Abstract
Objective. Angiogenesis is critical for successful pregnancy. An anti-angiogenic state has been implicated in preeclampsia, fetal growth restriction and fetal death. Increased maternal plasma concentrations of the anti-angiogenic factor, soluble vascular endothelial growth factor receptor (sVEGFR)-1, have been reported in women with preeclampsia and in those with fetal death. Recent observations indicate that an excess of sVEGFR-1 and soluble endoglin (sEng) is also present in the amniotic fluid of patients with preeclampsia. The aim of this study was to determine whether fetal death is associated with changes in amniotic fluid concentrations of sVEGFR-1 and sEng, two powerful anti-angiogenic factors.
Study design. This cross-sectional study included patients with fetal death (n = 35) and controls (n = 129). Fetal death was subdivided according to clinical circumstances into: (1) unexplained (n = 25); (2) preeclampsia and/or placental abruption (n = 5); and (3) chromosomal/congenital anomalies (n = 5). The control group consisted of patients with preterm labor (PTL) who delivered at term (n = 92) and women at term not in labor (n = 37). AF concentrations of sVEGFR-1 and sEng were determined by ELISA. Non-parametric statistics and logistic regression analysis were applied.
Results. (1) Patients with a fetal death had higher median amniotic fluid concentrations of sVEGFR-1 and sEng than women in the control group (p < 0.001 for each); (2) these results remained significant among different subgroups of stillbirth (p < 0.05 for each); and (3) amniotic fluid concentrations of sVEGFR-1 and those of sEng above the third quartile were associated with a significant risk of unexplained preterm fetal death (adjusted OR = 10.8; 95%CI 1.3–89.2 and adjusted OR 87; 95% CI 2.3–3323, respectively).
Conclusion. Patients with an unexplained fetal death at diagnosis are characterized by an increase in the amniotic fluid concentrations of sVEGFR-1 and sEng. These observations indicate that an excess of anti-angiogenic factors in the amniotic cavity is associated with unexplained fetal death especially in preterm gestations.
Introduction
Fetal death continues to be a major challenge in obstetrics [Citation1–5]. While several conditions (e.g. chromosomal/congenital anomalies, infection, preeclampsia, placental abruption, diabetes, intrauterine growth restriction (IUGR), cord accident, etc.) are associated with stillbirth [Citation1,Citation2,Citation6–8], the precise etiology, in many cases, is difficult to be determined. In as many as 25–60% cases of fetal death, the cause of stillbirth is unexplained [Citation1]. IUGR has been proposed to be responsible for the majority of ‘unexplained’ fetal death cases [Citation9]. Since fetal growth and placental development requires a balance between angiogenic and anti-angiogenic processes [Citation10–12], examining factors participating in angiogenesis may help to elucidate mechanisms of disease in a subset of patients with fetal death.
The main regulator for angiogenesis is vascular endothelial growth factor (VEGF)-signaling system [Citation13,Citation14]. VEGF exerts its biological effects through two high-affinity tyrosine kinase receptors [VEGF receptor (VEGFR)-1 and VEGFR-2] and plays a crucial role in both physiologic and pathologic angiogenesis [Citation13,Citation14]. The binding-affinity of VEGFR-1 to VEGF is 10-fold higher than that of VEGFR-2, whereas the kinase activity of VEGFR-1 is 10-fold weaker than that of VEGFR-2. Thus, VEGFR-2 is the major mediator of the mitogenic, angiogenic, permeability enhancing, and endothelial survival properties of VEGF [Citation13,Citation14]. In contrast, VEGFR-1 has a dual role for angiogenesis [Citation15]: (1) as a negative regulator by sequestration of VEGF (to prevent binding of VEGF to VEGFR-2), and (2) as a positive regulator by inducing activation and migration of monocytes/macrophages or promoting angiogenesis in pathologic conditions (e.g. lung cancer) [Citation15,Citation16]. Both VEGF receptors are present in two forms; the membranous and the soluble forms.
Another important regulator for angiogenesis is the transforming growth factor (TGF)-β signaling system [Citation17,Citation18]. Endoglin (Eng), a transmembrane co-receptor for TGF-β1 and TGF-β3, is highly expressed on proliferating endothelial cells [Citation19], vascular smooth muscle cells [Citation20], syncytiotrophoblast, endometrial stromal cells, activated monocytes [Citation21] and erythroid precursors [Citation18,Citation22]. This protein modulates the action of TGF-β1 as well as TGF-β3, and is essential for vascular homeostasis [Citation18,Citation23]. Expression of Eng increases during angiogenesis, wound healing and inflammation [Citation18]. The soluble form of this protein, sEng, has potent anti-angiogenic activity on endothelial cells [Citation23].
An anti-angiogenic state has been implicated in several obstetrical syndromes with perturbation of uteroplacental blood supply including preeclampsia,[Citation23–39] fetal growth restriction,[Citation40–43] placental abruption, [Citation44] ‘mirror syndrome’ [Citation45], twin-to-twin transfusion syndrome[Citation46] and fetal death [Citation47,Citation48]. Recent observations indicate that an excess of soluble vascular endothelial growth factor receptor-1 (sVEGFR-1) and sEng is present in both maternal blood and amniotic fluid of patients with preeclampsia [Citation23,Citation29,Citation30, Citation32,Citation49–51]. The purpose of this study was to determine if fetal death is associated with changes in the concentrations of sVEGFR-1 and sEng in amniotic fluid.
Patients and methods
Study design
A cross-sectional study was conducted by searching our clinical database and bank of biologic samples. This study included only singleton pregnancies. Amniocentesis was performed in 35 patients with fetal death. Since amniocenteses could not be performed in normal pregnant women without clinical indications, the control group (n = 129) included (1) patients with spontaneous preterm labor with intact membranes (PTL) who underwent amniocentesis and delivered at term without intra-amniotic infection/inflammation (n = 92); and (2) women at term not in labor without intra-amniotic infection (n = 37). Women in the control group had a normal pregnancy outcome without complications (except for PTL) and those who delivered small for gestational age (SGA) neonates were excluded.
Clinical definition
Fetal death was defined as a lack of fetal heart activity after 20 week of gestation diagnosed by ultrasound examination. This group was sub-classified according to clinical circumstances into: (1) unexplained fetal death (n = 25); (2) fetal death with preeclampsia and/or placental abruption (n = 5); and (3) fetal death with a known chromosome abnormality or major malformation (n = 5). Fetuses in the latter group included those with trisomy 21 (n = 1), non-immune hydrops fetalis (n = 3), and a cardiovascular defect with single umbilical artery (n = 1). Preeclampsia was defined as hypertension (systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg on at least two occasions, 4 h to 1 week apart) and proteinuria (≥300 mg in a 24-h urine collection or one dipstick measurement ≥2+) diagnosed after 20 weeks of gestation. Placental abruption was diagnosed based on clinical presentation (vaginal bleeding and abdominal pain) and the presence of a retroplacental blood clot.
Patients were considered to have a normal pregnancy outcome if they did not have any medical, obstetrical, or surgical complications, and delivered a term neonate (≥37 weeks) without complications. PTL was defined by the presence of regular of uterine contractions occurring at a frequency of at least two every 10 min associated with cervical change before 37 completed weeks of gestation that required hospitalization. Intra-amniotic inflammation was defined as an amniotic fluid interleukin-6 concentration ≥2.6 ng/ml [Citation52]. Intra-amniotic infection was defined as a positive amniotic fluid culture for micro-organisms. The diagnosis of SGA was based on an ultrasonographic estimated fetal weight and confirmed by a birthweight below the 10th percentile for gestational age according to the reference range proposed by Alexander et al. [Citation53] and Gonzalez et al. [Citation54] depending on the ethnicity of the patients.
The collection and utilization of the samples was approved by the Human Investigation Committees of the Sotero del Rio Hospital, Santiago, Chile (a major affiliate of the Catholic University of Santiago), and Wayne State University, (Detroit, MI) and the Institutional Review Boards of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD/NIH/DHHS). Many of these samples have been used in previous studies.
Sample collection and immunoassay
Amniotic fluid samples were obtained by transabdominal amniocentesis performed for genetic indications, evaluation of the microbial status of the amniotic cavity and/or assessment of fetal lung maturity in patients approaching term. Samples of amniotic fluid were transported to the laboratory in a sterile capped syringe and cultured for aerobic/anaerobic bacteria and genital mycoplasmas. Samples were centrifuged and stored at –70°C. Amniotic fluid concentrations of sVEGFR-1 and sEng were determined by sensitive and specific immunoassays obtained from R&D Systems (Minneapolis, MN). Briefly, the immunoassay utilized the quantitative sandwich technique and their concentrations were determined by interpolation from the standard curves. The inter- and intra-assay coefficients of variation obtained were: sVEGFR-1: 1.4% and 3.9%, respectively, and sEng: 2.3% and 4.6%, respectively. The sensitivities of the assays were: sVEGFR-1: 16.97 pg/ml, and sEng: 80 pg/ml.
Statistical analysis
Kolmogorov Smirnov and Shapiro-Wilk were used to test for normal distribution of the data. Kruskal Wallis with post hoc Mann–Whitney U test was utilized to determine the differences of the median among and between groups. The results were presented without and with adjustment for multiple comparisons using Holm method. Contingency tables, Chi-square and Fischer's exact test were employed for comparisons of proportions. Spearman correlation was used to examine the relationship between two continuous variables. Logistic regression (backward, step-wise) was applied to determine the association between the presence of fetal death and amniotic fluid concentrations of sVEGFR-1 and sEng while adjusting for potential confounders. Analysis was conducted with SPSS V.15 (SPSS Inc., Chicago, IL). A p value of <0.05 was considered significant.
Results
Demographic and clinical characteristics of women with fetal death and those with a normal pregnancy are displayed in . Women in the fetal death group had a lower median gestational age at amniocentesis than those in the control group (p = 0.03). The median amniotic fluid concentration of sVEGFR-1 and that of sEng were significantly higher in women with fetal death than that of women in the control group (p < 0.001 for each comparison; and ). Amniotic fluid concentrations of sVEGFR-1 and sEng in the control group decreased as a function of gestational age (sVEGFR-1: Spearman's ρ −0.3; p = 0.003 and sEng: Spearman's ρ −0.2; p = 0.02).
Table I. Demographic and clinical characteristics of the study population.
Figure 1. Amniotic fluid concentrations of sVEGFR-1 in the control and fetal death groups. Fetal death had a significantly higher median amniotic fluid concentration of sVEGFR-1 than the control (median 429.5 ng/ml, range 83.4-2366.3 ng/ml vs median 85.1 ng/ml, range 0.1–595.6 ng/ml; respectively; p < 0.001). The y-axis is in logarithmic scale.
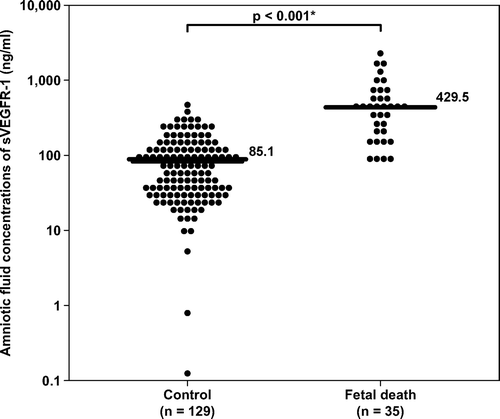
Figure 2. Amniotic fluid concentrations of sEng in the control and fetal death groups. Fetal death had a significantly higher amniotic fluid concentration of sEng than the control (median 2501 pg/ml, range 115–110,850 pg/ml, vs. median 256 pg/ml, range 0–12,775 pg/ml; respectively; p < 0.001). The y-axis is in logarithmic scale. Ten patients in the control group had amniotic fluid sEng concentrations below the limit of detection (data not show). *p < 0.05.
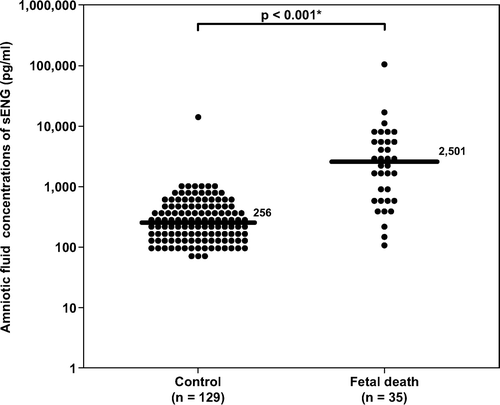
describes the demographic and clinical characteristics according to the subgroups of fetal death. The majority of fetal deaths were categorized as unexplained [71% (25/35)]. When the fetal death group was subdivided according to clinical circumstances, women with unexplained fetal death (n = 25), those with fetal death with preeclampsia and/or placental abruption (n = 5), and those with fetal death and major chromosomal/congenital anomalies (n = 5) had a significantly higher median amniotic fluid concentration of sVEGFR-1 than women in the control group (p < 0.001, p = 0.036 and p = 0.004, respectively; ). The results remained significantly different after adjusting for multiple comparisons using Holm method (p < 0.001, 0.036 and 0.008, respectively). The association between amniotic fluid concentrations of sVEGFR-1 (per each ng/ml increase) and the presence of fetal death in each subgroup remained significant after adjusting for gestational age at amniocentesis ().
Table II. Demographic and clinical characteristics of normal pregnant women and subgroups of fetal death.
Table III. Odds ratio for the associations between the presence of fetal death and amniotic fluid concentrations of sVEGFR-1 (per each ng/ml increase).
Figure 3. Amniotic fluid concentrations of sVEGFR-1 in the control group, unexplained fetal death, fetal death with preeclampsia and/or placental abruption, and fetal death with chromosomal or congenital anomalies. Unexplained fetal death (median 442 ng/ml, range 84–2366 ng/ml), fetal death with preeclampsia and/or placental abruption (median 202 ng/ml, range 83–830 ng/ml) and fetal death with major chromosomal and/or congenital anomalies (median 289 ng/ml, range 96–567 ng/ml) had a significantly higher median amniotic fluid concentration of sVEGFR-1 than the control group (median 85.1 ng/ml, range 0.1–595.6 ng/ml; p < 0.001, p = 0.04 and p = 0.004; respectively). The results remained significantly different after adjusting for multiple comparisons using Holm method (p < 0.001, 0.04 and 0.008 respectively). The y-axis is in logarithmic scale. *p < 0.05.
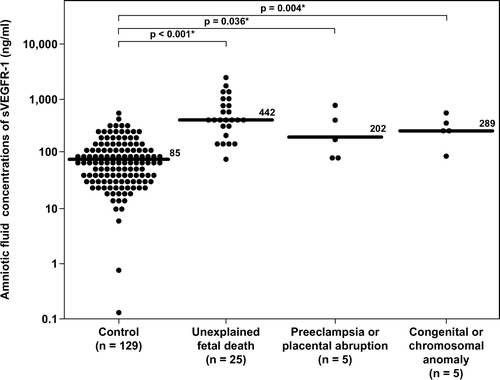
Similar results were obtained for amniotic fluid concentrations of sEng. The median amniotic fluid concentration of sEng in women with unexplained fetal death, in those with fetal death associated with preeclampsia and/or placental abruption, and in those with fetal death and major chromosomal/ congenital anomalies was significantly higher than that of women in the control group (p < 0.001, p = 0.005, and p = 0.001, respectively; ). The results remained significantly different after adjusting for multiple comparisons using Holm method (p < 0.001, 0.005 and 0.002, respectively). The association between amniotic fluid concentrations of sEng (per each ng/ml increase) and the presence of fetal death in each subgroup remained significant after adjusting for gestational age at amniocentesis ().
Table IV. Odds ratio for the associations between the presence of fetal death and amniotic fluid concentrations of sEng (per each ng/ml increase).
Figure 4. Amniotic fluid concentrations of sEng in the control, unexplained fetal death, fetal death with preeclampsia and/or placental abruption, and fetal death with chromosomal or congenital anomalies. Unexplained fetal death (median 1827 pg/ml, range 115–9726 pg/ml), fetal death with preeclampsia and/or placental abruption (median 3542 pg/ml, range 202–110,850 pg/ml) and fetal death with major chromosomal and/or congenital anomalies (median 2962 pg/ml, range 397–7650 pg/ml) had a significantly higher median amniotic fluid concentration of sEng than the control group (median 256 pg/ml, range 0–12,775 pg/ml p < 0.001; p = 0.005 and p = 0.001; respectively). The results remained significantly different after adjusting for multiple comparisons using Holm method (p < 0.001, 0.005 and 0.002 respectively). The y-axis is in logarithmic scale. Ten patients in the control group had amniotic fluid sEng concentrations below the limit of detection (data not show). *p < 0.05.
To examine whether changes in amniotic fluid sVEGFR-1 and sEng concentration in unexplained fetal death vary with gestational age at which fetal death was diagnosed patients with unexplained fetal death were stratified into those who had amniocentesis performed before and after term gestation. The clinical characteristics of women with PTL who delivered at term, women at term without labor and those with unexplained fetal death at preterm and term gestation are displayed in . Women with preterm unexplained fetal death had a lower median gestational age at amniocentesis and a higher rate of SGA neonates than women with PTL who delivered at term (p = 0.02 and p < 0.001; respectively). Unexplained preterm fetal death had a significantly higher median amniotic fluid concentration of sVEGFR-1 and sEng than women with PTL who delivered at term (p < 0.001 for each comparison; and ). There was no significant difference in the median amniotic fluid concentrations of sVEGFR-1 or sEng between patients with unexplained preterm fetal death with and without SGA [sVEGFR-1; SGA (n = 11): median 553 ng/ml, range 167–2366 ng/ml vs. without SGA (n = 9): median 442 ng/ml, range 162–1021 ng/ml; p = 0.3, and sEng; SGA: median 2895 pg/ml, range 376–8278 pg/ml vs. without SGA: median 2501 pg/ml, range 161–9726 pg/ml; p = 0.6).
Table V. Demographic and clinical characteristics of women with PTL who delivered at term, women at term not in labor and those with unexplained fetal death at preterm and term gestation.
Figure 5. Amniotic fluid concentrations of sVEGFR-1 in unexplained fetal death at preterm, term gestation and their control groups. Unexplained fetal death, both at preterm and at term gestation, had a significantly higher median amniotic fluid concentration of sVEGFR-1 than their respective control groups (preterm: median 461 ng/ml, range 162–2366 ng/ml vs. median 100 ng/ml, range 0.1–595 ng/ml; respectively; p < 0.001 and term: median 336 ng/ml, range 84–946 ng/ml vs. median 63 ng/ml, range 10–373 ng/ml; respectively; p = 0.003). The y-axis is in logarithmic scale. *p < 0.05.
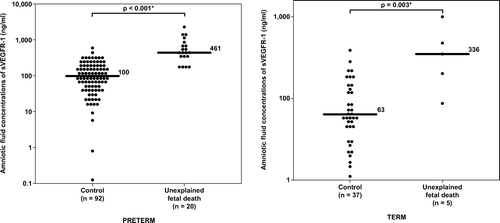
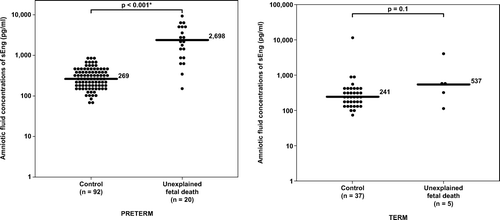
Figure 6. Amniotic fluid concentrations of sEng-1 in unexplained fetal death at preterm, term gestation and their control groups. Unexplained preterm fetal death had a significantly higher median amniotic fluid concentration of sEng than the control group (median 2698 pg/ml range 161–9726 pg/ml vs. median 269 pg/ml, range 0–871 pg/ml; respectively; p < 0.001). In contrast, there was no significant difference in the median amniotic fluid concentrations of sEng between unexplained fetal death at term and the control group (median 537 pg/ml, range 115–4076 pg/ml vs. median 241 pg/ml, range 0–12,775 pg/ml; respectively; p = 0.1). The y-axis is in logarithmic scale. Nine patients in the preterm control group and one patient in the term control group had amniotic fluid sEng concentrations below the limit of detection (data not show). *p < 0.05.
To examine the association between amniotic fluid sVEGFR-1 and sEng concentrations and the presence of unexplained preterm fetal death, amniotic fluid concentrations of sVEGFR-1 and sEng in patients with unexplained preterm fetal death and those with PTL who delivered at term were stratified into quartiles. Among preterm patients, the prevalence of fetal death in each quartile of sVEGFR-1 concentrations was 0% (0/28), 0% (0/28), 14% (4/28), and 57% (16/28), respectively. Similarly, the prevalence of fetal death in each quartile of sEng concentrations was 3.6% (1/28), 0% (0/28), 3.6% (1/28), and 64% (18/28), respectively. Amniotic fluid concentrations of sVEGFR-1 or those of sEng above the third quartile (>75th percentile, or >211.8 ng/ml and >580.5 pg/ml, respectively) were associated with unexplained preterm fetal death after adjusting for gestational age at amniocentesis (weeks), maternal age (years), smoking, African-American ethnicity, nulliparity, duration of sample storage (years) and the presence of SGA (adjusted OR 10.8; 95%CI 1.3–89.2 and adjusted OR 87; 95% CI 2.3–3323, respectively; reference: below the 1st quartile).
Unexplained fetal death at term had a higher median amniotic fluid concentration of sVEGFR-1 and sEng than women at term without labor. However, the difference reached statistical significance only for sVEGFR-1, but not for sEng (p = 0.003 and p = 0.1, respectively; and ).
Among women with fetal death, there was no significant relationship between amniotic fluid concentrations of sVEGFR-1 or sEng and amniotic fluid concentrations of glucose or white blood cell counts (p > 0.05 for each). While the amniotic fluid concentrations of sEng correlated with amniotic fluid red blood cell counts (RBC) (Spearman's ρ 0.5; p = 0.007) and with neonatal birthweight (Spearman's ρ −0.6; p < 0.001), there was no correlation between amniotic fluid concentrations of sVEGFR-1 and RBC counts or neonatal birthweight (p > 0.05 for each). However, there was a significant relationship between amniotic fluid concentrations of sVEGFR-1 and those of sEng in both the fetal death and the control groups (fetal death: Spearman's ρ 0.5; controls: Spearman's ρ 0.4; p < 0.001 for each).
Discussion
Principal findings
(1) Fetal death was associated with higher amniotic fluid concentrations of sVEGFR-1 and sEng than pregnancies in the control group; (2) these results remained significant among different subgroups of stillbirth including unexplained fetal death, fetal death with preeclampsia and/or placental abruption, and fetal death with chromosomal or major congenital anomalies; and (3) amniotic fluid concentrations of sVEGFR-1 and those of sEng above the third quartile were associated with a significant risk of unexplained preterm fetal death.
Fetal death is associated with increased amniotic fluid concentrations of sVEGFR-1
VEGF is essential for fetal development since the loss of even one copy of the VEGF gene leads to embryonic lethality [Citation10,Citation11]. Similarly, VEGFR-1 knock-out mice die due to overgrowth and dysfunction of blood vessels. This negative role of VEGFR-1 is dependent on its extra-cellular region, most likely by trapping VEGF to decrease VEGF function. Thus, the soluble form of VEGFR-1 is considered a natural antagonist of VEGF to control its activities.
Amniotic fluid is a rich source of growth factors including epidermal growth factors [Citation55], insulin-like growth factors [Citation56], hepatocyte growth factors [Citation57], stem cell factors [Citation58], fibroblast growth factors [Citation59], and VEGF [Citation60]. These growth factors may help to promote fetal growth [Citation7,Citation10,Citation11] and development [Citation58,Citation59,Citation61,Citation62]. During fetal life, VEGF is highly expressed in the lung, kidney, spleen and heart [Citation12]. VEGF and sVEGFR-1 are also present in human syncytiotrophoblast [Citation63–66] and amniotic fluid [Citation43,Citation49,Citation51,Citation60]. The findings that sVEGFR-1 concentrations in amniotic fluid were high during the first half of pregnancy and decreased during the third trimester correspond with the temporal changes in VEGF expression observed in the placenta [Citation66]. It is tempting to speculate that sVEGFR-1 in amniotic fluid serves to control the effects of VEGF for appropriate fetal growth and placental development. Indeed, during pregnancy, a state in which extensive angiogenesis is required [Citation67,Citation68], maternal plasma sVEGFR-1 concentrations increase substantially from the non-pregnant state and this elevation, in contrast to that of sVEGFR-1 concentration in the amniotic cavity, continued until term. Among the maternal compartment, fetal blood and amniotic fluid cavity, the highest concentration of sVEGFR-1 in normal pregnancy was observed in the latter [Citation43,Citation49].
An excess of sVEGFR-1 in amniotic cavity of patients with fetal death reported herein would indicate an imbalance of the VEGF-signaling system in the fetal compartment. The finding that fetal death was associated with increased amniotic fluid concentrations of sVEGFR-1 is consistent with the observation made by Tranquilli et al. that amniotic fluid concentrations of VEGF in the midtrimester of pregnancies with subsequent fetal death tended to be lower than normal pregnancies [Citation60]. This group of investigators also reported similar changes of amniotic fluid concentrations of VEGF in pregnant women who carried a Down syndrome fetus [Citation69]. The current study also found an increase of sVEGFR-1 concentrations in amniotic cavity of patients who carried a fetus with major chromosomal/congenital anomalies.
The finding that an unexplained fetal death at preterm gestation is associated with an elevation of sVEGFR-1 concentrations in the amniotic fluid is novel. Since an increase in plasma concentrations of sVEGFR-1 has been observed in several obstetrical syndromes associated with perturbation of the utero-placental blood supply including preeclampsia, [Citation23–35,Citation70–73] and fetal growth restriction [Citation40–43], an elevation of this protein in amniotic fluid could also represent an anti-angiogenic state in the fetal compartment of pregnancies with fetal death. Consistent with this hypothesis, placental lesions, suggestive of poor utero-placental perfusion (e.g. poor vascularization of the villi, occlusion of blood vessels, placental infarction and intravascular thrombi), were frequently found in pregnancies with fetal death especially at preterm gestation. Kidron et al. also found that more than 75% of the placentas from pregnancies with fetal death show abnormalities in maternal or fetal vascular supply [Citation74].
Although sVEGFR-1 could be released from monocytes or endothelial cells after stimulations with endotoxin or tumor necrosis factor-α [Citation75,Citation76], the current study did not observe a relationship between amniotic fluid concentrations of sVEGFR-1 and amniotic fluid white blood cell counts suggesting that the increase in amniotic concentrations of sVEGFR-1 in fetal death observed herein was not a consequence of intra-amniotic infection/inflammation. The lack of correlation between amniotic fluid sVEGFR-1 concentrations and amniotic fluid RBC counts also argues against traumatic amniocentesis as a cause of this elevation.
Fetal death is associated with increased amniotic fluid concentrations of sEng
Genetic deletion studies indicate that Eng is essential for fetal development. Three main functions of Eng during fetal life include: (1) vascular remodeling by function as a mediator for recruitment and differentiation of endothelial vascular smooth muscle cell and pericytes; (2) haematopoiesis in the yolk sac; and (3) embryonic heart development [Citation77]. The mutation of two copies of the Eng gene leads to embryonic lethality by disorganization of blood vessels and cardiac anomalies [Citation77], while the loss of one copy of the Eng gene leads to arterio-venous malformation, a condition similar to hereditary hemorrhagic telangiectasia type I in humans [Citation78]. Endoglin is also present in two forms; the membranous and the soluble forms. While the soluble form of VEGFR-1 is generated by alternative splicing of the VEGFR-1 gene, sEng is likely to be generated by proteolytic cleavage of the membrane-bound form [Citation23]. Eng differentially modulates TGF-β functions with complex mechanisms according to various cell types. In most cells (including endothelial cells), Eng expression counteracts TGF-β, a potent inhibitor of cell proliferation, to facilitate endothelial cell proliferation and differentiation. In contrast, the soluble form of Eng inhibits endothelial nitric oxide synthase and has a potent anti-angiogenic activity [Citation23].
The function of sEng in amniotic fluid is unknown. The findings that amniotic fluid concentrations of sEng decreased as a function of gestational age indicate that the function of sEng in the amniotic cavity may be more prominent in the early than in the late gestation. In contrast to sVEGFR-1, the concentrations of sEng are much lower in the amniotic cavity than those in maternal and fetal blood [Citation50]. Since the changes of amniotic fluid concentrations of sEng are similar to the temporal changes of sEng protein expression in the placenta according to gestational age [Citation79], the placenta could be a source of sEng in the amniotic cavity.
A recent study demonstrated that exposure of villous explants to low oxygen tension (3% oxygen) resulted in increased expression of sEng [Citation79]. Moreover, higher sEng protein expression (by Western blot analysis) was observed in the placenta of fetuses with IUGR and those of smaller twins with discordant growth compared to those of controls and to those of the larger co-twins, respectively [Citation79]. The finding that fetal death was associated with increased amniotic fluid concentrations of sEng is also consistent with a previous report of increased amniotic fluid concentrations of this protein in preeclampsia, a condition associated with reduced utero-placental blood supply [Citation50]. These observations indicate that reduced placental perfusion and/or placental hypoxia may contribute to the increased protein expression of the sEng in the placenta as well as in the amniotic cavity of pregnancies with fetal death. Alternatively, since other sources of proteins in amniotic fluid are the fetal lungs, kidneys, and gastrointestinal tract, it is possible that sEng concentrations in amniotic fluid could be derived from the fetus. Recently, a role of Eng in fetal lung development has been proposed [Citation80]. At midgestation, Eng expression was observed in peri-tubular mesenchymal stem cells, in peri-canalicular vessels, and in the endothelial cells of peri-bronchial vessels, whereas this protein was observed in alveolar capillaries only at late gestation [Citation80]. Interestingly, no immunoreactive staining of Eng in alveolar capillaries was observed in fetuses with Down syndrome, those with cardiac defect, those with IUGR fetuses or those with alveolar capillary dysplasia [Citation80]. It is unlikely, however, that amniotic fluid concentrations of sEng reflect fetal lung development since amniotic fluid concentrations of sEng decreased as term approached.
Although activated monocytes could release sEng [Citation21,Citation81], the current study observed no significant relationship between amniotic fluid concentrations of sEng and amniotic fluid white blood cell counts. This finding suggests that the increase in amniotic concentrations of sEng in fetal death observed herein is not a consequence of intra-amniotic infection/inflammation. Although there was a correlation between amniotic fluid sEng concentrations and amniotic RBC counts, it is unlikely that this elevation results from traumatic amniocentesis because the correlation coefficient was small and amniotic fluid concentrations of sEng was not consistently elevated in cases with high RBC counts in amniotic fluid.
Finally, since each subgroup of fetal death in various clinical circumstances were associated with increased amniotic fluid concentrations of sVEGFR-1 and sEng, it is possible that the changes in amniotic fluid concentrations of anti-angiogenic factors observed herein might be either a terminal event of impending fetal death or a phenomenon after a fetal death has occurred.
Strength and limitation of the study
This is the first study that examines the changes in amniotic fluid concentrations of sVEGFR-1 and sEng in patients with fetal death. Furthermore, a perturbation of amniotic fluid sVEGFR-1 and sEng concentrations in women with fetal death was described and stratified by clinical circumstances and by the gestational age at which the fetal death had occurred.
Three potential limitations of this study are: (1) since amniocentesis could not be performed in normal pregnant women at preterm gestation without clinical indications, the control group in this study included women who presented with PTL without intra-amniotic infection/inflammation and delivered at term; (2) due to the small sample size of women with fetal death and chromosomal/congenital anomalies, and of those with preeclampsia/placental abruption, the changes of sVEGFR-1 and sEng concentrations in these subgroups of fetal death should be interpreted with caution; and (3) the temporal relationship between the changes in amniotic fluid concentrations of sVEGFR-1 or sEng and the death of a fetus could not be determined because of the cross-sectional nature of this study.
In conclusion, patients with an unexplained fetal death at the time of diagnosis are characterized by an increase in the amniotic fluid concentration of sVEGFR-1 and sEng. This observation indicates that unexplained fetal death, especially at perterm gestation, is associated with an excess of anti-angiogenic factors in amniotic fluid. If these alterations occur before the death of a fetus, evaluation of amniotic fluid concentrations of anti-angiogenic factors could be useful to assess the risk of stillbirth.
Acknowledgements
This research was supported by the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS.
Related Research Data
References
- Fretts RC. Etiology and prevention of stillbirth. Am J Obstet Gynecol 2005;193:1923–1935.
- Silver RM, Varner MW, Reddy U, Goldenberg R, Pinar H, Conway D, Bukowski R, Carpenter M, Hogue C, Willinger M, Dudley D, Saade G, Stoll B. Work-up of stillbirth: a review of the evidence. Am J Obstet Gynecol 2007;196:433–444.
- Silver RM. Fetal death. Obstet Gynecol 2007;109:153–167.
- Smith GC, Fretts RC. Stillbirth. Lancet 2007;370:1715–1725.
- Goldenberg RL, McClure EM, Belizan JM. Commentary: reducing the world's stillbirths. BMC Pregnancy Childbirth 2009;9 (Suppl 1):S1.
- Blackwell S, Romero R, Chaiworapongsa T, Refuerzo J, Gervasi MT, Yoshimatsu J, Espinoza J, Berman S, Yoon BH. Unexplained fetal death is associated with changes in the adaptive limb of the maternal immune response consistent with prior antigenic exposure. J Matern Fetal Neonatal Med 2003;14:241–246.
- Richani K, Romero R, Soto E, Espinoza J, Nien JK, Chaiworapongsa T, Refuerzo J, Blackwell S, Edwin SS, Santolaya-Forgas J, et al Unexplained intrauterine fetal death is accompanied by activation of complement. J Perinat Med 2005;33:296–305.
- Blackwell S, Romero R, Chaiworapongsa T, Kim YM, Bujold E, Espinoza J, Camacho N, Hassan S, Yoon BH, Refuerzo JS. Maternal and fetal inflammatory responses in unexplained fetal death. J Matern Fetal Neonatal Med 2003;14:151–157.
- Gardosi J, Mul T, Mongelli M, Fagan D. Analysis of birthweight and gestational age in antepartum stillbirths. Br J Obstet Gynaecol 1998;105:524–530.
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996;380:439–442.
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380:435–439.
- Cheung CY. Vascular endothelial growth factor: possible role in fetal development and placental function. J Soc Gynecol Investig 1997;4:169–177.
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003;9:669–676.
- Shibuya M. Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. BMB Rep 2008;41:278–286.
- Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis 2006;9:225–230.
- Hiratsuka S, Maru Y, Okada A, Seiki M, Noda T, Shibuya M. Involvement of Flt-1 tyrosine kinase (vascular endothelial growth factor receptor-1) in pathological angiogenesis. Cancer Res 2001;61:1207–1213.
- Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res 2008;102:637–652.
- ten DP, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008;11:79–89.
- Raab U, Lastres P, Arevalo MA, Lopez-Novoa JM, Cabanas C, de la Rosa EJ, Bernabeu C. Endoglin is expressed in the chicken vasculature and is involved in angiogenesis. FEBS Lett 1999;459:249–254.
- Adam PJ, Clesham GJ, Weissberg PL. Expression of endoglin mRNA and protein in human vascular smooth muscle cells. Biochem Biophys Res Commun 1998;247:33–37.
- O'Connell PJ, McKenzie A, Fisicaro N, Rockman SP, Pearse MJ, d'Apice AJ. Endoglin: a 180-kD endothelial cell and macrophage restricted differentiation molecule. Clin Exp Immunol 1992;90:154–159.
- Perlingeiro RC. Endoglin is required for hemangioblast and early hematopoietic development. Development 2007;134:3041–3048.
- Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, et al Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006;12:642–649.
- Torry DS, Wang HS, Wang TH, Caudle MR, Torry RJ. Preeclampsia is associated with reduced serum levels of placenta growth factor. Am J Obstet Gynecol 1998;179:1539–1544.
- Taylor RN, Grimwood J, Taylor RS, McMaster MT, Fisher SJ, North RA. Longitudinal serum concentrations of placental growth factor: evidence for abnormal placental angiogenesis in pathologic pregnancies. Am J Obstet Gynecol 2003;188:177–182.
- Reuvekamp A, Velsing-Aarts FV, Poulina IE, Capello JJ, Duits AJ. Selective deficit of angiogenic growth factors characterises pregnancies complicated by pre-eclampsia. Br J Obstet Gynaecol 1999;106:1019–1022.
- Koga K, Osuga Y, Yoshino O, Hirota Y, Ruimeng X, Hirata T, Takeda S, Yano T, Tsutsumi O, Taketani Y. Elevated serum soluble vascular endothelial growth factor receptor 1 (sVEGFR-1) levels in women with preeclampsia. J Clin Endocrinol Metab 2003;88:2348–2351.
- Tsatsaris V, Goffin F, Munaut C, Brichant JF, Pignon MR, Noel A, Schaaps JP, Cabrol D, Frankenne F, Foidart JM. Overexpression of the soluble vascular endothelial growth factor receptor in preeclamptic patients: pathophysiological consequences. J Clin Endocrinol Metab 2003;88:5555–5563.
- Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003;111:649–658.
- Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, et al Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004;350:672–683.
- Chaiworapongsa T, Romero R, Espinoza J, Bujold E, Mee KY, Goncalves LF, Gomez R, Edwin S. Evidence supporting a role for blockade of the vascular endothelial growth factor system in the pathophysiology of preeclampsia. Young Investigator Award. Am J Obstet Gynecol 2004;190:1541–1547.
- Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, et al Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006;355:992–1005.
- Romero R, Nien JK, Espinoza J, Todem D, Fu W, Chung H, Kusanovic JP, Gotsch F, Erez O, Mazaki-Tovi S, et al A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J Matern Fetal Neonatal Med 2008;21:9–23.
- Chaiworapongsa T, Romero R, Gotsch F, Espinoza J, Nien JK, Goncalves L, Edwin S, Kim YM, Erez O, Kusanovic JP, et al Low maternal concentrations of soluble vascular endothelial growth factor receptor-2 in preeclampsia and small for gestational age. J Matern Fetal Neonatal Med 2008;21:41–52.
- Erez O, Romero R, Espinoza J, Fu W, Todem D, Kusanovic JP, Gotsch F, Edwin S, Nien JK, Chaiworapongsa T, et al The change in concentrations of angiogenic and anti-angiogenic factors in maternal plasma between the first and second trimesters in risk assessment for the subsequent development of preeclampsia and small-for-gestational age. J Matern Fetal Neonatal Med 2008;21:279–287.
- Vatten LJ, Eskild A, Nilsen TI, Jeansson S, Jenum PA, Staff AC. Changes in circulating level of angiogenic factors from the first to second trimester as predictors of preeclampsia. Am J Obstet Gynecol 2007;196:239–236.
- Robinson CJ, Johnson DD, Chang EY, Armstrong DM, Wang W. Evaluation of placenta growth factor and soluble Fms-like tyrosine kinase 1 receptor levels in mild and severe preeclampsia. Am J Obstet Gynecol 2006;195:255–259.
- Wikstrom AK, Larsson A, Eriksson UJ, Nash P, Norden-Lindeberg S, Olovsson M. Placental growth factor and soluble FMS-like tyrosine kinase-1 in early-onset and late-onset preeclampsia. Obstet Gynecol 2007;109:1368–1374.
- Unal ER, Robinson CJ, Johnson DD, Chang EY. Second-trimester angiogenic factors as biomarkers for future-onset preeclampsia. Am J Obstet Gynecol 2007;197:211–214.
- Crispi F, Llurba E, Dominguez C, Martin-Gallan P, Cabero L, Gratacos E. Predictive value of angiogenic factors and uterine artery Doppler for early- versus late-onset pre-eclampsia and intrauterine growth restriction. Ultrasound Obstet Gynecol 2008;31:303–309.
- Savvidou MD, Noori M, Anderson JM, Hingorani AD, Nicolaides KH. Maternal endothelial function and serum concentrations of placental growth factor and soluble endoglin in women with abnormal placentation. Ultrasound Obstet Gynecol 2008;32:871–876.
- Chaiworapongsa T, Espinoza J, Gotsch F, Kim YM, Kim GJ, Goncalves LF, Edwin S, Kusanovic JP, Erez O, Than NG, et al The maternal plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated in SGA and the magnitude of the increase relates to Doppler abnormalities in the maternal and fetal circulation. J Matern Fetal Neonatal Med 2008;21:25–40.
- Park CW, Park JS, Shim SS, Jun JK, Yoon BH, Romero R. An elevated maternal plasma, but not amniotic fluid, soluble fms-like tyrosine kinase-1 (sFlt-1) at the time of mid-trimester genetic amniocentesis is a risk factor for preeclampsia. Am J Obstet Gynecol 2005;193:984–989.
- Signore C, Mills JL, Qian C, Yu K, Lam C, Epstein FH, Karumanchi SA, Levine RJ. Circulating angiogenic factors and placental abruption. Obstet Gynecol 2006;108:338–344.
- Espinoza J, Romero R, Nien JK, Kusanovic JP, Richani K, Gomez R, Kim CJ, Mittal P, Gotsh F, Erez O, et al A role of the anti-angiogenic factor sVEGFR-1 in the ‘mirror syndrome’ (Ballantyne's syndrome). J Matern Fetal Neonatal Med 2006;19:607–613.
- Kusanovic JP, Romero R, Espinoza J, Nien JK, Kim CJ, Mittal P, Edwin S, Erez O, Gotsch F, Mazaki-Tovi S, et al Twin-to-twin transfusion syndrome: an antiangiogenic state? Am J Obstet Gynecol 2008;198:382–388.
- Espinoza J, Chaiworapongsa T, Romero R, Kim YM, Kim GJ, Nien JK, Kusanovic JP, Erez O, Bujold E, Goncalves LF, et al Unexplained fetal death: another anti-angiogenic state. J Matern Fetal Neonatal Med 2007;20:495–507.
- Rana S, Rauh-Hain A, Karumanchi A, Thadhani R. First-trimester serum angiogenic factors and the risk of intrauterine fetal demise. Am J Obstet Gynecol 2007;197:S12.
- Staff AC, Braekke K, Harsem NK, Lyberg T, Holthe MR. Circulating concentrations of sFlt1 (soluble fms-like tyrosine kinase 1) in fetal and maternal serum during pre-eclampsia. Eur J Obstet Gynecol Reprod Biol 2005;122:33–39.
- Staff AC, Braekke K, Johnsen GM, Karumanchi SA, Harsem NK. Circulating concentrations of soluble endoglin (CD105) in fetal and maternal serum and in amniotic fluid in preeclampsia. Am J Obstet Gynecol 2007;197:176.
- Vuorela P, Helske S, Hornig C, Alitalo K, Weich H, Halmesmaki E. Amniotic fluid – soluble vascular endothelial growth factor receptor-1 in preeclampsia. Obstet Gynecol 2000;95:353–357.
- Yoon BH, Romero R, Moon JB, Shim SS, Kim M, Kim G, Jun JK. Clinical significance of intra-amniotic inflammation in patients with preterm labor and intact membranes. Am J Obstet Gynecol 2001;185:1130–1136.
- Alexander GR, Himes JH, Kaufman RB, Mor J, Kogan M. A United States national reference for fetal growth. Obstet Gynecol 1996;87:163–168.
- Gonzalez RP, Gomez RM, Castro RS, Nien JK, Merino PO, Etchegaray AB, Carstens MR, Medina LH, Viviani PG, Rojas IT. A national birth weight distribution curve according to gestational age in Chile from 1993 to 2000. Rev Med Chil 2004;132:1155–1165.
- Scott SM, Buenaflor GG, Orth DN. Immunoreactive human epidermal growth factor concentrations in amniotic fluid, umbilical artery and vein serum, and placenta in full-term and preterm infants. Biol Neonate 1989;56:246–251.
- Merimee TJ, Grant M, Tyson JE. Insulin-like growth factors in amniotic fluid. J Clin Endocrinol Metab 1984;59:752–755.
- Kurauchi O, Itakura A, Ando H, Kuno N, Mizutani S, Tomoda Y. The concentration of hepatocyte growth factor (HGF) in human amniotic fluid at second trimester: relation to fetal birth weight. Horm Metab Res 1995;27:335–338.
- Heidari Z, Isobe K, Goto S, Nakashima I, Kiuchi K, Tomoda Y. Characterization of the growth factor activity of amniotic fluid on cells from hematopoietic and lymphoid organs of different life stages. Microbiol Immunol 1996;40:583–589.
- Hirai C, Ichiba H, Saito M, Shintaku H, Yamano T, Kusuda S. Trophic effect of multiple growth factors in amniotic fluid or human milk on cultured human fetal small intestinal cells. J Pediatr Gastroenterol Nutr 2002;34:524–528.
- Tranquilli AL, Giannubilo SR, Bezzeccheri V, Ciavattini A, Scagnoli C, Mazzanti L. Amniotic levels of nitric oxide and vascular endothelial growth factor in pregnancy with subsequent intrauterine fetal death. Eur J Obstet Gynecol Reprod Biol 2004;114:162–165.
- Mulvihill SJ, Hallden G, Debas HT. Trophic effect of amniotic fluid on cultured fetal gastric mucosal cells. J Surg Res 1989;46:327–329.
- Kong W, Yee LF, Mulvihill SJ. Hepatocyte growth factor stimulates fetal gastric epithelial cell growth in vitro. J Surg Res 1998;78:161–168.
- Charnock-Jones DS, Sharkey AM, Boocock CA, Ahmed A, Plevin R, Ferrara N, Smith SK. Vascular endothelial growth factor receptor localization and activation in human trophoblast and choriocarcinoma cells. Biol Reprod 1994;51:524–530.
- Clark DE, Smith SK, Sharkey AM, Charnock-Jones DS. Localization of VEGF and expression of its receptors flt and KDR in human placenta throughout pregnancy. Hum Reprod 1996;11:1090–1098.
- Cooper JC, Sharkey AM, McLaren J, Charnock-Jones DS, Smith SK. Localization of vascular endothelial growth factor and its receptor, flt, in human placenta and decidua by immunohistochemistry. J Reprod Fertil 1995;105:205–213.
- Ahmed A, Li XF, Dunk C, Whittle MJ, Rushton DI, Rollason T. Colocalisation of vascular endothelial growth factor and its Flt-1 receptor in human placenta. Growth Factors 1995;12:235–243.
- Banks RE, Forbes MA, Searles J, Pappin D, Canas B, Rahman D, Kaufmann S, Walters CE, Jackson A, Eves P, et al Evidence for the existence of a novel pregnancy-associated soluble variant of the vascular endothelial growth factor receptor, Flt-1. Mol Hum Reprod 1998;4:377–386.
- Chaiworapongsa T, Romero R, Kim YM, Kim GJ, Kim MR, Espinoza J, Bujold E, Goncalves L, Gomez R, Edwin S, et al Plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated prior to the clinical diagnosis of pre-eclampsia. J Matern Fetal Neonatal Med 2005;17:3–18.
- Tranquilli AL, Bezzeccheri V, Scagnoli C, Mazzanti L, Garzetti GG. Amniotic levels of vascular endothelial growth factor and nitric oxide at the second trimester in Down's syndrome. J Matern Fetal Neonatal Med 2003;13:28–31.
- Vatten LJ, Eskild A, Nilsen TI, Jeansson S, Jenum PA, Staff AC. Changes in circulating level of angiogenic factors from the first to second trimester as predictors of preeclampsia. Am J Obstet Gynecol 2007;196:239–236.
- Robinson CJ, Johnson DD, Chang EY, Armstrong DM, Wang W. Evaluation of placenta growth factor and soluble Fms-like tyrosine kinase 1 receptor levels in mild and severe preeclampsia. Am J Obstet Gynecol 2006;195:255–259.
- Wikstrom AK, Larsson A, Eriksson UJ, Nash P, Norden-Lindeberg S, Olovsson M. Placental growth factor and soluble FMS-like tyrosine kinase-1 in early-onset and late-onset preeclampsia. Obstet Gynecol 2007;109:1368–1374.
- Unal ER, Robinson CJ, Johnson DD, Chang EY. Second-trimester angiogenic factors as biomarkers for future-onset preeclampsia. Am J Obstet Gynecol 2007;197:211–214.
- Kidron D, Bernheim J, Aviram R. Placental findings contributing to fetal death, a study of 120 stillbirths between 23 and 40 weeks gestation. Placenta 2009;30:700–704.
- Tsao PN, Chan FT, Wei SC, Hsieh WS, Chou HC, Su YN, Chen CY, Hsu WM, Hsieh FJ, Hsu SM. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit Care Med. 2007;35:1955–1960.
- Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, et al Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007;115:1789–1797.
- Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science 1999;284:1534–1537.
- Lebrin F, Mummery CL. Endoglin-mediated vascular remodeling: mechanisms underlying hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med 2008;18:25–32.
- Yinon Y, Nevo O, Xu J, Many A, Rolfo A, Todros T, Post M, Caniggia I. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: hypoxic regulation via transforming growth factor-beta 3. Am J Pathol 2008;172:77–85.
- Barresi V, Grosso M, Vitarelli E, Granese R, Barresi G. Endoglin (CD105) immuno-expression in human foetal and neonatal lungs. Histol Histopathol 2008;23:701–708.
- Rajakumar A, Michael HM, Rajakumar PA, Shibata E, Hubel CA, Karumanchi SA, Thadhani R, Wolf M, Harger G, Markovic N. Extra-placental expression of vascular endothelial growth factor receptor-1, (Flt-1) and soluble Flt-1 (sFlt-1), by peripheral blood mononuclear cells (PBMCs) in normotensive and preeclamptic pregnant women. Placenta 2005;26:563–573.