Abstract
Rationale: Leukotrienes have been implicated in the pathogenesis of acute exacerbations of COPD, but leukotriene modifiers have not been studied as a possible therapy for exacerbations. Objective: We sought to test the safety and efficacy of adding oral zileuton (a 5-lipoxygenase inhibitor) to usual treatment for acute exacerbations of COPD requiring hospitalization. Methods: Randomized double-blind, placebo-controlled, parallel group study of zileuton 600 mg orally, 4 times daily versus placebo for 14 days starting within 12 hours of hospital admission for COPD exacerbation. Primary outcome measure was hospital length of stay; secondary outcomes included treatment failure and biomarkers of leukotriene production. Main Findings: Sixty subjects were randomized to zileuton and 59 to placebo (the study was stopped short of enrollment goals because of slow recruitment). There was no difference in hospital length of stay (3.75 ± 2.19 vs. 3.86 ± 3.06 days for zileuton vs. placebo, p = 0.39) or treatment failure (23% vs. 27% for zileuton vs. placebo, p = 0.63) despite a decline in urinary LTE4 levels in the zileuton-treated group as compared to placebo at 24 hours (change in natural log-transformed ng/mg creatinine −1.38 ± 1.19 vs. 0.14 ± 1.51, p < 0.0001) and 72 hours (−1.32 ± 2.08 vs. 0.26 ± 1.93, p<0.006). Adverse events were similar in both groups. Principal Conclusions: While oral zileuton during COPD exacerbations that require hospital admission is safe and reduces urinary LTE4 levels, we found no evidence suggesting that this intervention shortened hospital stay, with the limitation that our sample size may have been insufficient to detect a modest but potentially meaningful clinical improvement.
INTRODUCTION
In 2000, COPD exacerbations were responsible for 726,000 hospitalizations and 119,000 deaths in the United States (Citation1). Patients admitted to U.S. hospitals for COPD in 2002 had a mean length of stay (LOS) of 5.1 days and accounted for $15,400 in charges per patient (Citation2). Usual management of acute exacerbations of COPD (AECOPD) consists of bronchodilators, corticosteroids, and antibiotics and, with the exception of the use of noninvasive ventilation, has not changed in the last 30 years.
Leukotrienes are inflammatory mediators that are derived from arachidonic acid, initially through 5-lipoxygenase enzymatic activity. Subsequent enzymatic steps generate either LTB4, which mediates neutrophil and T-cell chemotaxis, or the cysteinyl leukotrienes (LTC4, LTD4, LTE4), which cause smooth muscle contraction, tissue edema, inflammatory cell chemotaxis, and mucus production (Citation3). Our rationale for a clinical trial of leukotriene blockade in AECOPD included: 1) prior clinical studies which implicate both of these classes of leukotrienes in the pathophysiology of AECOPD (Citation4–8), and 2) cross-sectional studies, which have found increased serum and exhaled breath condensate LTB4 levels in patients with COPD despite corticosteroid use (Citation9, 10) (suggesting that any benefit of LTB4 blockade might be additive to those of corticosteroids).
Despite these data, clinical trials of anti-leukotriene therapy in COPD have focused exclusively on stable disease (Citation11, 12). To date, there are no published randomized trials of anti-leukotriene therapy in AECOPD. However, a prior randomized controlled trial demonstrated rapid improvements in FEV1 and a reduction in treatment failure with the use of leukotriene receptor blockade for the treatment of asthma exacerbations in the emergency department (Citation13).
Based on studies of mediators of inflammation in COPD, clinical trials of anti-leukotriene therapy in stable COPD, and a clinical trial of anti-leukotriene therapy in exacerbations of asthma, we hypothesized that anti-leukotriene therapy would provide a novel therapeutic approach to AECOPD requiring hospitalization. Our primary goal was to reduce hospital LOS. As prior data implicates both cysteinyl leukotrienes and LTB4 in AECOPD, we studied pharmacologic inhibition of 5-lipoxygenase with zileuton, because this agent should block production of both of these classes of leukotrienes. To determine whether our intervention had the intended biological effect, we measured urinary LTE4 and serum LTB4 levels in our subjects.
MATERIALS AND METHODS
Study design
We performed a double-blind, placebo-controlled, parallel group study with subjects randomized to receive either zileuton tablets 600 mg or placebo tablets orally, 4 times per day for 14 days (, ClinicalTrials.gov identifier NCT00493974). Subjects were enrolled at 12 U.S. hospitals. After obtaining informed consent, the enrollment visit included spirometry (if the patient's condition allowed) within 2 h following an inhaled β-agonist bronchodilator. Inpatient follow-up took place at 24 and 72 h, every 48 h thereafter during the remainder of the hospitalization and on the day of discharge. At admission, we recommended a standardized approach to therapy, which included albuterol and ipratropium bromide, a 14-day course of systemic corticosteroids beginning with at least 40 mg of prednisone per day (or its equivalent) and a course of an antibiotic selected on the basis of local resistance patterns to S. pneumonia, H. influenza, and M. catarrhalis.
Figure 1 Study schematic. If patients remained hospitalized, they were visited as inpatients at the 7-, 14- and 30-day visits.
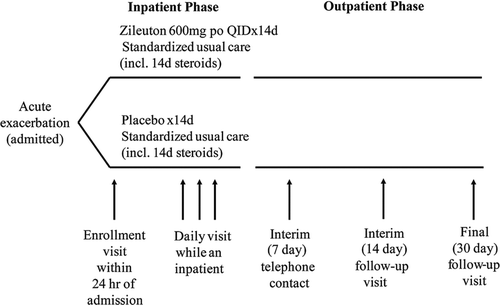
After discharge, subjects were followed by telephone contact at day 7, and by outpatient clinic visits at day 14 and day 30. Urinary LTE4 levels were measured at enrollment, 24 and 72 h, and serum LTB4 levels were measured at enrollment and at 30 days. The study was approved by local Institutional Review Boards and all patients provided written informed consent. The principal investigator (PGW) had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis, including any adverse effects. The study was approved by the Institutional Review Board (IRB) at each of the participating sites Additional detail on IRB approvals and study contacts is provided in the supplementary methods section.
Study population
Male and female patients, age >45 years, were eligible if they had an admitting diagnosis of AECOPD (defined as an acute increase in dyspnea, sputum volume, and/or sputum purulence without other attributable cause). Other inclusion criteria were ≥10 pack-years smoking history and an FEV1 < 60% predicted at time of inclusion or an inability to perform spirometry due to dyspnea. We required informed consent be completed within 12 h of the decision to admit the subject and study medication was administered as soon as possible after consent (i.e., after establishing eligibility and using the existing inpatient pharmacy procedures). Exclusion criteria, randomization procedures and criteria for discontinuation of study medication are provided in the supplementary methods section.
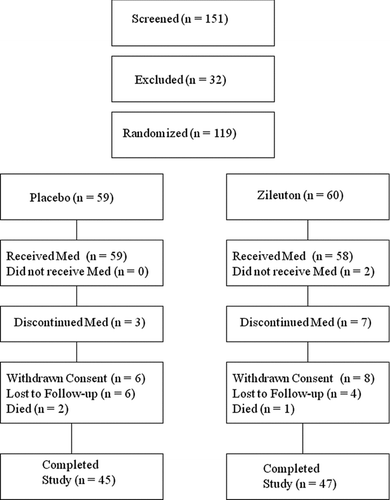
Figure 2 Consort flow chart.
Pre-specified endpoints
The primary outcome was hospital LOS which was recorded in days after review of the discharge summary. Secondary outcomes included: 1) treatment failure at or before 30 days (defined as death, intubation, readmission to a hospital for COPD, urgent visit to an outpatient or emergency department provider for symptoms of COPD or intensification of therapy including second course of antibiotics for COPD, and/or a second course of systemic steroids for COPD); 2) change in FEV1, FEV1/FEV6 and breathlessness during hospitalization; and 3) change in urinary LTE4 (from baseline to 24 and 72 h) or serum LTB4 levels (from baseline to 30 days).
Biomarker analyses
Urinary LTE4 levels were measured using UPLC/ESI-MS/MS at Vanderbilt University (Milne Laboratory). Serum LTB4 levels were measured using a commercially available forward sequential competitive binding assay (LTB4 Parameter Assay Kit, R&D Systems, Minneapolis, MN) performed in duplicate per manufacturer's instructions. Additional details are provided in the supplementary methods section.
Table 1 Subject characteristics by intervention group
Statistical methods
The pre-specified approach to analyzing LOS was to compare the two groups with or without truncating at 10 days to temper the influence of long hospital stays as these are frequently due to non-medical causes or to problems that are unrelated to COPD. Groups were compared using the Wilcoxon rank sum test. The study was designed to enroll a total of 520 subjects (one-half randomized to each arm) to achieve 90% power to detect a mean 0.75-day difference in hospital LOS between the 2 groups. Pre-specified secondary analyses applied Cox regression for analysis of LOS while controlling for potential confounders. Treatment failure in the two groups was compared using the chi-square test. Differences in improvement in spirometric outcomes, urinary LTE4 levels and serum LTB4 levels were compared using the Student's t-test (biomarker levels were natural log-transformed for normality). Data are expressed ± 1SD, unless otherwise stated. A p-value <0.05 was taken as statistically significant.
Table 2 Inpatient therapy by visit day
RESULTS
Subjects
The study was terminated early on the recommendation of the Data and Safety Monitoring Board due to inability to meet anticipated recruitment goals. This was related to a lower than expected rate of emergency department visits and hospitalizations than suggested by the literature and by epidemiologic data collected from the participating centers. A total of 151 subjects were screened for this study and 32 were excluded because they did not meet inclusion criteria (n = 24) or did not consent (n = 8, ). The remaining 119 subjects were randomized; 59 to placebo and 60 to zileuton. Subjects did not differ with respect to baseline characteristics, although spirometric evaluation was performed in only approximately a quarter of subjects because of patient dyspnea ().
Figure 3 There was no statistically significant difference in hospital length of stay (the primary outcome) between groups (hazard ratio 0.96; 95% confidence interval [95% CI], 0.64–1.42).
![Figure 3 There was no statistically significant difference in hospital length of stay (the primary outcome) between groups (hazard ratio 0.96; 95% confidence interval [95% CI], 0.64–1.42).](/cms/asset/ccdfa417-aa6d-4d17-b464-7f0bede8f1ac/icop_a_540273_f0003_b.jpg)
Overall, subjects were consented a mean of 5.7±4.5 h after admission. No differences were observed in other treatments during the hospitalization (). A similar number of subjects in both groups completed the final study visit (45/59, 76% randomized to placebo [6 withdrew, 6 were lost to follow-up and 2 died] versus 47/60, 78% randomized to zileuton [8 withdrew, 4 were lost to follow-up and 1 died], ). Study drug was administered a mean of 14.1±6.9 h after admission in the zileuton group and 15.4±7.8 h after admission in the placebo group (p = 0.38).
Hospital length of stay
When analyzed without truncation, the average LOS was 3.92 ± 2.80 days for zileuton vs. 4.76 ± 5.84 days for placebo, p = 0.39. When truncated at 10 days, the mean LOS was 3.75 ± 2.19 days for zileuton vs. 3.86 ± 3.06 days for placebo, p = 0.39. Although the proportion of subjects who remained hospitalized was numerically greater with zileuton than placebo until day 5 and then greater with placebo thereafter (), Cox proportional hazards analysis controlling for age, gender and pack-years showed no effect of the intervention on LOS (Hazard ratio 0.96; 95% confidence interval [95% CI], 0.64–1.42). Post-hoc power calculations indicated that our sample size provided approximately 80% power to detect a difference between the groups of 1.2 days LOS (e.g., 4.2 in placebo, 3.0 in zileuton). If these results were to be used as pilot data to develop sample size estimates for another prospective clinical trial (alpha = 0.05 two-sided, power = 80%), the total sample size required would range from 946 subjects (using untruncated LOS) to 17,544 total subjects (using truncated LOS data).
Table 3 Treatment failure by intervention group
Secondary outcomes
We found no difference in treatment failure at 30 days between intervention and placebo with 14/60 (23%) experiencing any treatment failure in the zileuton group vs. 16/59 (27%) in the placebo group, p = 0.63 (). We found no between group differences in change in spirometric measures although baseline data were sparse due to dyspnea (change in post-BD FEV1 from baseline to 14 days of 390 ± 140 ml with zileuton [n = 13] vs. 220 ± 100 ml with placebo [n = 12], p = 0.33). Available spirometric data at the 30-day visit did not differ between groups ().
Table 4 Spirometric data at the 30-day visit
Biomarker measurements
Urinary LTE4 levels were measured at enrollment, 24 h and 72 h. The within-person change in urinary LTE4 levels in subjects treated with zileuton demonstrated a statistically significantly decline as compared to the change observed in placebo treated subjects at 24 hours (change in natural log-transformed ng/mg creatinine -1.38 ± 1.19 [n = 49 zileuton] vs. 0.14 ± 1.51 [n = 46 placebo], p<0.0001) and at 72 hours (−1.32 ± 2.08 [n = 27 zileuton] vs. 0.26 ± 1.93 [n = 26 placebo], p<0.006, respectively) (, top).
Figure 4 Urinary LTE4 and serum LTB4 levels. (A) Urinary LTE4 levels declined with zileuton as compared to placebo at 24 h (p<0.001) and 72 h (p = 0.006). (B) There was no statistically significant difference in the change in serum LTB4 levels at 30 days between treatment groups (p = 0.19), despite a non-significant decline in geometric mean LTB4 level of 40% with zileuton.
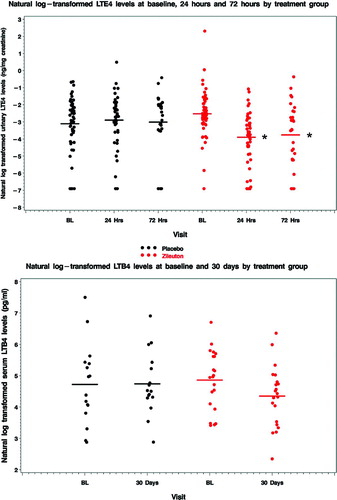
These changes reflected a 75% reduction in geometric mean LTE4 levels at 24 hours (0.08 to 0.02 ng/mg creatinine) and a 71% reduction at 72 h (0.08 to 0.023 ng/mg creatinine) with zileuton. By comparison, geometric mean LTE4 levels were relatively constant in the placebo group (0.044, 0.054 and 0.049 ng/mg creatinine at enrollment, 24 hours and 72 hours, respectively). Serum LTB4 levels were measured at enrollment and at 30 days. There was no statistically significant difference in the change in LTB4 level at 30 days between treatment groups (−0.51 ± 1.15 [n = 21 zileuton] vs 0.02±1.24 pg/ml [n = 16 placebo], p = 0.19) (, bottom), despite a non-significant decline in geometric mean LTB4 level of 40% with zileuton (from 128 to 77 pg/ml) versus a 3% increase with placebo (from 112 to 115 pg/ml).
Adverse events
We found no significant difference in total adverse events or in serious adverse events between groups (). There were 3 deaths in the placebo group and 1 in the zileuton group. We found no significant differences in AST or ALT at baseline, or at day 14 between treatment groups, nor any differences in change in white blood cell count or the development of clinical pneumonia during the study (3 in the zileuton group vs. 2 with placebo).
Table 5 Serious adverse events
DISCUSSION
The important findings of this study were that, despite biomarker analyses indicating that zileuton decreased urinary LTE4 levels at 24 and 72 hours, we found no clinically or statistically significant effect of zileuton on hospital LOS or on any of our pre-specified secondary clinical outcomes. Although our enrollment fell short of projected values, a post-hoc power calculation indicated that our sample size conferred 80% power to detect a difference in LOS of 1.2 days, an effect comparable to that observed with the use of systemic steroids (Citation14). Because zileuton was typically added to concomitant steroid therapy in our trial, it is unlikely that zileuton would result in such a substantial benefit.
Thus, although zileuton had the intended biological effect on cysteinyl leukotriene production, our sample size was insufficient to detect a modest but potentially meaningful clinical improvement. On the other hand, should these data be used to design another prospective trial, the required sample size would be between ∼1,000 and 17,000 subjects (using untruncated and truncated LOS data, respectively).
Several laboratory and clinical studies support a key role for leukotrienes in the pathogenesis of inflammation in stable COPD. Expression of two LTB4 receptors, BLT1 and PPARα, is increased in alveolar macrophages and airway wall leukocytes in surgical lung specimens from patients with COPD (Citation15). LTB4 levels are elevated in exhaled breath condensate (Citation9, Citation16), sputum (Citation16) and serum (Citation10) in patients with stable COPD as are plasma levels of the cysteinyl leukotriene, LTC4 (Citation8). To date, 2 clinical trials of anti-leukotriene therapy in stable COPD have been published (Citation11, 12).
The first was a randomized single-blind controlled trial that compared daily therapy with montelukast to placebo in 117 patients over 2 months (Citation11). Significant improvements in FVC, FEV1, PaO2 and St. George's Respiratory Questionnaire (SGRQ) scores (p < 0.05) were observed in the montelukast group with no comparable improvements in the placebo group, but between-group comparisons were not reported. The second was a small (n = 17) phase II randomized, double-blind, placebo-controlled, parallel-group trial of the effect of an oral leukotriene synthesis inhibitor BAYx1005 (500 mg twice daily) on sputum biomarkers (Citation12). Sputum LTB4 levels were reduced, but no differences were observed in sputum myeloperoxidase concentration or neutrophil chemotaxis between the 2 treatment arms.
Observational studies also support a possible role for leukotrienes in AECOPD. Two prior studies found that blood LTE4 levels were elevated in acute exacerbations and that levels decreased with treatment (Citation4, Citation17). Similarly, sputum and exhaled breath condensate LTB4 levels are elevated in AECOPD and decline with treatment (Citation5–7). In an accompanying in vitro study, Crooks and colleagues (Citation6) showed that LTB4 contributed approximately 30% of total neutrophil chemotactic activity in the sputum of these subjects during acute exacerbations.
Despite this rationale and clear evidence of effects of the leukotriene inhibitor on leukotriene levels, we did not find that the use of zileuton, when added to usual treatment of AECOPD, adds clinical benefit. In addition to the sample size issues discussed above, an important step in interpreting a negative clinical trial outcome is to document that the intervention achieved its intended pharmacologic effect. We found that treatment with zileuton was associated with a rapid reduction in urinary LTE4 levels as compared to placebo, indicating that the intended pharmacologic effect was achieved. The effect of zileuton on LTB4 production during the acute exacerbations is uncertain. Serum samples were only available at enrollment and at the end of the study (30 days) so the effect on LTB4 levels during the AECOPD could not be assessed.
The subjects whom we enrolled had relatively severe COPD at baseline which might have influenced the likelihood that leukotriene antagonism would be beneficial during an exacerbation. Overall, approximately half of our subjects were using home supplemental oxygen at baseline, and half reported history of a hospital visit for COPD in the last year (). Available spirometric data from the 30-day visit (in recovery) suggest that more than half of the subjects had GOLD stage III and IV disease (). Thus, our strategy of studying patients admitted to the hospital with AECOPD yielded patients with relatively severe COPD at baseline, and this could limit the absolute benefit of interventions made during exacerbations through a “ceiling effect”. However, our decision to study inpatients was based on a desire to reduce the high cost and health impact of those acute exacerbations which require hospitalizations.
In summary, our data indicate that 5-lipoxygenase inhibition reduces cysteinyl leukotriene levels in hospitalized patients with AECOPD. Whether this pharmacologic effect leads to improvements in clinically relevant outcomes (hospital LOS and treatment failure) is uncertain because early termination of the study limited our statistical power to detect modest but clinically meaningful improvements. Finally, we found that 5-lipoxygenase inhibition had no discernable adverse effects in AECOPD. These safety data, obtained in relatively sick patients with acute exacerbations of lung disease, may inform the development of further clinical trials of this strategy in stable COPD or in other inflammatory diseases.
DECLARATION OF INTEREST
SMS received a grant of $90,000 from Forrest Laboratories; JLC $10,001-$50,001 in capitation for a clinical trial from Boehringer Ingelheim; DEN received honoraria or advisory fees from Boehringer Ingelheim, Pfizer, AstraZeneca, GlaxoSmithKline, Nycomed, Forest Research Institute, Adams Respiratory Therapeutics, and Sepracor within the past 3 years. MTD has served on advisory boards and as a speaker for GlaxoSmithKline (<$10,000 in 2009) and Boehringer Ingelheim (<$5000 in 2009). He has received contracted research support from Aeris, Allegro, Altana/Nycomed/Forest, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Pfizer, and Roche. GJC Grant support: GlaxoSmithKline, Boehringer Ingelheim, Novartis, Respironics, Forest. Advisory groups: Dey, Boehringer Ingelheim, GlaxoSmithKline; MKH received Lecture Fees: Glaxo- SmithKline, CSL Behring, Boehringer Ingelheim, Pfizer. Consulting: Novartis, Nycomed.
FJM has received speaking, consultancy and steering committee fees from GlaxoSmithKline and Nycomed. He has received speaking and consulting fees from Medimmune/Astra Zeneca, Boehringer Ingelheim (BI) and Schering. He has received consulting and steering committee fees from Actelion. He has received consulting and DSMB fees from Novartis. He has received consulting fees from Forest/Almirall, Roche, Bayer and HLS. He has received advisory board fees from Merck, Pearl, UBC, Mpex, Talecris, Comgenix, and BoomComm. He has received lecture fees from France Foundation, NACE, MedEd, Potomac, fb Communications, Pfizer, Vox Medic, the American Lung Association, WebMD, epocrates and HIT Global. He has received royalities from Associates in Medical Marketing, and Castle Connolly. He has received sponsored grants from the NIH. His institution has received sponsored grants from BI.
ACKNOWLEDGEMENTS
All authors participated in study design, study enrollment procedures, review of data and manuscript preparation; drafted or revised the article for intellectual content; and provided final approval of the version to be published. Dr. Woodruff takes primary responsibility for the overall manuscript. The COPD Clinical Research Network is supported by a Cooperative Agreement from the Division of Lung Diseases of the National Heart, Lung, and Blood Institute. At some sites General Clinical Research Centers (GCRC) were utilized; their M01 grants from the National Center for Research Resources are listed. Members of this Network, with their personnel and grant support are: Brigham and Women's Hospital (Affiliated Sites: Fallon Clinic, VA Boston Healthcare System) –J.J. Reilly, Jr, (PI), G.R. Washko (Co-PI), M.L. Moy (Investigator), S. Peterson, C. Mayo, A. McDonald, K. Allain, K. Matthess, V. Danilack (Coordinators). Grant HL074428, GCRC Grant RR02635. Denver Health Medical Center (Affiliated Sites: National Jewish Medical & Research Center, University of Colorado) –R.K. Albert (PI), B. Make (Co-PI), M. Schwarz, C.Welsh (Investigators), M. Gilmartin, C.Verano, J. Binford (Coordinators). Grant HL074409, GCRC Grant RR00051. Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center –R. Casaburi, (PI), G.R. Mason, (Investigator), R. Kiledjian (Coordinator). Grant HL074407, GCRC Grant RR00425. Minnesota Veterans Research Institute, Minneapolis (Affiliated Sites: HealthPartners Research Foundation, Mayo Clinic) D.E. Niewoehner (PI), C. McEvoy, K.R. Rice, P.D. Scanlon (Co-PIs), D. Stuber, A. Fabbrini, D. Deno, K. Timm, P. Neuenfeldt, L. Loes, A. Adams (Coordinators). Grant 1U10-HL074416. Temple University –G.J. Criner (PI), W. Chatila, N. Marchetti, V. Kim, G.E. D'Alonzo, S.L. Krachman, F. Cordova, K. Brennan, N. Patel, J. Mamary (Investigators), C. Grabianowski, H. Smith (Coordinators). Grant HL074408. University of Alabama at Birmingham –M.T. Dransfield (Co-PI), J.A.D. Cooper (Co-PI), W.C. Bailey, P. O'Reilly (Investigators), N. Seabron, S. Tidwell, B. Martin, D. Brewton, D. Davis (Coordinators). Grant HL074418. University of California, San Francisco –S.C. Lazarus (PI), H.A. Boushey, P.G. Woodruff (Investigators), R. Sakurai, M. Dyjak, C. Nguyen (Coordinators). Grant 1U10-HL074431. University of Maryland, Baltimore –S.M. Scharf (PI), M. Alattar, P. Amelung, M. Cowan, J. Hanson, J.D. Hasday, A. Iacono, C. Shanholtz, N. Todd, A. Verceles (Investigators), T. Fitzgerald (Coordinator). Grant HL074441, GCRC Grant RR16500. University of Michigan, Ann Arbor –F.J. Martinez (PI), J.L. Curtis, M.K. Han, S.E. Gay (Investigators), D. Thompson, K. Baptist, C. Flaherty (Coordinators). Grant HL074422. University of Pittsburgh –F. Sciurba (PI), C. Atwood, J. Bon (Investigators), J. Roguskie, K. Hartwig , L. Kniolek (Coordinators). Grant HL074439, GCRC Grant RR00056. University of Minnesota (Data Coordinating Center) –J.E. Connett (PI), N.R. Anthonisen (Steering Committee Chair), C.Wendt (Co-PI), S. Harnden, W. Patrek, H. Voelker (Coordinators). Grant 1U10-HL074424. Data and Safety Monitoring Board –B.B. Bender, S.F. Kelsey, J.R. Landis, B.Phillips, G.M. Turino, R. Veatch, A. Waldo, A.Wanner. Protocol Review Committee –H.W. Kelly, J. Maurer, A.J. McSweeny, R.M. Senior, E.A. Thom, P.D. Wagner, R.L. ZuWallack; NHLBI –G. Weinmann (Director, Airway Biology & Disease Program), T. Croxton, A. Punturieri (Program Officers), M.P. Stylianou (Biostatistician).
REFERENCES
- Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic Obstructive Pulmonary Disease Surveillance—United States, 1971–2002: CDC; 2002. MMWR Surveillance Summary No. 51(SS06).
- Merrill CT, Elixhauser A. Hospitalization in the United States, 2002. Rockville, MD: Agency for Healthcare Research and Quality, 2005 HCUP Fact Book No. 6. AHRQ; 2005 No. 05- 0056.
- Peters-Golden M. Expanding roles for leukotrienes in airway inflammation. Curr Allergy Asthma Rep 2008; 8(4):367–373.
- Shindo K, Hirai Y, Fukumura M, Koide K. Plasma levels of leukotriene E4 during clinical course of chronic obstructive pulmonary disease. Prostaglandins Leukot Essent Fatty Acids 1997; 56(3):213–217.
- Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)-antitrypsin deficiency (PiZ). Am J Respir Crit Care Med 1999; 160(6):1968–1975.
- Crooks SW, Bayley DL, Hill SL, Stockley RA. Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. Eur Respir J 2000; 15(2):274–280.
- Biernacki WA, Kharitonov SA, Barnes PJ. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003; 58(4):294–298.
- Nannini LJ, Jr., Flores DM. Bronchodilator effect of zafirlukast in subjects with chronic obstructive pulmonary disease. Pulm Pharmacol Ther 2003; 16(5):307–311.
- Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhaled leukotrienes and prostaglandins in COPD. Thorax 2003; 58(7):585–588.
- Seggev JS, Thornton WH, Jr., Edes TE. Serum leukotriene B4 levels in patients with obstructive pulmonary disease. Chest 1991; 99(2):289–291.
- Celik P, Sakar A, Havlucu Y, Yuksel H, Turkdogan P, Yorgancioglu A. Short-term effects of montelukast in stable patients with moderate to severe COPD. Respir Med 2005; 99(4):444–450.
- Gompertz S, Stockley RA. A randomized, placebo-controlled trial of a leukotriene synthesis inhibitor in patients with COPD. Chest 2002; 122(1):289–294.
- Camargo CA, Jr., Smithline HA, Malice MP, Green SA, Reiss TF. A randomized controlled trial of intravenous montelukast in acute asthma. Am J Respir Crit Care Med 2003; 167(4):528–533.
- Niewoehner DE, Erbland ML, Deupree RH, Collins D, Gross NJ, Light RW, Anderson P, Morgan NA. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med 1999; 340(25):1941–1947.
- Marian E, Baraldo S, Visentin A, Papi A, Saetta M, Fabbri LM, Maestrelli P. Up-regulated membrane and nuclear leukotriene B4 receptors in COPD. Chest 2006; 129(6):1523–1530.
- Kostikas K, Gaga M, Papatheodorou G, Karamanis T, Orphanidou D, Loukides S. Leukotriene B4 in exhaled breath condensate and sputum supernatant in patients with COPD and asthma. Chest 2005; 127(5):1553–1559.
- Pinto-Plata VM, Livnat G, Girish M, Cabral H, Masdin P, Linacre P, Dew R, Kenney L, Celli BR. Systemic cytokines, clinical and physiological changes in patients hospitalized for exacerbation of COPD. Chest 2007; 131(1):37–43.