ABSTRACT
This study evaluated the safety and efficacy of arformoterol and formoterol over 6-months in subjects with COPD. In a multi-center, 6-month randomized, double-blind, double-dummy trial, subjects with COPD (mean FEV1 1.21 L, ∼41.0%% predicted) were randomized to receive either nebulized arformoterol (15μg BID [[n == 149]][[ARF 15]], 25μg BID [[n == 147]][[ARF 25]]), or racemic formoterol (12μg BID [[n == 147]][[FORM]]) delivered by DPI. The proportion of subjects with any post-treatment adverse event for ARF 15, ARF 25 μg, and FORM was 67.8%%, 76.2%% and 66.7%%, respectively, and those with at least one COPD exacerbation was 32.2%%, 30.6%%, and 22.4%%, respectively. Pulmonary function improved for all treatment groups and was maintained throughout the study. Mean change from baseline at 6-months for ARF 15, ARF 25 and FORM in peak FEV1 was 0.30L, and 0.34L, and 0.26L, respectively, in 24-hour trough FEV1 was, 0.10L, 0.14L, and 0.09L, and in inspiratory capacity was, 0.20L, 0.37L, and 0.23L. Dyspnea, (mean Transition Dypsnea Index (TDI) focal score) improved in all treatment arms (ARF 15: 1.4, ARF 25: 1.5, and FORM: 1.4) at 6 months, as did rescue short-acting β2-agonists use (mean range: −1.1 to −1.3 actuations/day) and ipratropium bromide (mean range: −0.3 to −0.8 actuations/day). Health status, measured by St George's Respiratory Questionnaire, improved from baseline at 6-months in all treatment groups (mean change: −3.7 to −6.8). In this 6-month study, arformoterol and formoterol were well-tolerated, and their use was associated with improvement in pulmonary function and health status in subjects with COPD with no apparent development of tolerance.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a multi-component disease characterized by airflow obstruction that is poorly reversible (Citation1, 2). It affects approximately 24 million people in the United States, and is currently the fourth-leading cause of death and is expected to be the third-leading cause of death by 2020 (Citation3,4). Evidence-based guidelines recommend the use of inhaled long-acting bronchodilators including long-acting β2-agonists (LABAs) for the initial maintenance treatment in patients with COPD (Citation2,3,Citation5). Nonetheless, multiple studies have found that physicians under-prescribe long-acting bronchodilators for maintenance treatment of patients with moderate to very severe COPD (Citation6–11). Although not recommended by the guidelines, (Citation5) many physician prescribe short-acting β-agonists (SABAs) as regular treatment for this patient population (Citation6–9).
Several factors may influence the prescription of such medications to patients with COPD. Recent concerns have been raised about the safety of prolonged use of LABAs in patients with asthma (Citation12). Furthermore, some studies suggest that the long-term use of β2-agonists can be associated with diminished efficacy over time (e.g., tolerance) in patients with COPD (Citation13–20). Few studies have investigated the long-term safety (at least 6-months) and efficacy of racemic formoterol (Citation19–22). Until recently, long-acting bronchodilators were administered primarily by metered dose inhalers (MDI) or dry powdered inhalers (DPI).
However, some patients with COPD, in particular those who cannot manipulate hand-held devices and those who cannot achieve a sufficient inspiratory flow for adequate drug delivery may have difficulty using these devices (Citation23). A nebulized delivery in such situations may prove superior to these more common delivery methods (Citation23). Arformoterol, the (R,R)-isomer of racemic formoterol, has recently become available as a nebulized formulation for the maintenance treatment of COPD (Citation15). Previous trials have shown that arformoterol significantly improved airway function at the end of the dosing interval (trough FEV1) and significantly improved post-dose FEV1 when compared with placebo and salmeterol (Citation13,Citation15,Citation16).
Only one previously published study investigated the long-term safety and efficacy of arformoterol using a dose (50 μg) that was higher than the approved and marketed dose (15 μg) (Citation13,Citation24). In this study, we examined the safety and efficacy of nebulized arformoterol 15 μg BID, 25 μg BID, and racemic formoterol over 6 months in subjects with COPD.
METHODS
This was a 6-month double-blind, double-dummy, multicenter (62 sites), randomized, active-controlled, parallel group study designed to examine the long-term safety and efficacy of twice daily arformoterol 15 μg and 25 μg and racemic formoterol 12 μg. This study was conducted according to the Declaration of Helsinki (Citation25) and approved by appropriate IRBs. A written informed consent was obtained from all subjects. The study (## NCT00250679) is posted in clinicaltrials.gov.
Study subjects
Subjects were 35 years of age or older, had a ≥ 15 pack-year smoking history, and had a physician-diagnosis of COPD (including emphysema and/or chronic bronchitis) with baseline breathlessness severity grade (Modified Medical Research Council [[MMRC (Citation26)]] Dyspnea Scale Score) of ≥ 2. Eligible subjects were also required to have pre-dose baseline FEV1 ≤ 65%% predicted, pre-dose FEV1 > 0.70L, and a pre-dose FEV1/FVC ratio of ≤ 70%%. Subjects were excluded if they had history of asthma, life-threatening or unstable respiratory illness, upper or lower respiratory tract infection within 30 days prior to screening, or malignancy. Subjects were also excluded if they had pre-existing cardiovascular conditions or other co-morbidities that may have precluded them from completing the study.
Study design
The primary endpoint for this study was the frequency of adverse events, including COPD exacerbations over 6 months. Secondary endpoints included change in pulmonary function (FEV1, inspiratory capacity) and the extent of use of rescue short-acting inhaled bronchodilators. Other secondary endpoints included dyspnea measured by Transition Dypsnea Index (TDI) (Citation27), exercise tolerance assessed by the 6-minute walk test (Citation28), and health status measured by the St George's Respiratory Questionnaire (SGRQ)(Citation29).
Baseline values were obtained prior to the first dose of study medication. Medical event calendars were completed daily. Approximately 7 to 14 days after screening, subjects were randomized in a 1:1:1 ratio to 26 weeks of one of three treatment regiments: (Citation1) nebulized arformoterol (Brovana, Sepracor, Inc., Marlborough, Massachusetts, USA) 15 μg BID plus placebo DPI BID (ARF 15), (Citation2) nebulized arformoterol 25 μg BID plus placebo DPI BID (ARF 25), or (Citation3) nebulized placebo plus racemic formoterol (Foradil® AerolizerTM; Schering, Kenilworth, New Jersey, USA) DPI BID (FORM). To facilitate an assessment of potential treatment differences by race, the randomization schedule was stratified by Caucasian, Black or Other. Subjects self-administered nebulized arformoterol or the nebulized placebo using the PARI LC PLUSTM nebulizer and TrekTM compressor (PARI Respiratory Inc., Richmond, Virginia, USA) followed by formoterol or placebo using the AerolizerTM Inhaler DPI, respectively. The nebulized medication/placebo was administered first followed by DPI administration. Subjects administered medication twice (approximately 12 hours apart) during the day at about 8 AM (± 1 hour) and 8 PM (± 1 hour).
During the study period, the use of long-acting bronchodilators other than the study medications was prohibited. Inhaled and oral corticosteroids (≤ 10 mg/day dose of prednisone or equivalent), short-acting xanthines and leukotriene inhibitors were allowed if subjects were on a stable regimen for ≥ 14 days prior to study entry. During the trial, up to two 14-day courses of oral corticosteroids separated by at least 6 weeks were permitted for treatment of COPD exacerbations. Short-acting β2-agonists (SABAs) [[racemic albuterol MDI (VENTOLIN®, GlaxoSmithKline, Research Triangle Park, North Carolina, USA) and levalbuterol (Xopenex®, Sepracor Inc., Marlborough, Massachusetts, USA)]] were supplied to be used as rescue medication, and ipratropium bromide MDI (ATROVENT®, Boehringer Ingelheim, Ridgefield, CT) as supplemental medication to be used as needed during the study. Study medication and rescue and supplemental medication were required to be withheld for ≥ 8 hours and ≥ 6 hours, respectively, prior to clinic visits.
To evaluate compliance, subjects completed daily diaries recording study medication use. In addition, investigators maintained a log of all study drug that was dispensed, used and returned, and compliance was calculated separately for the UDV and the DPI. Subjects were contacted by telephone on weeks 1, 2, 6, 9, 16, 19, and 22 to discuss study drug adherence, adverse events, concomitant medication and recording of information in the medical events calendar. Clinic visits were scheduled at randomization and at weeks 3, 13, and 26 (end of treatment). At each clinic visit COPD questionnaires and medical event calendars were collected and vital signs, heart rate, MMRC dyspnea scale, and spirometry were evaluated. Blood samples for laboratory evaluation were collected at screening, at randomization, and week 26.
Spirometry was performed in accordance to the current American Thoracic Society Guidelines (Citation30) prior to administering the study medication, immediately post-dose, and 30 minutes, 1, 2, 4, 6, 23, and 24 hours post-dose. All reported FEV1 values were the highest of three acceptable maneuvers. Inspiratory capacity was measured pre-dose and 2-hours post-dose at randomization and at weeks 3, 13, and 26.
Baseline Dyspnea Index (BDI) was assessed at randomization and TDI was evaluated before administering the study medication at weeks 13 and 26 (Citation27). The 6-minute walk test (Citation28) and SGRQ (Citation29) were evaluated at randomization and at weeks 13, and 26 prior to administering the study medication. The six minute walk test was also performed 3 to 4 hours post-dose.
Statistical analysis
Safety and efficacy summaries were based on the Intent-to-Treat (ITT) population, defined as randomized subjects who received at least one dose of study medication. In accordance with the ICH E9 Guidance for Industry document (Statistical Principles for Clinical Trials), safety and tolerability data from this study were analyzed descriptively (Citation31). Significance testing was not performed on any safety comparisons, except for the key safety endpoint of COPD exacerbation. The sample size was outside of statistical considerations and was not powered to detect specific efficacy differences between treatment groups.
Continuous efficacy outcomes were summarized using descriptive statistics. For categorical variables, statistical summaries included counts of subjects and percentages. Confidence intervals for means were based on the t-distribution. In general, missing observations were treated as missing at random, and no imputation was performed. Last observation carried-forward was used for the calculation of FEV1 AUC0–6 parameters to impute the 6-hour FEV1 value if missing.
Subgroup analyses were prospectively defined by race (Caucasian and Black) and baseline disease severity defined as baseline percent predicted FEV1 values: < 30%%, 30%% to < 50%%, and ≥50%%.
Treatment-emergent adverse events were summarized by MedDRA system organ class and preferred term (Citation32). In addition, certain prospectively defined composite adverse events were assessed including any adverse event categorized as follows: An event-based COPD exacerbation was defined as an increase in symptoms that necessitated any change in baseline medications other than bronchodilators or required subjects to have additional medical attention (Emergency Department, urgent care physician visit or hospitalization). Symptom-based COPD exacerbation included event-based COPD exacerbations in addition to reported adverse events associated with a worsening or new respiratory symptom. Cardiovascular adverse events were classified based on MedDRA terms into arrhythmic and ischemic events.
RESULTS
Of the 1014 subjects screened, 444 were randomized and 443 received at least one dose of medication (ITT population) (). Baseline characteristics and demographics were similar among the three treatment groups (). Baseline characteristics were also similar among treatment subgroups when stratified by disease severity (FEV1< 30%% predicted, 30%% to < 50%% predicted, and 50%% to 65%% predicted) and by race (Supplemental Tables and ). Of the subjects in the ITT population 311 (70%%) completed the 26 weeks study, with similar frequency of discontinuation among the three treatment groups (). The most common reasons for discontinuation were adverse events (9.7%%) and voluntary withdrawal (10.8%%). Both the nebulized and DPI active treatment groups reported good average compliance (95%%–100%%) with the protocol-specified treatments. Blacks discontinued from the study due to adverse events at a slightly higher frequency than Caucasians (Supplemental Table 4). Prior to screening, inhaled or oral corticosteroids were used by 35.4 to 40.1%% of subjects in the three treatment arms, and the long acting inhaled anticholinergic tiotropium by 11.6 to 20.1%% of subjects.
Figure 1. Consort diagram indicating the number of subjects enrolled (top), randomized to each treatment arm (middle) and either completed or withdrew from the study (bottom). Reasons for withdrawing from the study are indicated (bottom).
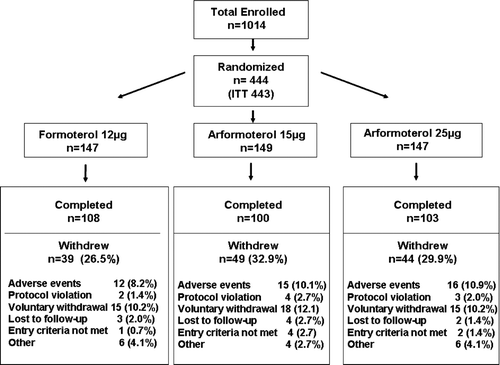
Table 1. Demographics and baseline characteristics
Safety outcomes
Adverse events
The overall frequency of adverse events was similar among the three treatment groups. The majority of subjects reported at least one adverse event () and about one-third reported a respiratory event (34.2%% for ARF 15, 34.0%% for ARF 25, and 27.2%% for FORM). Less than 10%% of the subjects in each treatment group reported a cardiovascular adverse event. The frequencies of serious adverse events were low among the three treatment groups (6.8 to 13.4%%; ). One subject treated with ARF 25 died of myocardial infarction, although this was not considered by the investigator as related to the study medication.
Table 2. Summary of adverse events
COPD exacerbations
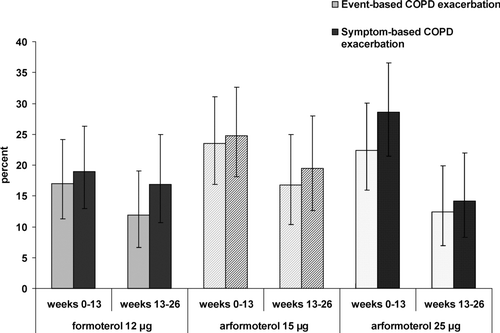
Over the 6-month treatment period event-based COPD exacerbations were observed to be less frequent in subjects treated with FORM (22.4%%) compared with either ARF 15 μg (32.2%%) or 25 μg (30.7%%) (). Using the more inclusive symptom-based definition of exacerbation, the proportion with exacerbations was also numerically less frequent in subjects treated with FORM (27.9%%) versus ARF 15 μg (35.6%%), and 25 μg (38.1%%). The frequency of both event-based and symptom-based COPD exacerbations appeared to be lower in the last 13 weeks of the study than in the first 13 weeks (Figure 2).
Figure 2. Percent of subjects with event-based and/or symptom-based COPD exacerbations over time. Bars represent the proportion of subjects treated with FORM (solid), ARF 15 (stripe) and ARF 25 (speckled) with event-based (grey) and/or symptom-based (black) COPD exacerbations. The 95% CI for the proportions are indicated.
β-adrenergic mediated adverse effects
The occurrence of β-adrenergic mediated treatment effects, including dizziness, tremor, nervousness, insomnia, and paresthesia was low (ARF 15; 10.7%%, ARF 25; 15.6%%, and FORM; 17.0%%). There was no meaningful difference in mean pre-dose glucose and potassium serum concentrations over the 26-week study period. Mean change [[SD]] in ventricular heart rate 2-hours post-dose from baseline and at week 26 was similar among the three treatment groups (ARF 15: −2.8 [[10.3]] bpm, ARF 25: −1.5 [[10.0]] bpm, and FORM : −1.6 [[9.8]] bpm). The frequency of adverse events, COPD exacerbations, and β-adrenergic mediated effects were similar in the different disease severity groups regardless of treatment (Supplemental ).
Rescue/supplemental medication use
During the pre-randomization period, 75.2%% of subjects used ipratropium bromide at least once as supplemental medication with a mean use of 2.4 to 2.7 actuations per day. The use of ipratropium bromide declined over the treatment period for all treatment groups by a mean of −0.83 to −1.09 actuations per day. Mean reductions were observed by week 3 and were maintained during the remainder of the treatment period. During the pre-randomization period, 79.5%% of subjects used a SABA at least once and baseline SABA use averaged 2.9 actuations per day. The use of rescue SABA medication declined from baseline over the treatment period for all treatment groups by a mean of −1.12 to −1.32 actuations per day.
Pulmonary function outcomes
Mean change in trough FEV1 at the 24-hour time point following the morning dose improved similarly for all three treatment groups from baseline (). This improvement was seen following the first dose of the study medication (mean of 0.16 L to 0.17 L), with stable improvement (mean of 0.09 L to 0.114 L) maintained over weeks 3 to 26.
Table 3. Change in pulmonary function outcomes from study baseline
Mean change in time-normalized FEV1AUC0–6 post-dose improved from baseline for all treatment groups at each study visit (). These increases in FEV1AUC0–6 were sustained throughout the 26-week treatment period (). Following the first dose, mean peak improvement in FEV1 over 6 hours post-dose ranged between 0.29 L and 0.36 L for the three treatment groups (). The extent of this improvement from study baseline FEV1 level was maintained for all treatment groups over 26-weeks (). Mean trough FEV1, FEV1 AUC0–6, and peak FEV1, improved from baseline for all disease severity groups independent of treatment with the single exception of trough FEV1 in the 50 to 65%% predicted group (). Improvement from baseline continued to be evident after 26 weeks for all treatment groups, although the magnitude of improvement declined to some extent at 26 weeks for subjects with less severe baseline FEV1 compromise (i.e., the 30 to <50%% predicted and 50 to 65%% predicted groups) except for those with severe (30 to < 50%%) COPD treated with ARF 25 μg (A-C). This was especially true for mean trough FEV1 values.
Figure 3. Mean change in time normalized FEV1AUC0–6 from baseline. Bars represent the change in FEV1 AUC0–6 for FORM (white), ARF 15 (grey) and ARF 25 (black). Standard errors are indicated. **denotes that the mean difference between an arformoterol treatment group and FORM were significant (95%% CI for the difference between treatment groups mean did not include zero). ## indicates the mean difference between ARF 25 and ARF 15 was significant (95%% CI for the difference between treatment groups means did not include zero).
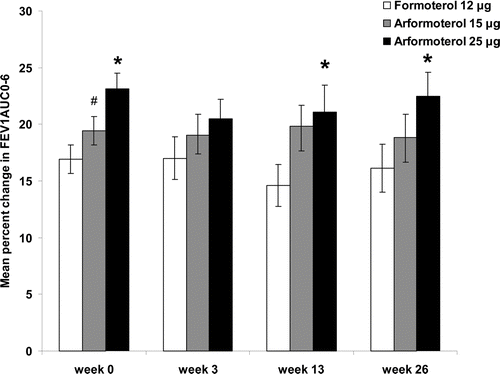
Figure 4. Pulmonary function outcomes stratified by baseline FEV1%% predicted (< 30%%, 30%% to < 50%%, and 50%% to 65%%). (A) Mean change in FEV1AUC0–6 (B) 6-hour peak change in FEV1, (C) 24-hour trough FEV1, and (D) pre-dose (trough) inspiratory capacity for patients with < 30%% (left), 30%% to < 50%% (middle), and 50%% to 65%% (right) percent predicted FEV1 at baseline for FORM (white), ARF 15 (grey), and ARF 25 (black). Standard error bars are indicated. **denotes that the mean difference between an arformoterol group and FORM was significant (95%% CI for the difference between treatment group means did not include zero). ## indicates the mean difference between ARF 25 and ARF 15 was significant (95%% CI for the difference between treatment groups means did not include zero).
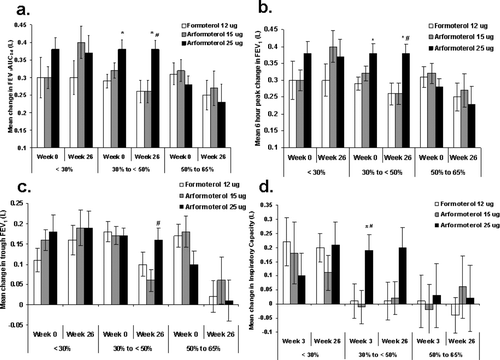
Mean change in pre-dose (trough) inspiratory capacity from baseline values over the 26-week treatment period consistently improved only for ARF 25 μg treatment group (). Mean change in inspiratory capacity 2 hours post-dosing improved similarly for all treatment groups following the first dose (). The extent of this improvement was sustained throughout the study. Trough inspiratory capacity improved from baseline only for subjects with very severe COPD and for those with severe (30 to < 50%% predicted) COPD treated with ARF 25 μg ().
Forced expiratory (FEV1) values improved to a similar extent in both Caucasian and Black COPD subjects, (Supplemental Table 5) although Blacks had less improvement in inspiratory capacity than Caucasians.
SYMPTOMS AND FUNCTIONAL OUTCOMES
Dyspnea, assessed by TDI focal scores, improved for all three treatment groups at weeks 13 and 26 (). At both weeks 13 and 26 approximately 50%% of the subjects in each treatment group had at least 1 unit improvement (the minimal clinically important difference [[MCID]]) in the TDI focal score.
Table 4. Change in dyspnea, 6 minute walk, and health status from baseline
All treatment groups had small improvements from baseline in median 6-minute walk distance (). These improvements were modest and unlikely to be clinically meaningful, and they did not change substantially between weeks 13 and 26. The median post-dose 6-minute walk distance was also only slightly greater than pre-dose median distance for all treatment groups, and again unlikely to be clinically meaningful. Mean SGRQ total scores improved from baseline for all treatment groups at weeks 13 and 26 (). FORM group had the greatest numerical mean improvement at both week 13 (−4.2 units versus −3.4 and −1.9 for ARF 15 and ARF 25 respectively) and at week 26 (−6.8 versus −3.7 for both ARF 15 and ARF 25). The proportion of subjects who had ≥ 4 unit (the MCID) improvement in SGRQ at week 26 was, 45.7%% for ARF15, 43.3%% for ARF25, and 56.7%% for FORM ().
DISCUSSION
This is the first study to compare the safety and efficacy of extended (up to 6 months) use of nebulized arformoterol compared with racemic formoterol. Overall, both doses of arformoterol, 15 μg and 25 μg, and racemic formoterol were well tolerated with pulmonary function improvement being maintained over the 6-month treatment period. The overall frequency of adverse events and serious adverse events were similar among treatment groups. The proportion of subjects with respiratory or cardiac adverse events was similar among the three treatment groups. Over the 6 month treatment period, COPD exacerbations resulting in a change in therapy or urgent visit (event-based definition) were observed less frequently in the FORM treatment group (33 of 147 subjects; 22.4%%) than in either the ARF 15 (48 of 149; 32.2%%) or in ARF 25 (45 of 147; 30.7%%) treated subjects. Whether this difference is real and related to pharmacologic properties inherent in the different therapies, due to unmeasured confounding factors not controlled at randomization, or chance is not clear. Nonetheless, the absence of differences for other safety outcomes for these treatment arms, and the lack of dose-response (higher exacerbation rate for ARF 15 versus ARF 25 μg) does not likely suggest a clear safety/tolerability difference for the 3 treatments.
Furthermore, the proportion of subjects with β2-adrenergic adverse effects such as tremor, insomnia, dizziness etc. was low and similar among the three treatment groups. During this 6-month study, the frequency of serious adverse events in all three treatment groups was low and was similar to prior reports (Citation15,16,Citation13,Citation21). Pulmonary function improvement following treatment was maintained over the 6-month treatment period in the 3 treatment groups.
The use of LABAs for maintenance treatment in COPD is currently recommended by all evidence based guidelines (Citation2,3). However, recent studies in asthma have raised questions regarding the long-term safety of these medications in asthma (Citation12,Citation33). Less is known regarding the safety of regular LABAs use in COPD. Data from the recently published TORCH trial, revealed that the three year use of the LABA, salmeterol, either as monotherapy or in combination with fluticasone, was not associated with an increased risk of death or exacerbation frequency (Citation17). The current study supports the safety of formoterol and arformoterol when used for maintenance therapy in COPD.
Some prior studies investigating the regular LABA use (13–17,Citation34) have demonstrated a small loss in the improvement in pre-dose (trough) FEV1 from baseline after a few weeks following the initiation of medications, while other studies failed to show such changes (Citation35,36). Clinically meaningful tolerance, measured by loss of bronchodilator response and disease control, was not observed in this study in any of the treatment groups. However, the extent of improvement observed in trough FEV1 on the first day diminished by week 3, but remained improved from baseline and did not decline further following weeks 3 in all three treatment groups. Of interest, there was little or no reduction in the magnitude of mean peak improvement in FEV1 or FEV1 AUC 0–6 from the first day of dosing to week 26. The fact that a decrease in trough FEV1 but not other pulmonary function measures is often detected with regular LABA therapy (Citation13–17, Citation34) suggests that this outcome may be a particularly sensitive indicator of tolerance.
The frequency of COPD exacerbations, a key measure of disease stability, did not increase during the course of the study; in fact, rate estimates decreased between the first and second half of the treatment period for all treatment groups (Figure 2). This phenomenon was also observed in the prior 12 week (Citation15,16) and 52 week (Citation13) arformoterol treatment trials. Higher exacerbation rates early in this and previous trials could have been contributed to by subjects with more severe disease discontinuing participation in trial after experiencing an exacerbation. This would result in the profile of subjects who completed the trial being less exacerbation-prone, with corresponding decrease in the observed rate late in the trial. Similarly, a possible beneficial impact of treatments on exacerbation risk that accrued over a long duration of treatment could also have contributed to this finding. Moreover, the sustained decrease in the use of rescue medication and supplemental medication for all three treatment groups further indicates that there was no worsening in the course of the disease.
Dyspnea (BDI/TDI) improved by more than 1 unit for approximately 50%% of subjects, a finding consistent with results seen in prior arformoterol (Citation11,Citation13) and formoterol studies (13–15,37,38). Health status, a measure that may be a better predictor of patient response to treatment, hospitalization (Citation39) and risk of death than FEV1 (Citation40) improved in all treatment arms, as was the case in prior studies (14,15,19,34). The use of FORM was associated with a mean change in the total SGRQ score of −6.8 which exceeds the metric (Citation4 unit improvement) that has been proposed as the minimum clinical important difference (MCID) for this outcome (Citation41). Improvement for both arformoterol doses also approached this cutoff (3.7 units). For all treatments the SGRQ symptom domain score had the greatest improvement after 6-months of treatment (mean change from baseline: FORM μg: −8.4, ARF 15: −6.1, and ARF 25 −7.0) compared with the other SGRQ sub-scores as has been noted in previous formoterol (Citation14,Citation42) and arformoterol (Citation15) reports.
Multiple genetic, behavioral, socioeconomic, and underlying disease factors, may impact how a patient with COPD responds to therapy. In general, subjects with very severe COPD at the start of the study had a greater absolute and percent improvement in airway function (particularly notable for trough FEV1 and inspiratory capacity) measures following 6-months of therapy than COPD subjects with less severe disease. Subjects with severe COPD treated with ARF 25 also had the greatest sustained improvements in pulmonary function, suggesting a possible dose-response effect. This differential and greater improvement for subjects with most severe disease is supportive of findings from a prior study that investigated the use of arformoterol and salmeterol in COPD (Citation16).
Several limitations must be taken into account when considering our study results. This study did not include a placebo arm. Hence, safety and efficacy outcomes of regular LABA therapy relative to as-needed bronchodilator therapy were not evaluated. However, two large prior 12-week efficacy and safety trials (Citation13, 14) were conducted using the two BID arformoterol dose regimens included in this trial and an additional 50 μg QD regimen, along with a placebo treatment group. The safety profile in those trials for subjects in each of the two BID arformoterol treatment groups did not differ from those in the placebo group. In contrast, the primary purpose of this trial was to make a comparative assessment of longer term safety and efficacy among the LABA treatments. As long-acting bronchodilator treatments are widely endorsed by independent guidelines (Citation5) as effective for use in COPD patients whose disease severity is consistent with the entry criteria for this trial, no placebo group was included.
Nonetheless, safety signals among the 3 treatment groups in this trial did not occur with a greater frequency over 6 months than reported in prior clinical trials (Citation13,Citation15,16,Citation22,Citation21). A longer study enriched for exacerbation-prone subjects would have increased the ability of the study to quantify possible exacerbation-risk differences among the treatments. As stated above, a substantial number of subjects (about 30%% for each treatment group) discontinued prior to completion of the 6-month trial. Subjects who withdrew early from the trial may have differed from those who completed in such a way as to impact outcomes measured at weeks 13 and 26, such as early versus late exacerbation rates.
In summary, this randomized trial supports the long-term safety and efficacy of the arformoterol and racemic formoterol in COPD patients (Citation13,Citation19,Citation21,Citation37,38,Citation40). These results add to a growing body of evidence that support the importance of long-acting bronchodilators in controlling symptoms and optimizing functional status of patients with COPD.
Declaration of interest
Dr. Hanania has received research grant support from Sepracor, GlaxoSmithKline, Boehringer Ingelheim, Novartis, and Dey. Dr. Hanania has also served as a consultant to Sepracor, GlaxoSmithKline, Novartis, & Dey. Dr. Donohue has received research grants from Sepracor and Novartis and served on advisory boards and as a consultant to both. Arformoterol is manufactured by Sepracor and formoterol by Novartis. Dr. Nelson has consultant arrangements with Genentech/Novartis, Abbott Laboratories, MediciNova, AstraZeneca, Amgen, GlaxoSmith-Kline, Schering-Plough, Dyson, Sepracor and Nycomed. He has also received grant/research support from Shering-Plough, Novartis, Genentech, Ception, and AstraZeneca. Lastly, he is a member of the Speakers' Bureau for GlaxoSmithKline. Mr. Sciarappa is a full-time employee of Sepracor Inc. which markets arformoterol (brand name: Brovana). Dr. Goodwin is a full-time employee of Sepracor Inc. which markets arformoterol (brand name: Brovana). Dr. Baumgartner is a previous employee of Sepracor Inc. Dr. Hanrahan is a former employee of Sepracor Inc. Dr. Hanrahan was the Senior Medical Director responsible for the design and conduct of this trial. Drs N.A. Hanania, J.F. Donohue and H. Nelson participated as investigators in this study. K. Sciarappa PhD and E. Goodwin PhD are Sepracor employees. Dr. J. P. Hanrahan and Dr. R.A. Baumgartner are former Sepracor employees. Support for this study provided by Sepracor Inc. Data were compiled and statistical analyses were performed by Sepracor Inc. and were available to all authors. All authors had access to the data and were involved in the manuscript preparation.
REFERENCES
- Agusti A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don't know (but should). Proc Am Thorac Soc 2007; 4:522–525.
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van WC, Zielinski J. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary. Am J Respir Crit Care Med 2007; 176:532–555.
- Celli BR and MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004; 23:932–946.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001; 163:1256–1276.
- Foster JA, Yawn BP, Maziar A, Jenkins T, Rennard SI, Casebeer L. Enhancing COPD management in primary care settings. Med Gen Med 2007; 9:24.
- Miravitlles M, Murio C, Tirado-Conde G, Levy G, Muellerova H, Soriano JB, Ramirez-Venegas A, Ko FW, Canelos-Estrella B, Giugno E, Bergna M, Cherrez I, Anzueto A. Geographic differences in clinical characteristics and management of COPD: the EPOCA study. Int J Chron Obstruct Pulmon Dis 2008; 3:803–814.
- Bourbeau J, Sebaldt RJ, Day A, Bouchard J, Kaplan A, Hernandez P, Rouleau M, Petrie A, Foster G, Thabane L, Haddon J, Scalera A. Practice patterns in the management of chronic obstructive pulmonary disease in primary practice: the CAGE study. Can Respir J 2008;15:13–19.
- Barr RG, Celli BR, Martinez FJ, Ries AL, Rennard SI, Reilly JJ, Jr., Sciurba FC, Thomashow BM, Wise RA. Physician and patient perceptions in COPD: the COPD Resource Network Needs Assessment Survey. Am J Med 2005; 118:1415.e9–1415.e17.
- Tsagaraki V, Markantonis SL, Amfilochiou A. Pharmacotherapeutic management of COPD patients in Greece–adherence to international guidelines. J Clin Pharm Ther 2006; 31:369–374.
- Sivori ML and Raimondi GA. [[Survey of chest physicians regarding COPD diagnosis and treatment]]. Medicina (B Aires) 2004; 64:113–119.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Donohue JF, Hanania NA, Sciarappa K, Goodwin E, Grogan DR, Baumgartner RA, Hanrahan JP. Arformoterol and salmeterol in the treatment of chronic obstructive pulmonary disease: A one year evaluation of safety. Thera Adv Respir Dis 2008; 2:37–48.
- Gross NJ, Nelson HS, Lapidus RJ, Dunn L, Lynn L, Rinehart M, is-Mize K. Efficacy and safety of formoterol fumarate delivered by nebulization to COPD patients. Respir Med 2008; 102:189–197.
- Baumgartner RA, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Hanrahan JP. Nebulized arformoterol in patients with COPD: a 12-week, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled trial. Clin Ther 2007; 29:261–278.
- Hanrahan J, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Donohue JF, Menjoge S, Kesten S. Tolerance to bronchodilating effects of salmeterol in COPD. Respir Med 2003; 97:1014–1020.
- Donohue JF, van Noord JA, Bateman ED, Langley SJ, Lee A, Witek TJ, Jr., Kesten S, Towse L. A 6-month, placebo-controlled study comparing lung function and health status changes in COPD patients treated with tiotropium or salmeterol. Chest 2002; 122:47–55.
- Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Yates JC, Vestbo J. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Donohue JF, Hanania NA, Fogarty C, Campbell SC, Rinehart M, Denis-Mize K. Long-term safety of nebulized formoterol: Results of a twelve-month open-label clinical trial. Therapeutic Advances in Respiratory Disease 2008; 2:199–208.
- Szafranski W, Cukier A, Ramirez A, Menga G, Sansores R, Nahabedian S, Peterson S, Olsson H. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J 2003; 21:74–81.
- Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J 2003; 22:912–919.
- Dolovich MB, Ahrens RC, Hess DR, Anderson P, Dhand R, Rau JL, Smaldone GC, Guyatt G. Device selection and outcomes of aerosol therapy: Evidence-based guidelines: American College of Chest Physicians/American College of Asthma, Allergy, and Immunology. Chest 2005; 127:335–371.
- BrovanaTM (arformoterol tartrate) Inhalation Solution [[package insert]]. 2007.
- World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA 1997; 277:925–926.
- Fletcher C. Standardised questionnaire on respiratory symptoms: a statement prepared and approved by the MRC Committee on the Aetiology of Chronic Bronchitis (MRC breathlessness score). BMJ 1960; 2:1665.
- Mahler DA, Weinberg DH, Wells CK, Feinstein AR. The measurement of dyspnea. Contents, interobserver agreement, and physiologic correlates of two new clinical indexes. Chest 1984; 85:751–758.
- Sciurba F, Baumgartner RA, Sciarappa K, Benzo RP, Hanrahan JP. Degree of exercise limitation in COPD: impact on 6-minute walk in response to long-acting beta-agonist (LABA) treatment (Abstract). Eur Respir J 2007; 30:76S.
- Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation. The St. George's Respiratory Questionnaire. Am Rev Respir Dis 1992; 145:1321–1327.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, Van Der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J. Standardisation of spirometry. Eur Respir J 2005; 26:319–338.
- FDA and Center for Drug Evaluation and Research. Guidance for industry: E9 statistical priciples for clinical trials. 1998;1–43.
- MedDRA and MSSO. The medical dictionary for regulatory activities. http://www meddramsso com/MSSOWeb/index htm; 2008.
- Salpeter SR, Buckley NS, Ormiston TM, Salpeter EE. Meta-analysis: effect of long-acting beta-agonists on severe asthma exacerbations and asthma-related deaths. Ann Intern Med 2006; 144:904–912.
- Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, Anderson J, Maden C. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003; 361:449–456.
- Hanania NA, Kalberg C, Yates J, Emmett A, Horstman D, Knobil K. The bronchodilator response to salmeterol is maintained with regular, long-term use in patients with COPD. Pulm Pharmacol Ther 2005; 18:19–22.
- Stockley RA, Chopra N, Rice L. Addition of salmeterol to existing treatment in patients with COPD: a 12 month study. Thorax 2006; 61:122–128.
- Campbell M, Eliraz A, Johansson G, Tornling G, Nihlen U, Bengtsson T, Rabe KF. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Rossi A, Kristufek P, Levine BE, Thomson MH, Till D, Kottakis J, Della CG. Comparison of the efficacy, tolerability, and safety of formoterol dry powder and oral, slow-release theophylline in the treatment of COPD. Chest 2002; 121:1058–1069.
- Osman IM, Godden DJ, Friend JA, Legge JS, Douglas JG. Quality of life and hospital re-admission in patients with chronic obstructive pulmonary disease. Thorax 1997; 52:67–71.
- Halpin DM, Peterson S, Larsson TP, Calverley PM. Identifying COPD patients at increased risk of mortality: Predictive value of clinical study baseline data. Respir Med 2008; Nov; 102(11):1615–1624.
- Schunemann HJ, Griffith L, Jaeschke R, Goldstein R, Stubbing D, Guyatt GH. Evaluation of the minimal important difference for the feeling thermometer and the St. George's Respiratory Questionnaire in patients with chronic airflow obstruction. J Clin Epidemiol 2003; 56:1170–1176.
- D’Urzo AD, De Salvo MC, Ramirez-Rivera A, Almeida J, Sichletidis L, Rapatz G, Kottakis J. In patients with COPD, treatment with a combination of formoterol and ipratropium is more effective than a combination of salbutamol and ipratropium: A 3-week, randomized, double-blind, within-patient, multicenter study. Chest 2001; 119:1347–1356.