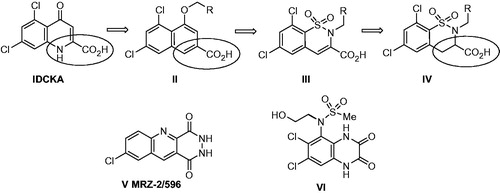
Based on the speculations mentioned above, we expected that the H-bond acceptor in the position 4 of naphthalene II would allow to reduce the polarity of compounds by removing the carboxyl group, while not affecting the affinity for the NMDA receptor. The necessary H-bond acceptor can be included in structure II by using carboxamide or sulfonamide group. In both cases, the structure may be derivatised on nitrogen atom. However, compounds containing carboxamide group in this position (4-oxo-quinazolidine-2-carboxylic acids) were inactive in [3H]-Gly displacement at 100 µM concentrationCitation15. Surprisingly, sulfonamides were highly active in corresponding benzo-1,2,4-thiadiazine-3-carboxylic acid 1,1-dioxide seriesCitation14. The role of the sulfonamide group in this particular class of compounds has never been explained. However, combination of sulfonamide and carboxy functions has already been known to be of great importance in this type of structuresCitation15,Citation16. Both modifications described above have been designed based on a common pharmacophore model of 5,7-dichlorokynurenic acid (I, DCKA)Citation13, including NH as a presumable H-donor and specific substituents as in structure 1. The surprising loss of activity, when the sulfonyl group was replaced by carbonylCitation15, prompted us to evaluate carba-analogs III possessing the same sulfonamide fragment and carboxyl group as crucial pharmacophores. The efficiency of sp2 CH, rather than NH, for the development of new GlyB ligands has already been provedCitation12,Citation17. Altogether, this information provided a highly promising background to identify novel potent and less polar ligands for the NMDA receptor glycine binding site, using structure III and, possibly structure IV.
Materials and methods
Chemistry
Commercial grade reagents and anhydrous solvents were used as received. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury BB 200 MHz and Varian Mercury plus 400 (400 MHz) (Ex Varian, Agilent Technologies, Santa Clara, CA) spectrometer. Chemical shift values are given in parts-per-million (ppm) relative to tetramethylsilane (TMS) with the residual solvent proton resonance as internal standard (CDCl3, 7.26 ppm; DMSO-d6, 2.50 ppm). LC–MS analyses were performed on Shimadzu CBM – 20A (Kyoto, Japan) with Applied Biosystems API 2000 (Foster City, CA) mass detector. Elemental analyses were carried out on a Carlo Erba Instruments (Milan, Italy) EA1108 analyser. Thin layer chromatography was performed on DC Alufolien, Kieselgel 60 F254 (Merck, Darmstadt, Germany) plates; UV detection. Flash column chromatography was carried out using silica gel (Merck silica gel 60). Melting points were determined on a Stanford Research System MPA100 (Sunnyvale, CA) Automated Melting Point Apparatus. The purity of all compounds was determined by HPLC on a Shimadzu CBM – 20A (HPLC column: Phenomenex (Torrance, CA), Gemini C18, 50 × 2 mm, 5 µm, solvent: MeCN; flow rate: 0.3 mL/min; column temperature: 25 °C; mobile phase: 0.1% HCOOH in water/MeCN; detection: UV at 254 nm; injection volume: 3 µL). Method I – gradient: from 10% MeCN/0.1% HCO2H to 90% MeCN/0.1% HCO2H in 12 min, hold 3 min 90% MeCN/0.1% HCO2H. Method II – gradient: from 40% MeCN/0.1% HCO2H to 90% MeCN/0.1% HCO2H in 12 min, hold 3 min 90% MeCN/0.1% HCO2H. Method III – gradient: from 10% MeCN/0.1% HCO2H to 95% MeCN/0.1% HCO2H in 7 min, hold 3 min 95% MeCN/0.1% HCO2H.
4-Chloro-2-methylbenzene-1-sulfonyl chloride (2a)
To a mixture of ClSO3H (26.0 mL, 395 mmol) and SOCl2 (2.3 mL, 31.6 mmol) stirred at 0 °C was slowly added 1-chloro-3-methylbenzene (1a) (18.7 mL, 158 mmol) and stirred for 1 h. Then stirring was continued at room temperature for 19 h. The black reaction mixture was poured into ice. The solid precipitated was filtered, washed with water and dried to give the title compound (30.67 g, 86%) as a white solid; mp > 41 °C (decomp.). TLC: Rf = 0.24 (petroleum ether). 1H NMR (200 MHz, CDCl3), δ ppm: 2.77 (s, 3H), 7.37–7.42 (m, 2H), 7.99 (d, J = 8.2 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 20.2, 126.9, 130.2, 133.2, 140.0, 141.3, 141.7.
4,5-Dichloro-2-methylbenzene-1-sulfonyl chloride (2b)
Prepared according to the procedure for compound 2a using 1,2-dichloro-3-methylbenzene (1b) (12 mL, 93.15 mmol) to give the title compound (22.99 g, 95%) as a white solid; mp 78–81 °C. TLC: Rf = 0.24 (petroleum ether). 1H NMR (200 MHz, CDCl3), δ ppm: 2.74 (s, 3H), 7.53 (s, 1H), 8.15 (s, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 19.7, 130.3, 131.2, 134.8, 137.5, 140.0, 141.7.
N-tert-butyl-4-chloro-2-methylbenzenesulfonamide (3a)
To a solution of 4-chloro-2-methylbenzenesulfonyl chloride (2a) (26.8 g, 119.1 mmol, 1 eq) in CH2Cl2 (250 mL) at 0 °C were added t-BuNH2 (14.94 mL, 142.9 mmol, 1.2 eq) and Et3N (33 mL, 238.2 mmol, 2 eq). The reaction mixture was stirred at room temperature for 22 h. Water was added (800 mL) and the mixture was extracted with CH2Cl2 (3 × 700 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated. The residue was washed with Et2O and filtered to give the title compound (22.39 g, 72%) as a white solid; mp 142–145 °C. TLC: Rf = 0.36 (Hex:EtOAc, 6:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.21 (s, 9H), 2.63 (s, 3H), 7.27 (m, 2H), 7.95 (d, J = 9.0 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 20.1, 30.1, 54.8, 126.2, 130.5, 132.1, 138.3, 138.7, 139.6. MS m/z 260 [M−H]−.
See Supplementary Material for the respective compounds:
N-tert-butyl-4,5-dichloro-2-methylbenzenesulfonamide (3b).
N-tert-butyl-2,4-dichloro-6-methylbenzenesulfonamide (3c).
2-(Tert-butyl)-5-chlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (4a)
A mixture of H5IO6 (155.96 g, 684.23 mmol, 8 eq) in MeCN (200 mL) was stirred vigorously at room temperature for 1 h, then CrO3 (0.86 g, 8.55 mmol, 0.1 eq) was added followed by acetic anhydride (64.8 mL, 684.2 mmol, 8 eq). The mixture was cooled to −4 °C then N-tert-butyl-4-chloro-2-methylbenzenesulfonamide (3a) (22.39 g, 85.53 mmol, 1 eq) was added in a small portions. After stirring at 0 °C for 30 min, the reaction mixture was allowed to reach room temperature and stirring was continued for 15 h. The solvent was removed under reduced pressure. Then water was added (200 mL), and the precipitate was filtered and dried to give the title compound (18.34 g, 78%) as a white solid; mp 138–141 °C. TLC: Rf = 0.38 (Hex:EtOAc, 6:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.76 (s, 9H), 7.77 (s, 2H), 7.95 (s, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 27.7, 61.7, 121.5, 124.8, 129.2, 134.5, 136.0, 140.9, 158.7. MS m/z 216 [M−57]− (tert-Bu).
See Supplementary Material for the respective compounds:
2-(Tert-butyl)-5,6-dichlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (4b).
2-(Tert-butyl)-5,7-dichlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (4c).
5-Chlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (5a)
A solution of compound 4a (18.3 g, 66.85 mmol) in TFA (70 mL) was heated at reflux for 70 h (TLC control). Then TFA was removed under reduced pressure. The residue was treated with dry Et2O and filtered to give the title compound (12.8 g, 88%) as a white solid; mp > 140 °C (decomp.). TLC: Rf = 0.23 (EtOAc:AcOH, 1:0.01). 1H NMR (200 MHz, DMSO-d6), δ ppm: 5.02 (s, 1H), 7.95–8.00 (m, 2H), 8.12 (d, J = 8.8 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 122.9, 124.5, 130.3, 135.0, 138.2, 139.3, 160.0. MS m/z 216 [M−H]−.
See Supplementary Material for the respective compounds:
5,6-Dichlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (5b).
5,7-Dichlorobenzo[d]isothiazol-3(2H)-one 1,1-dioxide (5c).
5-Chloro-2-(4-methoxybenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (6a)
To a solution of compound 5a (1.6 g, 7.35 mmol) in DMF (5 mL) was added NaH (60% in mineral oil, 0.29 g, 7.35 mmol) and heated at 80 °C for 30 min. Then 4-methoxybenzyl chloride (1.19 mL, 8.82 mmol) was added and heated at 80 °C for 16 h. The reaction mixture was allowed to reach room temperature, then water was added (100 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried over Na2SO4, filtered and evaporated. Purified by flash chromatography (petroleum ether:EtOAc, 4:1) to give the title compound (1.9 g, 75%) as a white solid; mp > 108 °C (decomp.). TLC: Rf = 0.33 (Hex:EtOAc, 4:1). 1H NMR (200 MHz, CDCl3), δ ppm: 3.79 (s, 3H), 4.84 (s, 2H), 6.87 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.8 Hz, 2H), 7.77–7.88 (m, 2H), 7.99 (s, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 42.5, 55.2, 114.0, 122.2, 125.3, 126.1, 129.1, 130.4, 134.8, 135.8, 141.2, 157.6, 159.6;. MS m/z 216 [M−121]− (elimination of p-metoxybenzyl (p-MeOBn)).
See Supplementary Material for the respective compounds:
5,6-Dichloro-2-(4-methoxybenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (6b).
5,7-Dichloro-2-(4-methoxybenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (6c).
4-Chloro-2-(hydroxymethyl)-N-(4-methoxybenzyl)-benzenesulfonamide (7a)
To a solution of compound 6a (1.84 g, 5.44 mmol) in THF (20 mL) was added solution of NaBH4 (0.68 g, 19.95 mmol) in water (20 mL) and stirred at room temperature for 17 h. The solvent was removed under reduced pressure and the aqueous solution was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was washed with hexane to give the title compound (1.779 g, 95%) as a white solid; mp 93–98 °C. TLC: Rf = 0.25, (Hex:EtOAc, 2:1). 1H NMR (400 MHz, CDCl3), δ ppm: 3.76 (s, 3H), 4.03 (d, J = 6.0 Hz, 2H), 4.93 (s, 2H), 5.53 (t, J = 6.0 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 9.2 Hz, 2H), 7.37 (dd, J = 8.4, 2.0 Hz, 1H), 7.48 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 47.0, 55.3, 63.1, 114.0, 127.7, 128.1, 129.3, 131.0, 131.3, 136.4, 139.2, 140.1, 159.3. MS m/z 340 [M−H]−.
See Supplementary Material for the respective compounds:
4,5-Dichloro-2-(hydroxymethyl)-N-(4-methoxybenzyl)-benzenesulfonamide (7b).
2,4-Dichloro-6-(hydroxymethyl)-N-(4-methoxybenzyl)-benzenesulfonamide (7c).
Ethyl 2-(4-chloro-2-(hydroxymethyl)-N-(4-methoxybenzyl)phenylsulfonamido)acetate (8a)
To a solution of compound 7a (4.37 g, 12.8 mmol) in THF (30 mL) was added NaH (60% in mineral oil, 0.57 g, 14.2 mmol) and heated at 50 °C for 30 min. Then ethyl bromoacetate (1.56 mL, 14.08 mmol) was added and heating was continued at the same temperature for 24 h. The solvent was removed under reduced pressure. To the residue water was added (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. Purified by flash chromatography (petroleum ether:EtOAc, 4:1) to give the title compound (3.541 g, 64%) as a colorless oil. TLC: Rf = 0.33 (Hex:EtOAc, 4:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.17 (t, J = 6.8 Hz, 3H), 3.16 (t, J = 7 Hz, 1H), 3.80 (s, 3H), 3.88 (s, 2H), 4.05 (q, J = 6.8 Hz, 2H), 4.52 (s, 2H), 4.92 (d, J = 7.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.8 Hz, 2H), 7.39 (dd, J = 8.2, 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.98 (d, J = 8.6 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 14.0, 45.9, 50.8, 55.3, 61.7, 61.8, 114.2, 126.1, 127.7, 130.2, 131.0, 131.3, 135.5, 139.8, 142.4, 159.7, 168.9. MS m/z low ionization. Purity (HPLC, 254 nm, Method I, tR = 9.47 min) 98%.
See Supplementary Material for the respective compounds:
Ethyl 2-(4,5-dichloro-2-(hydroxymethyl)-N-(4-methoxybenzyl)phenylsulfonamido)acetate (8b).
Ethyl 2-(2,4-dichloro-6-(hydroxymethyl)-N-(4-methoxybenzyl)phenylsulfonamido)acetate (8c).
Ethyl 2-(2,4-dichloro-6-formyl-N-(4-methoxybenzyl)phenylsulfonamido)acetate (9)
To a solution of compound 8c (0.48 g, 1.04 mmol, 1 equiv.) in dry THF (6 mL) was added MnO2 (0.36 g, 4.14 mmol, 4 equiv.) and stirred at room temperature for 16 h. Reaction was not complete, therefore 8 equiv. of MnO2 were added and stirring was continued for 22 h. The reaction mixture was filtered through celite, washed with EtOAc and the filtrate concentrated under reduced pressure to give the title compound (0.15 g, 31%) as a white solid, mp: 117–121 °C. Rf = 0.30 (Hex/EtOAc 6:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.21 (t, J = 7.2 Hz, 3H); 3.78 (s, 3H); 4.11 (q, J = 7.2 Hz, 2H); 4.54 (s, 2H); 6.79 (d, J = 8.8 Hz, 2H); 7.06 (d, J = 8.8 Hz, 2H); 7.45 (d, J = 2.0 Hz, 1H); 7.66 (d, J = 2.0 Hz, 1H); 10.52 (s, 1H). 13C NMR (100 MHz, CDCl3): δ = 14.0; 47.7; 51.9; 55.3; 61.6; 114.2; 125.8; 127.6; 130.1; 134.4; 134.6; 135.8; 139.5; 142.0; 159.7; 168.3; 189.5. MS: m/z 438]M−121]− (p-MeOBn).
Ethyl 6,8-dichloro-2-(4-methoxybenzyl)-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (10)
To a solution of compound 9 (0.14 g, 0.3 mmol, 1 equiv.) in dry THF (0.5 mL) was added NaH (60% in mineral oil, 3 mg, 0.08 mmol, 0.25 equiv.) and stirred at room temperature for 1 h. The solvent was removed under reduced pressure. To the residue was added water (20 mL) and it was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtred and concentrated under reduced pressure. The residue was triturated with hexane and filtered to give the title compound (0.12 g, 87%) as a white solid, mp: 91–95 °C. Rf = 0.21 (petroleum ether/EtOAc 6:1). 1H NMR (200 MHz, CDCl3), δ: 1.39 (t, J = 7.0 Hz, 3H); 3.68 (s, 3H); 4.38 (q, J = 7.0 Hz, 2H); 5.02 (s, 2H); 6.59 (d, J = 9.0 Hz, 2H); 6.90 (d, J = 9.0 Hz, 2H); 7.19 (d, J = 1.6 Hz, 1H); 7.34 (s, 1H); 7.49 ppm (d, J = 1.4 Hz, 1H). MS: m/z 440 [M−H]−, 320 [M−121]− (p-MeOBn). Anal. Calcd. for C19H17Cl2NO5SxH2O: C 49.58, H 4.16, N 3.04, found: C 49.21, H 3.78, N 3.05.
Ethyl 6,8-dichloro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (11)
A solution of compound 10 (1.64 g, 3.7 mmol) in TFA (8 mL) was stirred at room temperature for 6 h then it was concentrated under reduced pressure. Purification by flash chromatography gave the title product 11 (0.85 g, 71%) as a light yellow solid, mp: 173–177 °C. Rf = 0.35 (petroleum ether/EtOAc/AcOH 4:1:0.01). 1H NMR (200 MHz, CDCl3): δ = 1.41 (t, J = 7.2 Hz, 3H); 4.42 (q, J = 7.2 Hz, 2H); 7.05 (s, 1H); 7.42 (d, J = 2.2 Hz, 1H); 7.54 (d, J = 2.2 Hz, 1H); 7.86 ppm (s, 1H). 13C NMR (100 MHz, CDCl3): δ = 14.1; 63.3; 111.2; 127.5; 129.3; 130.2; 131.2; 131.5; 135.3; 138.3; 161.1. MS: m/z 320 [M−H]−. Anal. Calcd. for C11H9Cl2NO4Sx0.2C6H14: C 43.23, H 3.39, N 4.13, found: C 43.39, H 3.07, N 3.92.
Ethyl 2-(3-bromobenzyl)-6,8-dichloro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (12b)
To a solution of compound 11 (70 mg, 0.22 mmol, 1 equiv.) in DMF (1.5 mL) was added DIEA (0.06 mL, 0.33 mmol) and 3-bromobenzyl bromide (70 mg, 0.24 mmol) and the mixture was stirred at room temperature for 12 h. Then water (10 mL) was added and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated. Purification by column chromatography gave the title compound (100 mg, 95%) as a white solid, mp: 111–114 °C. Rf = 0.29 (petroleum ether:EtOAc, 4:1). 1H NMR (400 MHz, CDCl3): δ = 1.38 (t, J = 7.2 Hz, 3H); 4.37 (q, J = 7.2 Hz, 2H); 5.03 (s, 2H); 6.94 (d, J = 7.6 Hz, 1H); 7.00 (t, J = 7.6 Hz, 1H); 7.19 (s, 1H); 7.24 (d, J = 1.6 Hz, 1H); 7.27–7.28 (m, 1H); 7.39 (s, 1H); 7.54 ppm (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 14.2; 52.8; 62.6; 122.3; 123.8; 126.9; 127.8; 129.8; 130.9. 131.2; 131.2; 131.3; 132.5; 132.9; 134.2; 136.5; 137.9; 161.6. MS: low ionization. Anal. Calcd. for C18H14BrCl2NO4S: C 44.02, H 2.87, N 2.85, found: C 44.09, H 2.95, N 2.81.
Ethyl 6,8-dichloro-2-(3-chlorobenzyl)-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (12c)
Prepared according to procedure for compound 12b using 3-chlorobenzyl chloride. Yield 82%. White solid, mp: 113–116 °C. Rf = 0.39 (Hex/EtOAc 6:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.37 (t, J = 7.2 Hz, 3H); 4.37 (q, J = 7.2 Hz, 2H); 5.03 (s, 2H); 6.88–6.92 (m, 1H); 7.01–7.14 (m, 3H); 7.25 (d, J = 2.0 Hz, 1H); 7.39 (s, 1H); 7.54 ppm (d, J = 2.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm: 14.1; 52.8; 62.6; 123.6; 126.4; 127.8; 128.3; 128.4; 129.5; 131.1; 132.4; 132.9; 134.1; 136.3; 138.0; 161.6. MS m/z 447 [M+H]+ (low ionization).
6,8-Dichloro-2-(4-methoxybenzyl)-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (13a)
To a solution of compound 10 (20 mg, 0.05 mmol) in THF (1 mL) was added a solution of NaOH (2 mg, 0.05 mmol) in water (1 mL) and stirred at room temperature for 6 h. THF was removed under reduced pressure and the aqueous residue was acidified till pH∼2 by addition of 1 N HCl. The precipitate was filtered, washed with water and dried to give the title compound (12 mg, 53%) as a light brown solid, mp: 184–187 °C. 1H NMR (200 MHz, CDCl3): δ: 3.69 (s, 3H); 5.05 (s, 2H); 6.60 (d, J = 8.0 Hz, 2H); 6.94 (d, J = 8.0 Hz, 2H); 7.19 (s, 1H), 7.48 (s, 1H); 7.53 ppm (s, 1H). HPLC: (Method I) tR = 10.38 min, purity 98%. MS m/z 412 [M−H]−.
See Supplementary Material for the respective compounds:
2-(3-Bromobenzyl)-6,8-dichloro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (13b).
6,8-Dichloro-2-(3-chlorobenzyl)-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (13c).
Ethyl 2-(2-(bromomethyl)-4-chloro-N-(4-methoxybenzyl)phenylsulfonamido)acetate (14a)
To a solution of compound 8a (2.68 g, 6.26 mmol) in dry Et2O (20 mL) was added PBr3 (0.21 mL, 2.19 mmol) and stirred at room temperature for 4 h. The solvent was removed under reduced pressure. Purified by flash chromatography (petroleum ether:EtOAc, 4:1) to give the title compound (2.09 g, 68%) as a white solid; mp 112–115 °C. TLC: Rf = 0.29 (Hex:EtOAc, 4:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.18 (t, J = 7.2 Hz, 3H), 3.79 (s, 3H), 3.95 (s, 2H), 4.07 (q, J = 7.2 Hz, 2H), 4.49 (s, 2H), 4.91 (s, 2H), 6.80 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H), 7.38 (dd, J = 8.2, 2.4 Hz, 1H); 7.65 (d, J = 2.2 Hz, 1H), 7.98 (9 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 14.0, 28.4, 46.4, 51.0, 55.3, 61.4, 114.1, 126.0, 128.6, 130.3, 131.3, 133.1, 136.6, 138.3, 139.3, 159.6, 168.6.
See Supplementary Material for the respective compounds:
Ethyl 2-(2-(bromomethyl)-4,5-dichloro-N-(4-methoxybenzyl)phenylsulfonamido)acetate (14b).
Ethyl 2-(2-(bromomethyl)-4,6-dichloro-N-(4-methoxybenzyl)phenylsulfonamido)acetate (14c).
Ethyl 6-chloro-2-(4-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (15a)
To a solution of compound 14a (2.08 g, 4.24 mmol) in THF (20 mL) was added NaH (60% in mineral oil, 0.17 g, 4.25 mmol) and stirred at room temperature for 6 h. The solvent was removed under reduced pressure. To the residue water (30 mL) was added and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was separated by column chromatography (petroleum ether:EtOAc, 4:1) to give the title compound (1.38 g, 79%) as a white solid; mp 103–107 °C. TLC: Rf = 0.27 (Hex:EtOAc, 4:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.22 (t, J = 7.2 Hz, 2H), 3.09 (dd, J = 16.6, 7.0 Hz, 1H), 3.44 (dd, J = 17.0, 9.4 Hz, 1H), 3.78 (s, 3H), 4.02–4.26 (m, 3H), 4.30 (d, J = 15.0 Hz, 1H), 2.43 (d, J = 15.0 Hz, 1H), 6.77 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 7.26 (s, 1H), 7.39 (dd, J = 8.0, 2 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 14.0, 27.6, 51.6, 55.2, 57.2, 62.0, 113.8, 125.0, 126.7, 127.7, 129.1, 130.2, 136.1, 136.5, 138.3, 159.4, 169.5. MS low ionization.
See Supplementary Material for the respective compounds:
Ethyl 6,7-dichloro-2-(4-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (15b).
Ethyl 6,8-dichloro-2-(4-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (15c).
Ethyl 6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (16a)
To a solution of compound 15a (0.47 g, 1.15 mmol) in CH2Cl2 (1 mL) was added TFA (1 mL) and stirred at room temperature for 5 h. The solvent was removed under reduced pressure. Purified by column chromatography (petroleum ether:EtOAc, 2:1) to give the title compound (0.25 g, 75%) as a white solid; mp 135–138 °C. TLC: Rf = 0.44 (Hex:EtOAc, 2:1). 1H NMR (400 MHz, CDCl3), δ ppm: 1.33 (t, J = 7.2 Hz, 3H), 3.16 (dd, J = 16.8, 8.4 Hz, 1H), 3.43 (dd, J = 16.8, 6.0 Hz, 1H), 4.29 (q, J = 7.2 Hz, 2H), 4.62 (dd, J = 13.8, 7.6 Hz, 1H), 7.27 (d, J = 2 Hz, 1H), 7.41 (dd, J = 8.4, 1.6 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 14.1, 30.5, 54.4, 62.8, 125.2, 128.4, 129.2, 135.0, 136.5, 138.6, 169.4. MS m/z 290 [M+H]+.
Ethyl 6,7-dichloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (16b)
Prepared in 77% yield from compound 15b according to procedure for compound 16a. A light pink solid; mp 154–158 °C. TLC: Rf = 0.19 (Hex:EtOAc, 4:1). 1H NMR (200 MHz, CDCl3), δ ppm: 1.33 (t, J = 7.0 Hz, 3H); 3.13 (dd, J = 17.0, 8.0 Hz, 1H), 3.41 (dd, J = 17.0, 5.8 Hz, 1H), 4.29 (q, J = 7.0 Hz, 2H); 4.61 (dd, J = 8.0, 5.8 Hz, 1H), 7.39 (s, 1H), 7.93 (s, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 14.1, 29.8, 54.3, 63.0, 125.7, 131.0, 132.8, 137.0, 137.4, 169.3. MS m/z 324 [M+H]+. Anal. Calcd. for C11H11Cl2NO4S: C, 40.76; H, 3.42; N, 4.32. Found: C, 40.89; H, 3.47; N, 4.23.
Synthesis of 2-substituted ethyl 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylates 17 (general procedure A)
To a solution of ethyl 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide 16 (1 mmol) in DMF (2 mL) was added NaH (60% in mineral oil, 1 mmol) and it was stirred at room temperature for 30 min. Then the alkyl halogenide (1.1 mmol) was added and stirred at room temperature for 4–15 h (TLC control). To the reaction mixture water (10 mL) was added and it was extracted with EtOAc (3 × 10 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The product was purified by column chromatography (petroleum ether:EtOAc).
Ethyl 6-chloro-2-(3-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17a)
Prepared according to general procedure A using compound 16a and 3-methoxybenzyl bromide to give product 17a in 62% yield as a colorless oil; TLC: Rf = 0.33 (Hex:EtOAc, 4:1). 1H NMR (400 MHz, CDCl3), δ ppm: 1.22 (t, J = 7.2 Hz, 3H), 3.14 (dd, J = 16.8, 6.8 Hz, 1H), 3.46 (dd, J = 16.2, 9.8 Hz, 1H), 3.72 (s, 3H), 4.05–4.30 (m, 3H), 4.35 (d, J = 15.2 Hz, 1H), 4.48 (d, J = 15.2 Hz, 1H), 6.77–6.80 (m, 3H), 7.16 (t, J = 8.0 Hz, 1H), 7.28 (s, 1H), 7.40 (dd, J = 8.8, 2.0 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H). MS m/z 410 [M+H]+.
See Supplementary Material for the respective compounds 17:
Ethyl 6-chloro-2-(4-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17b).
Ethyl 6-chloro-2-(3-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17c).
Ethyl 2-(3-bromobenzyl)-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17d).
Ethyl 2-benzyl-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17e).
Ethyl 6-chloro-2-[(2-pyridinyl)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17f).
Ethyl 6-chloro-2-[(3-pyridinyl)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17g).
Ethyl 6-chloro-2-[(2-methyl-1,3-thiazolyl-4)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17h).
Ethyl 2-(tert-butoxycarbonylmethyl)-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17i).
Ethyl 6-chloro-2-(2-phenethyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17j).
Ethyl 6-chloro-2-[2-(3-methoxyphenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17k).
Ethyl 6-chloro-2-[2-(2-methoxyphenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17l).
Ethyl 6-chloro-2-[2-(2-fluorophenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17m).
Ethyl 6-chloro-2-[2-(3-methoxyphenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17n).
Ethyl 6-chloro-2-[2-(2-methoxyphenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17o).
Ethyl 6-chloro-2-[2-(3-fluorophenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17p).
Ethyl 6-chloro-2-[2-(4-chlorophenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17q).
Ethyl 6,7-dichloro-2-(3-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17r).
Ethyl 6,7-dichloro-2-(2-phenylethyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (17s).
Hydrolysis of ethyl 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylates 15, 16 and 17 (general procedure B).
To a solution of ethyl 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide (1 mmol) in THF (2 mL) was added a solution of LiOH·H2O or NaOH (1 mmol) in H2O (2 mL), then it was stirred at room temperature for 2–8 h (TLC control). The reaction mixture was acidified by 1 N HCl (∼pH 2) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was triturated with Et2O or hexane and washed with small amount of Et2O, CH2Cl2 or CHCl3. Dried in vacuo over P2O5 (room temperature).
6-Chloro-2-(4-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18a)
Prepared according to general procedure B using compound 15a and LiOH·H2O. Yield 62%. white solid; mp 179–181 °C. 1H NMR (400 MHz, CDCl3), δ ppm: 3.21 (dd, J = 16.2, 7.8 Hz, 1H), 3.43 (dd, J = 15.4, 9.6 Hz, 1H), 3.77 (s, 3H), 4.11 (dd, J = 9.4, 7.2 Hz, 1H), 4.21 (d, J = 14.6 Hz, 1H), 4.43 (d, J = 14.4 Hz, 1H), 6.77 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 7.29 (s, 1H), 7.43 (dd, J = 7.8, 1.6 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H). MS m/z 380 [M−H]−. Purity (HPLC, 254 nm, Method I, tR = 8.74 min) 99%. Anal. Calcd. for C17H16ClNO5S: C, 53.48; H, 4.22; N, 3.67. Found: C, 53.56; H, 3.98; N, 3.67.
See Supplementary Material for the respective compounds 18:
6,8-Dichloro-2-(4-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18b).
6-Chloro-2-(3-methoxybenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18c).
6-Chloro-2-(4-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18d).
6-Chloro-2-(3-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18e).
2-(3-Bromobenzyl)-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18f).
2-Benzyl-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18g).
6,7-Dichloro-2-(3-chlorobenzyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18h).
6-Chloro-2-(2-phenylethyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18i).
6-Chloro-2-[2-(3-methoxyphenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18j).
6-Chloro-2-[2-(2-methoxyphenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18k).
6-Chloro-2-[2-(2-fluorophenyl)-ethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18l).
6,7-Dichloro-2-(2-phenylethyl)-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18m).
6-Chloro-2-[2-(3-methoxyphenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18n).
6-Chloro-2-[2-(2-methoxyphenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18o).
6-Chloro-2-[2-(3-fluorophenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18p).
6-Chloro-2-[2-(4-chlorophenyl)-2-oxoethyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18q).
6-Chloro-2-[(2-pyridinyl)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18r).
6-Chloro-2-[(3-pyridinyl)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18s).
6-Chloro-2-[(2-methyl-1,3-thiazolyl-4)-methyl]-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18t).
2-(Tert-butoxycarbonylmethyl)-6-chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (18u).
6-Chloro-3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxide (19)
Prepared according to general procedure B using compound 16a and LiOH·H2O. White solid; mp 177–181 °C. 1H NMR (400 MHz, DMSO-d6), δ ppm: 3.08 (dd, J = 17.2, 12.0 Hz, 1H), 3.25 (dd, J = 17.6, 4.8 Hz, 1H), 4.42 (td, J = 12.0, 4.8 Hz, 1H), 7.52 (dd, J = 8.0, 2.0 Hz, 1H), 7.60 (d, J = 1.6 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 10.8 Hz, 1H). LC/MS m/z 260 [M−H]−. Purity (HPLC, 254 nm, Method II, tR = 7.35) 97%.
Binding experiments
For the evaluation of binding affinity of the test compounds on the glycine binding pocket of the NMDA receptor, [3H]-MDL-105,519 (GE Healthcare, Freiburg, Germany) displacement studies were performed using a 96-well plate robotic platform. MDL-105,519 is a selective, high affinity antagonist at the NMDA receptor glycine binding site.
Preparation of cortical membranes
Tissue preparation was performed according to Foster & WongCitation18 with some modificationsCitation19. For isolation of the cell membranes, rat cortices were homogenized in 20 volumes of ice-cold 0.32 M sucrose (Sigma-Aldrich, Taufkirchen, Germany) using a glass-Teflon homogenizer. The homogenate was centrifuged at 1000g for 10 min, the pellet was discarded and the supernatant centrifuged at 20 000g for 20 min. The resulting pellet was re-suspended in 20 volumes of distilled water and centrifuged for 20 min at 8000g. The supernatant and the buffy coat were then centrifuged three times (48 000g for 20 min) in the presence of 50 mM Tris-HCl, pH 8.0 (assay buffer). All centrifugation steps were carried out at 4 °C. After re-suspension in 5 volumes of 50 mM Tris-HCl, pH 7.5, the membrane suspension was frozen rapidly at −80 °C. On the day of assay, the membranes were thawed and washed four times by re-suspension in 50 mM Tris-HCl, pH 7.5 and centrifugation at 48 000g for 20 min. The final pellet was suspended in assay buffer. The amount of protein in the final membrane preparation was determined according to the method of Lowry et al.Citation20 with some modificationsCitation21. The final protein concentration used for our studies was 400 µg/mL.
[3H]MDL-105,519 displacement studies
A robotic system designed for binding assays (Tecan Deutschland GmbH, Crailsheim, Germany) was loaded with the membrane solution, solutions for bound control (buffer/DMSO 20%), unlabeled glycine (1 mM) for evaluation of non-specific binding, all compounds to be tested (at 20-fold concentrations), radioligand and respective 96-well plates. Before performing displacement studies, saturation experiments were performed to determine the equilibrium dissociation constant (Kd) of [3H]-MDL-105,519, which is a parameter for the affinity of the radioligand to the binding site. The protein/receptor concentration was held constant, whereas the amount of specific bound radioligand was determined using increasing concentrations of ligand. On the basis of the saturation experiments, a final [3H]-MDL-105,519 concentration of 2 nM was selected. First, the assay plates were loaded with membrane solution and were shaken at 4 °C. The mother plates were then prepared by pipetting the compounds into assay buffer/20% DMSO to obtain the desired final concentrations (dose response curve with five different concentrations, e.g. 10, 3, 1, 0.3 and 0.1 µM). After transferring radioligand into the assay plates, the compounds were added (including the bound and the non-specific binding control). The final DMSO concentration was 1%. The assay plates were incubated and shaken at 4 °C for 1 h, before the mixture was exhausted as rapidly as possible via a vacuum manifold using the Multiscreen HTS glass fibre (type B) filter plates (Millipore, Schwalbach, Germany) under a constant vacuum of 450 mbar. The membranes were washed four times with cold assay buffer (100 µL). A 50 µL of Ultima Gold scintillation cocktail (PerkinElmer, Rodgau-Jügesheim, Germany) was added to the wet filter plates and incubated at room temperature overnight before counting the disintegration per minutes using a liquid scintillation counter (MicroBeta, PerkinElmer, Rodgau-Jügesheim, Germany).
Analysis of data
For the evaluation of binding affinity of the test compound to the glycine B binding site and its potency to displace [3H]-MDL-105,519, the measured radioactivity of the radioligand alone is set as 100% bound control and the non-specific binding of the radioligand (which could not be displaced by glycine, 1 mM) represented the 0% control. The residual radioactivity after displacement of the test compound (n = 5) is then corrected with respect to the set controls.
Results and discussion
Chemistry
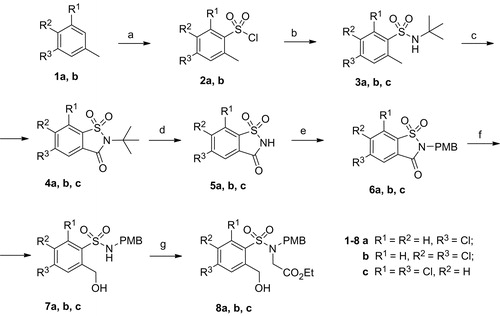
There are two main approaches for the synthesis of 2H-1,2-benzothiazine-3-carboxylates 10–12. One of them is based on the diazotation of an appropriate aniline and subsequent Meerwein arylation of acrylonitrileCitation22. Then sulfonation with chlorosulfonic acid gives the corresponding benzenesulfonyl chloride, which is cyclised to a derivative of 3,4-dihydro-2H-1,2-benzothiazine. First, we attempted the synthesis according to the procedure described above. Unfortunately, the Meerwein arylation of acrylonitrile with 3,5-dichlorobenzenediazonium chloride did not work well in our hands. All attempts to optimise the reaction conditions resulted in only a 9% isolated yield of corresponding 3-aryl-2-chloropropionitrile. Therefore, we focused on the second approach comprising a six-step synthesis from saccharinCitation23. This involved chemical transformation of saccharin derivatives to sulfonamides 8 is shown in Scheme 1. Further on, the hydroxyl group of compound 8 was transformed to bromide which, in turn, was cyclised in the presence of a base to the corresponding 3,4-dihydro-2H-1,2-benzothiazine. Finally, oxidation with NBS gave 2H-1,2-benzothiazine analogues of structure 10. Synthesis of necessary benzisothiazolones 5 was attempted in accordance with the previously described procedureCitation24. As phenylsulfonyl chlorides 2a and 2b were not commercially available, they were prepared by chlorosulfonation of toluene derivatives 1a and 1b. In the next step, sulfonyl chlorides 2 were treated with t-butyl amine to generate corresponding sulfonamides 3. The N-tert-butyl group is compatible with the oxidative cyclisation of compounds 3 with H5IO6 and CrO3 in acetonitrile to obtain benzisothiazoles 4 in high yieldCitation24. The cleavage of N-tert-butyl group and subsequent N-protection with p-methoxybenzyl group gave benzisothiazoles 6.
Scheme 1. Synthesis route to compounds 8a, b, c. Reagents and conditions: (a) SOCl2, ClSO3H, 0 °C – r.t.; (b) t-BuNH2, Et3N, CH2Cl2; (c) H5IO6, CrO3, (CH3CO)2O, MeCN, 0 °C – r.t.; (d) TFA; (e) PMBCl, NaH, DMF, 80 °C; (f) NaBH4, THF/H2O; (g) NaH, ethyl bromoacetate, THF, 50 °C.
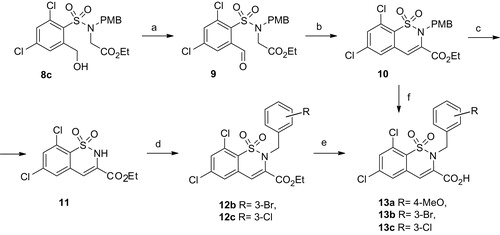
The next step was crucial since the authors of this synthetic pathway for the reductive opening of the isothiazolone ring used lithium aluminum hydride, which gave the product only with 41% yieldCitation24. It appeared that the formation of byproducts could be overcome by using sodium borohydride in THF/water (1:1). Thus, benzenesulfonamides 7 were obtained in 94–98% yield. Alkylation of sulfonamides 7 with ethyl bromoacetate in the presence of base gave compounds 8. The reported three-step transformation of 2-(hydroxymethyl)-benzenesulfonamide to 2H-1,2-benzothiazines included the hydroxide conversion to bromide, ring closure, and finally the oxidation of the ring systemCitation23. To prepare the crucial intermediate 10 from compound 8c we used a two-step procedure as shown in Scheme 2. Oxidation of benzylic hydroxyl group with MnO2 gave aldehyde 9, which was easily cyclised to benzothiazine 10 in the presence of base. The N-(p-methoxybenzyl) group of compound 17 was cleaved with trifluoroacetic acid to give benzothiazine 11 in good yield. Compound 11 was expected to be a versatile synthon for the parallel synthesis of N-substituted 2H-1,2-benzothiazine-3-carboxylic acids. Thus, alkylation of benzothiazine 11 with 3-bromobenzylbromide gave the expected ester 12b in 95% yield. Finally, alkaline hydrolysis of esters 10 and 12 by NaOH or LiOH was used to afford the desired carboxylic acids 13.
Scheme 2. Synthesis route to compounds 13a, b, c. Reagents and conditions: (a) MnO2, THF; (b) NaH, THF; (c) TFA; (d) DIEA, DMF, 3-ClC6H4CH2Cl or 3-BrC6H4CH2Br; (e) LiOH, THF/H2O; (f) NaOH, THF/H2O.
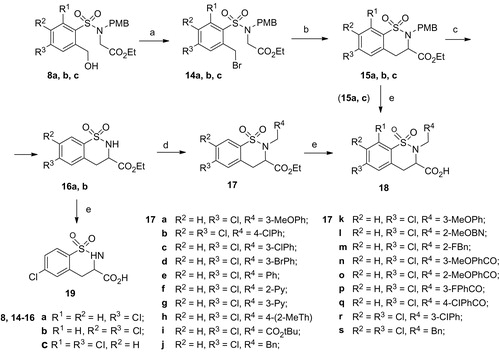
Compounds 18 were synthesized according to the previously described approachCitation22 shown in Scheme 3. Benzyl alcohols 8 were treated with PBr3 to give bromides 14, which were then cyclised to benzothiazines 15, using NaH for deprotonation. Cleavage of N-(p-methoxybenzyl) group with TFA gave 1,2-benzothiazines 16, which were used in N-alkylation reactions with different aryl- and heteroaryl-methyl halides. Then alkaline hydrolysis of ethyl esters 17 gave the desired carboxylic acids 18 in variable yields. The N-(p-methoxybenzyl) group containing compounds 18 was prepared by hydrolysis of the corresponding esters 15.
Scheme 3. Synthesis of target compounds 18 and 19. Reagents and conditions: (a) PBr3, Et2O; (b) NaH, THF; (c) TFA, CH2Cl2; (d) RCitation4CH2Hal, NaH, DMF; (e) LiOH.H2O or NaOH, THF/H2O.
Binding results
The affinity of benzothiazine compounds 13a, b, c to the glycine binding site of the NMDA receptor was determined by the displacement of radioligand [3H]MDL-105,519Citation25–27 from rat cortical membrane preparations. Surprisingly, all 2H-benzothiazine-3-carboxylic acids 13a, b, c were found completely inactive at a 10 µM concentration. Only compound 13b displayed weak affinity (Ki = 90 µM) for the glycine binding site.
The in vitro activity data of compounds 18 and 19 are shown in . Inhibition of radioligand binding at 10 µM concentration of substrate (I, %) is given for less potent compounds in cases when a correct dose-response curve has not been obtained. Roughly, figures close to 50% of radioligand displacement correspond to IC50 ∼ 10 µM in . Our first synthesized 3,4-dihydrobenzothiazine 18b showed a considerable activity at 10 µM concentration, replacing ca 50% of radioligand. It should be noted that the reference compound of kynurenic acid with two halogen atoms in positions 6 and 8 (DCKA), showed higher activity than its 6-monochloro analogueCitation13. In our case, 6,8-dichlorobenzothiazine 18b and monochloro derivative 18a displayed comparable activities. Therefore, the study was continued mainly with 6-chlorosubstituted benzothiazines. An argument in favour of this decision was based on the dramatic solubility diminishing effect after the introduction of the second halogen in position 8. Surprisingly, 6,7-dichlorobenzothiazines were better soluble, allowing the screening to be performed at concentration of at least 10 µM. We also tested 6,7-dichloro-1,2-benzothiazine-3-carboxylic acids 18h and 18m and both showed significantly lower activity, displacing only 45% and 20% of radioligand, respectively at 10 µM concentration. It is important to note that even 2-unsubstituted benzothiazine 19 displays moderate affinity for Gly B with Ki ∼ 10 μM. N-benzyl derivatives displayed higher affinity if the substituents in the phenyl ring were in the meta position (MeO, 18c; Cl, 18e and Br, 18f), regardless of the electronic properties of the substituent. Replacement of N-benzyl with N-phenethyl group resulted in lowered Gly B activity (18i, R4 = 3-MeOPh; Ki = 2.5 µM and 18j, R4 = 3-MeOBn; displacing only ∼40% of radioligand at 10 µM). However, in the case of unsubstituted N-benzyl and N-phenethyl derivatives (18g and 18i, respectively), this effect is not marked because of the reduced activity of both compounds. Actually, compound 18i is more active than 18g (62% and 41% displacement, respectively). The group of acetophenones (18n–18q) possessing additional hydrogen bond acceptor displayed significantly higher activity than N-phenethyl-benzothiazines. It is possible that the oxo group is involved in additional hydrogen bonding with the receptor. In this series of compounds the highest Gly B affinity was observed for compounds with methoxy group in meta and ortho positions of the N-benzyl group (18n and 18o, Ki = 1.1 μM and Ki = 1.7 μM, respectively). Considering the above-mentioned higher activity of the phenone group containing benzothiazines, one could assume that N-substitution with heteroaryl group, which may act as a hydrogen bond acceptor, should also result in potentially active compounds.
Table 1. Activity of compounds 18 and 19 in binding experiments.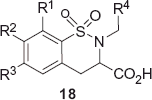
Nonetheless, N-heteroarylmethyl benzothiazines 18r, 18s and 18t were less active. Low activity of 2-pyridinyl derivative 18r may point to different hydrogen bonding vectors in comparison to the phenone group, if such an interaction occurs. To investigate if there is any functional importance of phenyl group, ester 18u was screened. Compound 18u was inactive, even though it possessed an oxygen functionality at the same distance from the benzothiazine core as corresponding acetophenone 18o.
Conclusions
In summary, a series of 2-substituted 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxides were synthesized and evaluated for their affinity to the glycine binding site of the NMDA receptor. The most active compounds displayed micromolar level of activity. The most attractive structures in the search for prospective NMDA receptor ligands were identified to be 2-arylcarbonylmethyl substituted 3,4-dihydro-2H-1,2-benzothiazine-3-carboxylic acid 1,1-dioxides. It has been demonstrated for the first time that the replacement of NH group in the ligand by sp3 CH2 is tolerated. This finding may pave the way for previously unexplored approaches for designing new ligands of the NMDA receptor.
Supplementary material available online
IENZ_1057722_Supplementary_data.pdf
Download PDF (118.8 KB)Acknowledgements
The authors thank Tanja Bauer and Sabine Denk for their expert technical assistance in performing the in vitro experiments.
Declaration of interest
The authors declare no conflicts of interest.
This work was financially supported by Merz Pharmaceuticals GmbH.
Related Research Data
References
- Watkins JC, Collingridge GL, eds. The NMDA receptor. 2nd ed. England: Oxford University Press; 1994:522
- Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010;62:405–96
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 1994;330:613–22
- Danysz W, Parsons CG. Glycine and N-methyl-d-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev 1998;50:597–664
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987;325:529–31
- Jansen M, Dannhardt G. Antagonists and agonists at the glycine site of the NMDA receptor for therapeutic interventions. Eur J Med Chem 2003;38:661–70
- Catarzi D, Colotta V, Varano F. Competitive Gly/NMDA receptor antagonists. Curr Top Med Chem 2006;6:809–21
- Nagata R, Katayama S, Ohtani K, Tanaka H, Shimago K. Tricyclic quinoxalinediones, aza-kynurenic acids, and indole-2-carboxylic acids as in vivo active NMDA-glycine antagonists. Curr Top Med Chem 2006;6:733–45
- Leeson PD, Iversen LL. The glycine site on the NMDA receptor: structure–activity relationships and therapeutic potential. J Med Chem 1994;37:4053–67
- Danysz W, Kozela E, Parsons CG, et al. Peripherally acting NMDA receptor/glycineB site receptor antagonists inhibit morphine tolerance. Neuropharmacology 2005;48:360–71
- Deur C, Agrawal AK, Baum H, et al. N-(6,7-dichloro-2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-N-alkylsulfonamides as peripherally restricted N-methyl-d-aspartate receptor antagonists for the treatment of pain. Bioorg Med Chem Lett 2007;17:4599–603
- Henrich M, Bauer A, Nagel J, et al. Glycine B antagonists. PCT Patent Appl WO 2010;139481 (09.12.2010)
- Leeson PD, Baker R, Carling RW, et al. Kynurenic acid derivatives. Structure–activity relationships for excitatory amino acid antagonism and identification of potent and selective antagonists at the glycine site on the N-methyl-d-aspartate receptor. J Med Chem 1991;34:1243–52
- Jimonet P, Audiau F, Aloup JC, et al. Synthesis and SAR of 2H-1,2,4-benzothiadiazine-1,1-dioxide-3-carboxylic acid derivatives as novel potent glycine antagonists of the NMDA receptor-channel complex. Bioorg Med Chem Lett 1994;4:2735–40
- Colotta V, Catarzi D, Varano F, et al. Synthesis and biological evaluation of a series of quinazoline-2-carboxylic acids and quinazoline-2,4-diones as glycine-NMDA antagonists: a pharmacophore model based approach. Arch Pharm (Weinheim) 1997;330:129–34
- Varano F, Catarzi D, Colotta V, et al. Synthesis of 2-substituted-6,8-dichloro-3,4-dihydro-3-oxo-2H-1,4-benzothiazine-1,1-dioxides and -1-oxides as glycine-NMDA receptor antagonists. Farmaco 1998;53:752–7
- Costa BM, Irvine MW, Fang G, et al. Structure–activity relationships for allosteric NMDA receptor inhibitors based on 2-naphthoic acid. Neuropharmacology 2012;62:1730–6
- Foster AC, Wong EH. The novel anticonvulsant MK-801 binds to the activated state of the N-methyl-d-aspartate receptor in the rat brain. Br J Pharmacol 1987;91:403–9
- Parsons CG, Danysz W, Quack G, et al. Novel systemically active antagonists of the glycine site of the N-methyl-d-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther 1997;283:1264–75
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265–75
- Hartree EF. Determination of protein: a modification of the Lowry method that gives a linear photometric response. Anal Biochem 1972;48:422–7
- Vidal A, Madelmont JC, Mounetou EA. Simple and efficient synthesis of methyl 3,4-dihydro-2-methyl-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide from saccharin sodium salt. Synthesis 2006;4:591–3
- Wells GJ, Tao M, Josef KA, Bihovsky R. 1,2-Benzothiazine 1,1-dioxide P2–P3 peptide mimetic aldehyde calpain I inhibitors. J Med Chem 2001;44:3488–503
- Xu L, Shu H, Liu Y, Zhang S, Trudell ML. Oxidative cyclization of N-alkyl-o-methyl-arenesulfonamides to biologically important saccharin derivatives. Tetrahedron 2006;62:7902–10
- Baron BM, Siegel BW, Harrison BL, Gross RS, Hawes C, Towers P. [3H]MDL 105,519, a high-affinity radioligand for the N-methyl-d-aspartate receptor-associated glycine recognition site. J Pharmacol Exp Ther 1996;279:62–8
- Baron BM, Harrison BL, Kehne JH, et al. Pharmacological characterization of MDL 105,519, an NMDA receptor glycine site antagonist. Eur J Pharmacol 1997;323:181–92
- Höfner G, Wanner K-Th. Characterization of the binding [3H]MDL 105,519, a radiollabelled antagonist for the N-methyl-d-aspartate-associated glycine site, to pig cortical brain membranes. Neurosci Lett 1997;226:79–82