Abstract
Objective: To explore the future potential of genetic screening to detect newborns at risk of childhood-onset hearing loss. Design: An expert led discussion of current and future developments in genetic technology and the knowledge base of genetic hearing loss to determine the viability of genetic screening and the implications for screening policy. Results and Discussion: Despite increasing pressure to adopt genetic technologies, a major barrier for genetic screening in hearing loss is the uncertain clinical significance of the identified mutations and their interactions. Only when a reliable estimate of the future risk of hearing loss can be made at a reasonable cost, will genetic screening become viable. Given the speed of technological advancement this may be within the next 10 years. Decision-makers should start to consider how genetic screening could augment current screening programmes as well as the associated data processing and storage requirements. Conclusion: In the interim, we suggest that decision makers consider the benefits of (1) genetically testing all newborns and children with hearing loss, to determine aetiology and to increase knowledge of the genetic causes of hearing loss, and (2) consider screening pregnant women for the m.1555A> G mutation to reduce the risk of aminoglycoside antibiotic-associated hearing loss.
| Abbreviations | ||
| DNA | = | Deoxyribonucleic acid |
| NGS | = | Next generation sequencing |
| NHS | = | National Health Service |
| PCR | = | Polymerase chain reaction |
| SNPs | = | Single nucleotide polymorphisms |
| TSC | = | Targeted sequence capture |
Congenital hearing loss is the most common birth defect in developed countries and the most prevalent sensorineural disorder (Hilgert et al, Citation2009). Permanent childhood hearing impairment of a moderate or greater degree (i.e. detection thresholds ≥ 40 decibels hearing level averaged across 0.5, 1, 2 and 4 kHz) is present at birth in about 1.6 per 1000 live births, of which approximately 1.0 in 1000 are bilateral impairments and 0.6 in 1000 are unilateral impairments (CitationNHS Newborn Hearing Screening Programme, 2011; Bamford et al, Citation2007). However, studies have shown that the prevalence of permanent childhood hearing impairment continues to increase through infancy, and by the school entry hearing screen (4–5 years of age) possibly affects 3.5 per 1000 children (Bamford et al, Citation2007). Causes of childhood-onset hearing loss may be either genetic or acquired.
It is estimated that at least two thirds of cases of childhood-onset hearing loss have a genetic cause (Hilgert et al, Citation2009), with the remaining third caused by environmental factors e.g. cytomegalovirus infection, meningitis, acquired conductive loss, and the impact of extracorporeal membrane oxygenation (ECMO). Interest in genetic screening for hearing loss is increasing (Wu et al, Citation2011). The rationale for screening babies to identify the risk of future hearing loss from a genetic cause, is to prepare the family and child, enabling them to consider use of parallel communication strategies in advance of hearing deterioration (albeit that the uncertainty of future hearing loss may limit the numbers prepared to adopt this approach), and ultimately may enable interventions to be tailored to specific causes of hearing loss. Genetic screening could also have a role in identifying newborns with hearing loss too mild to be detected with current screening programmes. Genetic screening for a specific mitochondrial mutation during pregnancy could offer a strategy of minimizing hearing loss in babies from exposure to avoidable risk factors such as neonatal use of aminoglycoside antibiotics.
Since March 2006, all babies in England have been offered hearing screening through the NHS Newborn Hearing Screening Programme. Within 4–5 weeks of birth the automated otoacoustic emission test is carried out to identify moderate, severe, and profound hearing impairment. If the test does not show a clear response, the automated otoacoustic emission test may be repeated, or the baby will have an automated auditory brainstem response screening test which records brain activity in response to sounds. In England, approximately 2% of babies screened are subsequently referred for full diagnostic assessment involving tympanometry and further auditory brainstem response testing (NHS Newborn Hearing Screening ProgrammeCitation, 2011).
Between March 2006 when the CitationNHS Newborn Hearing Screening Programme commenced and June 2011, more than 7300 babies with permanent childhood hearing impairment were identified. On average, of the 690 000 babies who are screened each year, around 1100 babies (1.6 per 1000 births) are found to have permanent childhood hearing impairment at birth (CitationNHS Newborn Hearing Screening Programme, 2011). Around 90% of babies who are identified with hearing loss are born into families with no history of the disorder.
Children who have a clear response to the newborn hearing screening tests (around 93%) are not generally retested again until they receive the school entry hearing screen (age 4–5). In the UK, 3.5 per 1000 children have permanent hearing impairment at school screen age, indicating for example in the birth cohort for Citation2010, in addition to the 1100 babies identified at birth, a further 1300 children (1.9 per 1000 live births) require identification after the newborn screen (Bamford et al, Citation2007). It is not clear however, what proportion of the 1.9 per 1000 births are accounted for by, (1) babies who initially passed the screen but in whom the losses present in the newborn period were too mild to be detected by the current newborn hearing screen; (2) true false negatives, i.e. cases with the target condition that were missed by the screen, and (3) cases of acquired hearing loss, whether with an underlying genetic cause i.e. genetic predisposition to hearing loss following aminoglycoside exposure, or a non-genetic cause. Within the 1.9 per 1000 births, there will also be programme false negatives, e.g. children with hearing loss who did not have a newborn hearing screen (although acceptance rates for newborn screening are generally high). The actual number of children therefore with childhood-onset hearing loss that could be expected to be identified as being at risk by genetic screening may only be around 60–70% of the 1300 children requiring identification after the newborn screen. Other countries have different experiences and the ‘gap’ may be smaller or larger.
Hearing impairment in an infant can have dramatic effects on language and educational progress. There is debate however, about the benefits of identification at an early age. Some evidence has shown that with early intervention and support, including cochlear implants for severe to profound hearing loss and hearing aids for mild to moderate hearing loss, a proportion of children can develop language within the range of normal hearing peers (Ching, Citation2012). If childhood-onset hearing loss ensues postlingually, interventions may have less impact.
Rationale for the review
The pressure to use genetic technology is growing in many areas of medicine (particularly in cancer and haematology). In some complex disorders, however, there appears to be misalignment between the pace at which information is being generated (aided by the rapid technological advances in genetic testing capabilities in the research setting) and the knowledge required to interpret the clinical significance of this information. In this paper the current knowledge on the genetics of hearing loss is considered along with the technological developments on the horizon to determine whether at some point in the future the convergence between these two factors will allow genetic screening in newborns to detect the likelihood of childhood-onset hearing loss to become a viable prospect that decision makers will need to address.
Genetic inheritance of hearing loss
Of the children who develop childhood-onset hearing loss with a genetic basis, the majority (around 70%) are non-syndromic, i.e. have no associated visible abnormalities of the external ear or any related medical problems other than hearing impairment, and arise predominantly from mutations inherited in an autosomal recessive pattern (Van Camp et al, Citation1997). In less than 1% of cases, inheritance is either X-linked (affecting predominantly males), or mitochondrial (Cryns & Van Camp, Citation2004). Even in cases of syndromic hearing loss, symptoms may not always be apparent at birth.
Unlike in some conditions where a common mutation in just one gene is responsible for the majority of cases (e.g. the deltaF508 deletion of three nucleotides in the CFTR gene in cystic fibrosis), hearing loss is genetically very heterogeneous involving mutations in many genes (Van Camp et al, Citation1997). It is therefore not possible to predict the risk of developing hearing loss from assessing a single or even a selection of the genes that have currently been identified.
The most frequent causative genes that have been identified in autosomal recessive non-syndromic hearing loss (ARNSHL), in order of frequency are GJB2, SLC26A4, MYO15A, OTOF, CDH23, and TMC1 (Hilgert et al, Citation2009; Hutchin et al, Citation2005). For each of these genes, at least 20 mutations have been reported (see ). GJB2, which encodes the gap junction protein connexin 26 regulating the passage of ions in and out of cells, is the most frequently reported gene involved in genetic hearing loss, yet still only accounts for a maximum of 10–20% of cases of childhood hearing loss in northern European populations (Hutchin et al, Citation2005). It is often claimed that mutations in GJB2 account for over 50% of cases of recessive genetic non-syndromic hearing loss, but this figure is misleading as it is dependent on the denominator used and the population it refers to. For example, mutations in GJB2 occur at a frequency of 50% in families with two or more siblings with hearing loss in Mediterranean countries such as Spain or Italy, but then drops to 30% in families with siblings with hearing loss in Northern Europe and then to 10–20% in families with one child with hearing loss (Lench et al, Citation1998; Zelante et al, Citation1997; Hutchin et al, Citation2005).
Table 1. Most common genes associated with autosomal recessive non-syndromic hearing loss and numbers of reported mutations per gene (adapted from Hilgert et al, Citation2009).
GJB2 is a small gene containing a single coding exon, but over 300 different mutations have been reported (Human Gene Mutation Database: www.hgmd.org). Specific mutations however, are more common in certain populations e.g. deletion of a G nucleotide at position 35 (c.35delG) is carried by 2–3% of the white population (Green et al, Citation1999), while the c.235delC mutation is common in Asian populations. Onset of hearing loss caused by mutations in GJB2 is nearly always pre-lingual, but not necessarily congenital. It is therefore possible that hearing may be normal at birth and then deteriorate rapidly during the first few months of life, although this is rare (Bitner-Glindzicz, Citation2002).
Mutations in the pendrin gene SLC26A4 are the second most frequent cause of ARNSHL, accounting for up to 3.5% of cases (Hutchin et al, Citation2005). SLC26A4, which codes for an anion transport protein, also underlies Pendred syndrome, which is one of the most common forms of syndromic hearing loss. It is under-diagnosed as it is characterized by late-onset thyroid symptoms and appears non-syndromic during childhood (Gardener et al, Citation2006). Beyond GJB2 and SLC26A4, the frequency of mutations in other genes involved in hearing loss is uncertain (Kothiyal et al, Citation2010), most having been described in small studies and large families.
As of January 2012, mutations in 68 genes for non-syndromic hearing loss had been identified and 35 in more ‘common’ forms of syndromic hearing loss. A further seven non-syndromic, and four syndromic gene mutations have been identified within mitochondrial DNA (Hereditary Hearing Loss Homepage: http://hereditaryhearingloss.org). Maternally-inherited hearing impairment due to mutations in the mitochondrial genome appears to be a rare cause of prelingual hearing loss, but the most common mitochondrial mutation, m.1555A> G, can predispose to irreversible hearing loss resulting from aminoglycoside exposure (Hutchin et al, Citation1993).
It is anticipated that over 500 genes, some containing hundreds of mutations, are likely to be involved in causing hearing loss and there is the added complication that the disorder could also result from interactions between several genes. For example, some people will have mutations in one gene but not necessarily experience hearing loss unless they also have another mutation in a different gene. Sometimes different mutations within the same gene can result in either dominant or recessive inheritance, further increasing the genetic complexity of this disorder (Rodriguez-Paris et al, Citation2010). Inevitably, this will make it difficult to determine whether a newborn will develop hearing loss, especially if novel mutations are present in a young asymptomatic child.
Analysis of mutations
The two main techniques currently used to analyse the presence of mutations in the genes involved in hearing loss are direct DNA sequencing, which identifies all mutations present in a gene, and hybridization-based techniques (using microarrays/chips), which identify selected mutations in a gene.
Direct sequencing
Direct (or full) sequencing involves sequentially determining the order of nucleotide bases in a strand of DNA typically produced by the polymerase chain reaction (a PCR “amplicon”). Mutations are identified by comparison of the patient's DNA sequence with a reference sequence. It is the reference (gold) standard of mutation detection as almost all types of mutations can be detected (except partial or full gene deletions). Non-coding regions of genes are not usually sequenced and so only coding region mutations, or those close by (e.g. in splice sites) are detected. The Sanger chain termination or dideoxy sequencing technique has remained the most commonly used direct sequencing method to date (see for details of the technique), however it is expensive, time consuming, and labour intensive and is not well suited for multigene analysis (Gardener et al, Citation2006; Metzker, Citation2005). The diagnostic process can take several months to fully sequence large genes. In the last few years, the Sanger method has been partially superseded by next generation sequencing (NGS), also called massively parallel sequencing, which retains the ability to fully sequence genes, but offers dramatic increases in cost-effective sequence throughput, and as such is poised to emerge as the dominant technology in genomics research (see section on future developments, and ) (Morozova & Marra, Citation2008; Biesecker, Citation2010; Ng et al, Citation2010).
Hybridization mutation chips
In contrast to direct sequencing, DNA chips hybridize sample DNA (the patient's) to short sequences of reference DNA (oligonucleotides) containing known specific mutations in genes (usually single nucleotide polymorphisms (SNPs), or insertions or deletions) attached to a microarray. While this technology cannot identify all mutations, it can simultaneously analyse in parallel many mutations in several genes in a single experiment. These multigene mutation chips are therefore cheaper and faster than direct sequencing, but are generally less sensitive and accurate and will miss mutations that have not previously been described and included on the chip.
Current state of genetic testing for hearing loss
While it is possible using currently available technology to determine if an asymptomatic newborn has a mutation in the genes known to be implicated in hearing loss, it is not always possible to say with any certainty for all of these genes if that person will go on to experience hearing loss in the future. Molecular genetic testing in the United Kingdom (UK) is therefore restricted to patients with known hearing loss in order to establish the cause. There are a limited number of centres in the UK that provide such genetic testing facilities, giving rise to geographical variation in services. Current UK health service recommendations are to fully sequence GJB2 in neonates with hearing loss and whose parents consent, and SLC26A4 in patients with suspected Pendred syndrome, or enlarged vestibular aqueducts. However, this is not always offered, and many other rarer genes associated with hearing loss are not tested. Full direct sequencing, as opposed to specific mutation analysis is necessary due to the large number of mutations in GJB2, and ethnic diversity in the UK.
Genetic population screening for hearing loss
If the technology is available to determine if a newborn has a genetic mutation linked with hearing loss, could population genetic screening of newborns to detect future hearing loss be an impending possibility? In recent years, population based screening has been proposed as one of the major strategies for translating advances in genetic knowledge and associated technologies into population health gains (Khoury et al, Citation2003). However, there are multiple issues regarding the clinical validity, utility, acceptability, feasibility, and equity in access to these genetic screening programmes to be considered before they can be implemented.
The use of criteria to appraise screening programmes and to guide policy decisions is a widely accepted practice in the field of public health. In 1968, Wilson and Junger developed the first criteria for the assessment of proposed screening programmes (CitationWilson & Junger, 1968). The application and appropriateness of these criteria to genetic screening has been challenged (Goel, Citation2001; Ross, Citation2006; Dhondt, Citation2007). In response, Andermann and colleagues carried out an extensive review of the screening criteria literature and consulted stakeholders to produce a decision support guide for genetic screening policy-making (Andermann et al, Citation2011). The 20 decision support criteria proposed by Andermann et al includes modified Wilson and Junger criteria and emerging criteria (). For a screening programme for genetic risk to be implemented, all of these criteria would need to be considered, but of these, criteria 13–20 are the most pertinent to this paper where the technological developments on the horizon are considered.
Table 2. Decision support criteria for genetic screening programmes (Andermann et al, Citation2011, with permission from S. Karger, Basel).
Criteria 19, ‘there should be a suitable screening test’ is unsurprisingly a key criterion. A shift from genetic testing of infants already known to have hearing loss to genetic screening in all newborns to identify those likely to develop hearing loss in the future, requires not only significant advances in technology with unit cost reduction, but also in the knowledge of the genetic basis of hearing loss.
Selective gene/mutation screening
Specific mutations within a limited selection of genes
Despite the large number of mutations in many genes that contribute to hearing loss, it has been proposed that it may be useful to select ‘hotspot’ mutations in frequently implicated genes such as GJB2 and SLC26A4 for newborn screening at a population level (Hutchin et al, Citation2005; Li et al, Citation2008; Khoury et al, Citation2003). SNP mutation chips capable of identifying a selection of common mutations in a limited number of genes for syndromic and non-syndromic hearing loss have been available for the last two to three years (see ). Most have been developed in the research setting, but several are now available commercially. They have been developed predominantly in the USA to complement newborn hearing screening programmes as a means of following up positive cases to determine the cause of hearing loss.
Table 3. Mutation chips for hearing loss.
Table 4. Developments in next generation sequencing technologies and specifications compared with automated Sanger sequencing (adapted from Wellcome TrustCitation, 2009).
The range of chips currently available can identify between 15–300 mutations in 4–31 genes. Most include the genes GJB2 and SLC26A4 as part of the gene selection, but only incorporate a small number (around 5–10) of the most common mutations within these genes. The chips are versatile in that the mutations detected can be expanded as new mutations are identified by research, or redesigned to detect common mutations in different ethnic populations. There are however, many genes associated with hearing loss that are not represented on these chips. There is a trade off between including a selection of mutations with the highest frequencies, but not so many as to make the time to result and the cost of the technology prohibitive.
Some of the limitations of mutation chips which have restricted their adoption in the UK include:
Hearing loss can result from any number of several hundred mutations in over potentially 500 genes, therefore the identification of mutations in 4–31 genes when they occur in such low frequency is of limited utility from a diagnostic perspective.
The lack of a straightforward link between the presence of a mutation in a gene implicated in hearing loss and a definitive diagnosis due to variability in penetrance (the proportion of individuals with a mutation who exhibit clinical symptoms) and mutation interactions. Indeed, it is not always necessarily known if a sequence change is a pathogenic mutation or natural variation.
The knowledge that even chips that have attempted to include all known mutations, for example the APEX array (Gardener et al, Citation2006; Rodriguez-Paris et al, Citation2010), are of limited use as it still only reflects a small selection of genes that could potentially be involved in hearing loss. As novel mutations continue to be discovered that are spread throughout the genome, these chips quickly become outdated.
A lack of identification of mutations by these chips does not necessarily exclude a genetic cause of hearing loss entirely.
Sequencing methods have advanced at such a pace that the role of SNP mutation chips when compared to direct DNA sequencing is now questionable.
There may be a case however, for genetic screening of all pregnant women for the mitochondrial m.1555A> G mutation. This point mutation, which is maternally inherited, has a population prevalence of 1.9 per 1000 (Bitner-Glindzicz et al, Citation2009) and is responsible for causing permanent profound hearing loss with a penetrance close to 100% in carriers following standard therapeutic doses of aminoglycoside antibiotics. As use of these antibiotics is restricted to serious infections, the frequency of aminoglycoside prescribing would have to be taken into account before considering the benefits of such a screening programme, but increasing antimicrobial resistance to newer antibiotics has led to a revived interest in the use of aminoglycosides (Durante-Mangoni et al, Citation2009). Hence, knowledge of the carrier status of m.1555A> G would prompt implementation of alternative therapies for infection control for a small number of babies and children, and potentially prevent unnecessary hearing loss. Several of the available mutation chips currently incorporate this mutation in the MTRNR1 (12SrRNA) gene (see ).
Wu et al (Citation2011) reported a preliminary genetic screening study in 1017 consecutive newborns. The screen targeted four point mutations associated with hearing loss in three genes (GJB2, SLC26A4, and mitochondrial m12SrRNA) commonly found in the Taiwanese population. Correlation of the results of the newborn genetic screen with the newborn hearing screen, revealed nine babies who passed the otoacoustic emission hearing screen at birth, but in whom genetic variants suggested potential for hearing loss. Audiological assessment in these babies at three months identified one with slight hearing loss and two with mild hearing loss. In the remaining six babies who continued with normal hearing, the authors recommended comprehensive audiological evaluation at one year in those with a definite diagnosis on newborn genetic screening, or genetic counselling and additional genetic study (full gene sequencing) in those with one mutated allele.
All mutations (full gene sequencing) within a limited selection of genes
Population screening of GJB2 mutations in newborns capturing all mutations within this gene by full sequence analysis is an alternative approach to identify multiple mutations. Ultimately this may not be helpful since hearing loss caused by GJB2 mutations is usually congenital in onset and the vast majority of those with these mutations will have already been detected by the newborn hearing screen. In addition to those who are homozygous or compound heterozygous for GJB2 mutations, many would be identified as carriers and, due to the genetic heterogeneity of hearing loss, there would still be very many babies with hearing loss who would not be detected by GJB2 mutation testing alone.
Future developments in mutation analysis
In recognition of the genetic complexity of hearing loss, the immediate future (i.e. next three to five years) of mutation analysis is moving towards full sequencing to capture all mutations in a large number of genes. This is being made possible by the significant advances relating to the capacity, speed, and cost of direct sequencing technologies, which is impacting on how much of the genome can be sequenced (Wright et al, Citation2011). Next generation sequencing is a more efficient way of directly sequencing DNA, giving rise to several hundred fold increases in speed and reductions in cost. It is already in widespread use within the research environment and has recently become available from a number of diagnostic service providers. Next generation sequencers differ mainly in read length capability (the number of bases that can be generated contiguously using a sequencing machine), speed, and total sequencing capacity (see ).
Short-read next generation sequencers (Illumina and SOLiD) are more suitable for re-sequencing, where the genome has already been sequenced, and it is possible to compare the newly sequenced strand against the reference to identify unique variations. The cost of next generation sequencing equipment is in the range of US$500 000–1 000 000 (equivalent to €340 000–680 000; £305 000–610 000), which will initially restrict use of this technology. Newer ‘benchtop’ models are coming on the market and should be significantly cheaper.
Targeted sequence capture is a technique designed to work with next generation sequencing. It enables specific genes and their upstream or downstream regulatory regions to be isolated from the genome so that they can be sequenced separately. Agilent SureSelect is a solution-based form of targeted sequence capture (www.genomics.agilent.com), while NimbleGen is a solid-phase form of targeted sequence capture (www.nimblegen.com). Once known, all of the genes involved in causing hearing loss, including all exons (coding regions of genes), exon/intron boundaries and promoter sequences could be fully sequenced on a diagnostic platform to produce a specific genetic test for hearing loss. OtoSCOPE (Otological Sequence Capture Of Pathogenic Exons) is the first massively parallel sequencing platform that utilizes targeted sequence capture and next-generation sequencing for genetic testing of hearing loss (Shearer et al, Citation2010). It has been developed by the University of Iowa and is being used in a research setting to fully sequence all exons of 57 genes associated with hearing loss. At this stage in its development various methods of targeted sequence capture and next generation sequencing are being compared to determine which combination has the greatest level of sequence coverage.
Otogenetics Corporation in the USA is currently offering a genetic mutation testing service using targeted sequence capture and Illumina next generation sequencing for the detection of variants in 84 known human genes associated with hearing loss for approximately US$500 (equivalent to €350; £300) per sample (Otogenetics CorporationCitation, 2011). However, their sensitivity when applied to different populations will need evaluation. The pace of next-generation sequencing development and uptake has been remarkable, and targeted sequence capture of the exome (sum total of all exons) is now a well established research technique. While both of these technologies are capable of fully sequencing the included genes associated with hearing loss, Otogenetics is available only for researchers, and OtoSCOPE is available privately, but only in a diagnostic rather than a screening capacity at a cost of approximately US$2000.
It is anticipated that the next five to ten years will see yet more improvements in the speed and cost of DNA sequencing. There is already discussion about ‘third generation’ DNA sequencing, which aims to increase the speed of sequencing and reduce costs even further. It is likely to be available in a research capacity by 2012 (some laboratories have early access to these machines), and with this comes the possibility of routine whole genome sequencing. Although sequencing the whole genome seems exhaustive, it could be more cost-effective than having to select the genes of interest (Wright et al, Citation2011). There is likely to be a ten-fold reduction in equipment costs compared with next generation sequencing which could enable use in regional laboratories. The eventual goal is to improve sequencing technology to such an extent that it becomes possible to produce the ‘$1000 genome’, which may then extend the potential of this technology to clinical applications, but only once knowledge of the impact of the sequence variants is known.
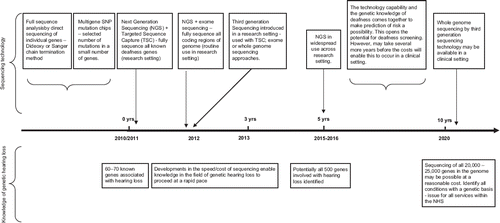
summarizes the timeline of anticipated technology developments in genetic screening for hearing loss for the next 10 years.
Figure 1. Timeline of developments in genetic screening for hearing loss.
Implications for screening policy
While the technology is on course to provide the means of comprehensively identifying the genetic mutations associated with hearing loss, this is just one of the many issues to consider prior to the decision to implement a genetic screening programme for this disorder as outlined by Andermann et al, (Citation2011).
Programme management level (benefit versus harm)
While a detailed evaluation of the evidence for the clinical utility of the early diagnosis and intervention in childhood-onset hearing loss is worthwhile, it is beyond the remit of this article. It is, as yet, uncertain how much additional benefit there would be to individuals and families, and also to education, health, and social services from knowing which and how many children are at risk of developing childhood-onset hearing loss.
The developmental, health, and psychosocial benefits of knowing ahead of time that a child's hearing may deteriorate are also unknown. While we touched on the potential to initiate parallel communication strategies with this knowledge, the uncertainty around the level of risk for individual children and families may preclude any preparatory measures, and in the era of cochlear implants the adoption of anything other than mainstream communication modes is not common. In addition, repeated hearing testing throughout childhood would be likely to engender parent and child concern.
The time frame of the potential deterioration (as some of the hearing loss might not occur for many years, even well into adulthood) would impact on this harm and benefit equation.
Indeed as some of these hearing losses will develop slowly, with a long window in which to make the diagnosis clinically, prior genetic screening may not actually speed up the diagnosis but will expose individuals and family to an uncertain period of concern. It is also unclear whether the additional 1.9 per 1000 live births currently detected with permanent childhood hearing impairment between newborn hearing screening and school entry are on average of lesser severity than those detected at birth. This is all before there is any consideration of the costs (to individual, families, and services) and the cost-effectiveness of any screening programme.
The key benefit of genetic screening may be in knowing which subset of individuals to monitor, so that interventions can be offered at a point where they can have maximum impact, in contrast to the existing chasm of years of lost opportunity following the newborn screen.
Clinical services level
At a clinical services level, while the incidence and prevalence of hearing loss is relatively low (1.6 per 1000 live births), its suitability for screening has been demonstrated with the prior existence of a national screening programme in the UK in recognition of the severity of the disorder and the potential for substantially better outcomes from intervention and educational support if early identification is achieved. The latency period of childhood-onset hearing loss however, means that the current screening strategy at birth is inadequate. An additional genetic screen may offer the potential to identify at an earlier stage some of the 1300 children per year in the UK (1.9 per 1000 live births) who have a genetic component to their childhood-onset hearing loss, but would not help identify those with acquired hearing loss. This may enable strategies such as preparing the family and child, developing parallel communication strategies, and minimizing exposure to known risk factors to be employed in advance of hearing deterioration. While no therapeutic interventions can be linked to a diagnostic test at present, knowledge of the specific cause of hearing loss may lead to tailored interventions in the coming years.
The potential clinical value of genetic screening lies in identifying those newborns at risk of developing hearing loss at a later stage so that they can monitored with repeat hearing testing. Routine hearing testing in childhood will still be essential to identify hearing loss arising from mutations outside the selected genetic screen and for acquired hearing loss. Genetic screening would therefore augment current hearing screening programmes rather than replace them. The need to offer genetic screening to all newborns in order to identify this relatively small ‘at risk’ group who could develop hearing loss through the presence of causative mutations could present a barrier to implementation, certainly from a cost perspective. Although babies identified with hearing loss at birth could be excluded from any such screening programme, genetic testing is already undertaken as part of the aetiological investigation of their hearing loss.
Given the genetic heterogeneity of hearing loss, it is not possible to give a definitive diagnosis of a genetic risk for future hearing loss using tests that screen for one or two genes associated with hearing loss, or mutation chips that identify only a selection of mutations in a small number of genes. Thus, categorization of an asymptomatic newborn into ‘screen positive’ or ‘screen negative’ will be far more complex for progressive childhood-onset hearing loss.
Laboratory testing level
The technology used to conduct population based genetic screening has to fulfil requirements in terms of its reliability to categorize into positive and negative cases with acceptable levels of sensitivity and specificity, and patient and societal acceptance. Next generation DNA sequencing is already becoming an established research technique and the introduction of third generation sequencing in the next two to three years introduces the possibility of sequencing an entire genome (within reasonable costs) within 10 years. These advances in sequencing technology are offering improved sensitivity and specificity of mutation analysis and have greatly contributed to the pace at which genetic knowledge of hearing loss is being accrued, to the point where clinical applications of this technology may be possible within the 10 year timeframe.
While DNA sequencing technology itself is evolving rapidly, there is a relative lack of commercial interest in developing genetic diagnostic assays for hearing loss compared to other conditions with a genetic basis. Possible reasons are that methods are easy to copy and adapt, which results in a weaker intellectual property position for industry; also there are currently no commercially produced therapeutic interventions that could be linked to a diagnostic test. Moreover, due to the significant ethnic variation in the UK, diagnostic tests for hearing loss that have been developed from population samples in other countries may not be applicable to the UK population.
In addition to the sensitivity and specificity of the technology, a further issue to consider is the need to standardize interpretation of genetic tests across the health service to ensure consistency. Currently, variation between clinical geneticists exists because the available literature is contentious and the data are simply not available, or are conflicting. Although the best means to ensure consistency is to put algorithms in place so that an automated informatics output could provide an interpretation, it will be some considerable time before this becomes a possibility. Much more information about the phenotypic consequences of genomic variants will be needed to develop robust predictions based upon sequence.
From a patient acceptability perspective, current methods of DNA analysis require between 2–5 mls of blood which would be unacceptable for a newborn screen, but with improving sequencing techniques, it is anticipated that sufficient DNA could be extracted from a drop of blood taken at the same time that blood is collected for the newborn bloodspot metabolic screen (the Guthrie test). The information obtained from genetic screening programmes however may pose wider issues of acceptability for the child, other family members, and society. Sequencing the whole genome will identify at birth all germ line mutations that could give rise to future genetic diseases. The ethics in relation to incidental findings are extensively documented in the literature (Nuffield Council on Bioethics, Citation2006), the predominant dilemma being if mutations associated with conditions such as Huntington's disease or familial cancer are identified at the same time as testing for hearing loss, is there a moral obligation to inform the patient, and/or their parents, even if this was not the purpose of the test?
Finally, there are the infrastructure requirements to consider with the implementation of a national genetic screening programme. Sequencing equipment and data analysis is expensive, with research centres sometimes contracting out to large central sequencing services. Portable (point of care) DNA sequencers are in development—these are only currently capable of analysing a few SNPs at a time; however, it is possible to envisage comprehensive targeted mutation screening in the future. Whole genome sequencing would probably need to be done at one of the large centres to manage the volume of data generated. The data processing and storage requirements would be substantial and likely to be prohibitive for currently-configured health services in the short term. The data storage capacity and basic information technology infrastructure within the UK health services needs to be addressed to enable targeted sequence capture or whole genome sequencing at a population level (Department of Health, Citation2003; Wright et al, Citation2011).
Conclusion
Genetics cannot be reliably used in a screening capacity for hearing loss until more is known about the genes involved and the clinical significance of the identified mutations. While it is anticipated that this knowledge could be determined within the next five years, potential interactions and the pathogenic significance of specific sequence variants i.e. potential variable penetrance into phenotypes, may take several more years to decipher. Only then will it be possible to determine with any certainty the value of a screening programme.
Decision-makers will need to resist the pressure to adopt genetic technologies until such a time when the potential benefits of obtaining this information are more certain, outweigh the harms, and can be obtained at a reasonable cost. Our view is that while genetic screening for predicting the risk of future hearing loss in newborns is not justified for this condition at this time or in the immediate future, its likely viability in the longer term indicates the need for decision-making bodies to start considering how genetic screening could be used to augment the current hearing testing screening programme.
The infrastructure and data processing requirements of a screening programme for childhood-onset hearing loss are likely to be prohibitive for a one-disease screening programme to implement, and instead may need a wider health service commitment to genetic screening for other conditions. In the 10-year timeframe, it is likely that this discussion will be taking place within the context of a much wider application of genetics in medicine. Information on hearing loss will be just one disorder among many others extracted from a bank of information obtained on an individual through whole genome sequencing. Cost-effectiveness in this context will therefore have to be considered differently to a standalone genetic screening programme.
In the interim, we suggest there is a role for genetic testing in all newborns who do not pass newborn hearing screening and in children identified with childhood-onset hearing loss to identify presently known genetic causes of their hearing loss and to increase our knowledge of the genetic causes of hearing loss. In doing so, this information could assist in establishing the prevalence and links between gene mutation and hearing loss in the UK. The potential for increased usage of aminoglycoside antibiotics also supports the case for a genetic screening programme of pregnant women for the m.1555A> G mutation which could avoid unnecessary cases of hearing loss.
Declaration of interest: The authors report no conflicts of interest.
The research was part of a strategic future planning exercise commissioned by Professor Adrian Davis on behalf of the NHS Newborn Screening Programme, and was conducted by the National Institute for Health Research (NIHR) Horizon Scanning Centre, which is a NIHR research programme funded by the Department of Health for England. The NIHR had no role in the study design, the collection, analysis, and interpretation of the data, the preparation of the manuscript, or in the decision to submit the article for publication. The views expressed in this paper are those of the authors and not necessarily those of the NHS, Department of Health or the funding bodies concerned. LLP, SS, and CP are funded by the National Institute for Health Research. MBG is supported by Great Ormond Street Hospital Children's Charity (GOSHCC). KPS and CL are supported by the Wellcome Trust (grant number 098051). SJD is funded by the Wellcome Trust (grant number 091092/Z/09/Z) and Deafness Research UK. NL is funded by Great Ormond Street Hospital, London. AD is funded by the Department of Health for England.
References
- Abe S., Yamaguchi T. & Usami S-I. 2007. Application of deafness diagnostic screening panel based on deafness mutation/gene database using Invader Assay. Genet Test, 11, 333–340.
- Andermann A., Blancquaert I., Beauchamp S. & Costea I. 2011. Guiding policy decisions for genetic screening: Developing a systematic and transparent approach. Public Health Genomics, 14, 9–16.
- Bamford J., Fortnum H., Bristow K., Smith J., Vamvakas G. . 2007. Current practice, accuracy, effectiveness, and cost effectiveness of the school entry hearing screen. Health Technol Assess, 11(32), 1–168.
- Biesecker L.G. 2010. Exome sequencing makes medical genomics a reality. Nat Genet, 42, 13–14.
- Bitner-Glindzicz M. 2002. Hereditary deafness and phenotyping in humans. Br Med Bull, 63, 73–94.
- Bitner-Glindzicz M., Pembrey M., Duncan A., Heron J., Ring S.M. . 2009. Prevalence of mitochondrial 1555A→G mutation in European children. NEJM, 306, 640–642.
- CGC Genetics. Molecular diagnosis of congenital deafness (syndromic and nonsysndromic) by Array CGC. Available at: www.CGCGenetics.com. Accessed 25/11/2010.
- Ching Y.C. 2012. The Longitudinal Outcomes of Children with Hearing Impairment (LOCHI) study. Available at: http://www.outcomes.nal.gov.au/Publications_Presentations_Newsletters/2012_Yr%203%20outcomes_FirstVoice%20meeting.pdf Accessed 21/07/2012.
- Cryns K. & Van Camp G. 2004. Deafness genes and their diagnostic applications. Audiol Neurootol, 9, 2–22.
- Department of Health. 2003. Our Inheritance, Our Future: Realising the Potential of Genetics in the NHS. London: HMSO.
- Dhondt J-L. 2007. Neonatal screening: from the ‘Guthrie age’ to the ‘genetic age’. J Inherit Metab Dis, 30, 418–422.
- Durante-Mangoni E., Grammatikos A., Utili R. & Falagas M.E. 2009. Do we still need aminoglycosides?Int J Antimicrob Agents, 33, 201–205.
- Gardener P., Oitmaa E., Messner A.Hoefsloot L.Metspalu A. . 2006. Simultaneous multigene mutation detection in patients with sensorineural hearing loss through a novel diagnostic microarray: A new approach for newborn screening follow-up. Pediatrics, 118, 985–994.
- Goel V. 2001. Appraising organised screening programmes for testing for genetic susceptibility to cancer. BMJ, 322, 1174–1178.
- Green G.E., Scott D.A., McDonald J.M., Woodworth G.G., Sheffield V.C. . 1999. Carrier rates in the Midwestern United States for GJB2mutations causing inherited deafness. JAMA, 281, 2211–2216.
- Hilgert N., Smith R.J. & Van Camp G. 2009. Forty six genes causing nonsyndromic hearing impairment: Which ones should be analysed in DNA diagnostics?Mutat Res, 681, 189–196.
- Hutchin T., Coy N.N., Conlon H., Telford E., Bromelow K. . 2005. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK: Implications for genetic testing. Clin Genet, 68, 506–512.
- Hutchin T., Haworth I., Higashi K., Fischel-Ghodsian N., Stoneking M. . 1993. A molecular basis for human hypersensitivity to aminoglycoside antibiotics. Nucleic Acids Res, 21, 4174–4179.
- Khoury M.J., McCabe L.L. & McCabe E.R.B. 2003. Population screening in the age of genomic medicine. NEJM, 348, 50–58.
- Kothiyal P., Cox S., Ebert J.Husami A., Kenna M.A. . 2010. High-throughput detection of mutations responsible for childhood hearing loss using resequencing microarrays. BMC Biotechnology, 10, 10.
- Lench N., Houseman M., Newton V., Van Camp G. & Mueller R. 1998. Connexin-26 mutations in sporadic non-syndromal sensorineural deafness. Lancet, 351, 415.
- Li C-X., Pan Q., Guo Y-G., Li Y., Gao H-F. . 2008. Construction of a multiplex allele-specific PCR-based universal array (ASPUA) and its application to hearing loss screening. Hum Mutat, 29, 306–314.
- Metzker M.L. 2005. Emerging technologies in DNA sequencing. Genome Res, 15, 1767–1776.
- Morozova O. & Marra M.A. 2008. Applications of next-generation sequencing technologies in functional genomics. Genomics, 92, 255–264.
- Ng S.B., Nickerson D.A., Bamshad M.J. & Shendure J. 2010. Massively parallel sequencing and rare disease. Hum Mol Genet, 19(R2), R119–124.
- NHS Newborn Hearing Screening Programme. NHS Newborn Hearing Screening Programme 2010–11 Annual report and 2009–10 Data Report. Available at: www.hearing.screening.nhs.uk/publications. Accessed 23/01/ 2012.
- Nuffield Council on Bioethics. 2006. Genetic Screening: A Supplement to the 1993 Report by the Nuffield Council of Bioethics. London: Nuffield Council of Bioethics.
- Otogenetics Corporation. Available at: www.otogenetics.com/index_htm_files/Deafness_80_genelist.pdf Accessed 07/06/2011.
- Partners Healthcare. Harvard Medical School. Otochip Test for hearing lossand Usher Syndrome. Available at: pcpgm.partners.org/lmm/tests/ hearing-loss/OtoChip. Accessed 25/11/2010.
- Pediatrix Medical Group. Soundgene Screening Panel. Available at: www.pediatrix.com/body.cfm?id = 2889&0TopID = 50. Accessed 17/12/2010.
- Rodriguez-Paris J., Pique L., Colen T., Roberson J., Gardener P. . 2010. Genotyping with a 198 mutation arrayed primer extension array for hereditary hearing loss: Assessment of its diagnostic value for medical practice. PLoS One, 5, e11804.
- Ross L.F. 2006. Screening for conditions that do not meet the Wilson and Junger Criteria: The case of Duchenne muscular dystrophy. Am J Med Genet, 140A, 914–922.
- Shearer A.E., DeLuca A.P., Hildebrand M.S., Taylor K.R., Gurrola II J. . 2010. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci USA, 107, 21104–21109.
- Siemering K., Manji S.S.M., Hutchinson W.M., Sart D.D., Phelan D. . 2006. Detection of mutations in genes associated with hearing loss using a microarray-based approach. J Mol Diagn, 8, 483–489.
- Van Camp G., Willems P.J., Smith R.J. 1997. Non-syndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet, 60, 758–764.
- Wellcome Trust. Genomics – the next generation. Available at: www.wellcome.ac.uk/News/2009/Features/WTX056032.htmAccessed 17/12/2010.
- Wilson J.M.G., Junger G. (1968). Principles and Practice of Screening for Disease. Geneva: World Health Organisation.
- Wright C., Burton H., Hall A., Moorthie S., Pokorska-Bocci A., . 2011. Next steps in the sequence. The implications of whole genome sequencing for health in the UK. Cambridge: PHG Foundation.
- Wu C-C., Hung C-C., Lin S-Y., Hsieh W-U., Tsao P-O., . 2011. Newborn genetic screening for hearing impairment: A preliminary study at a tertiary center. PLoS One, 6, e23314.
- Zelante L., Gasparini P., Estivill X., Melchionda S., D’Agruma L., . 1997. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol. Genet, 6, 1606–1609.