Abstract
Alpha-1 antitrypsin Deficiency (AATD) is a common, but under recognized metabolic genetic disease. Although many mutations in the alpha-1 antitrypsin (AAT) gene are described, the Z variant is the allele overwhelmingly associated with liver disease. PI*ZZ homozygotes occur in approximately 1 in 2,000-5,000 births in North American and European populations. The AAT protein is synthesized in large quantities by the liver, and then secreted into serum. Its physiologic function is to inhibit neutrophil proteases in order to protect host tissues from non-specific injury during periods of inflammation. The mutant Z gene of AAT directs the synthesis of a mutant protein which folds abnormally during biogenesis in the endoplasmic reticulum of hepatocytes and is retained intracellularly, rather than efficiently secreted. Intracellular proteolysis pathways, including the proteasome and autophagy, are activated as a response to the intracellular burden of misfolded protein. The lack of circulating anti-protease activity leaves the lung vulnerable to injury and the development of emphysema. The intracellular accumulation of AAT mutant Z protein within hepatocytes can cause liver injury, cirrhosis and hepatocellular carcinoma by triggering a cascade of chronic hepatocellular apoptosis, regeneration, and end organ injury. There is no specific treatment for PI*ZZ associated liver disease, other than standard liver supportive care and liver transplantation. There is a high degree of variability in the clinical manifestations among PI*ZZ homozygous patients, suggesting a strong influence of as yet poorly characterized, genetic and environmental disease modifiers. Studies of the processes of intracellular injury have led to a new era of rational therapeutic development.
Clinical liver disease
Liver disease associated with alpha-1 antitrypsin deficiency (AATD) has a complex pathophysiology, is highly variable between patients, and is under diagnosed. Alpha-1 antitrypsin (AAT) is a serum glycoprotein primarily synthesized in the liver and secreted into the serum. Smaller quantities are also produced in leukocytes, enterocytes and other tissues. AAT is secreted into the serum where its function is to inhibit neutrophil proteases released non-specifically during periods of inflammation (Citation1–4). Over 100 variant alleles of the AAT gene (SERPINA1) have been described but the overwhelming majority of patients with liver disease are homozygous for the Z mutant allele. Homozygosity for this autosomal co-dominant Z mutant of AAT, referred to as ZZ, PI*ZZ, or PI*ZZ in World Health Organization nomenclature, represents the classical form of AATD (Citation5).
The protein product of the mutant Z gene accumulates within hepatocytes rather than being efficiently secreted. This results in a lower, “deficient” level of protease inhibitor activity in serum. Within the hepatocyte, the accumulated Z mutant protein accumulates in the endoplasmic reticulum (ER), and may attain an altered conformation in which many AAT mutant Z molecules aggregate to form large polymers (). Pi*ZZ homozygous adults have a markedly increased risk of developing emphysema by a loss-of-function mechanism in which insufficient circulating AAT is available in the lung to inhibit connective tissue breakdown. A subgroup of Pi*ZZ homozygous children and adults may also develop liver disease and hepatocellular carcinoma via a gain-of-function mechanism in which the intracellular accumulation in the liver of AAT mutant Z protein triggers cell death and chronic liver injury (Citation6, 7).
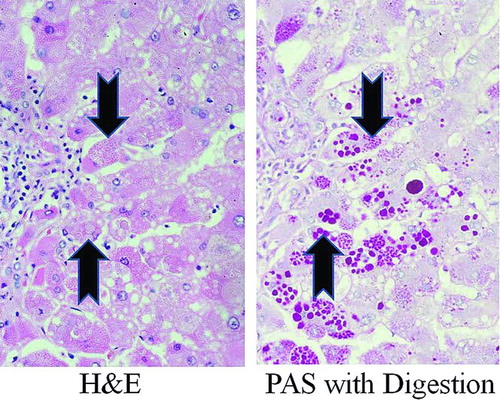
Figure 1. Photomicrograph of human Pi*ZZ liver. Human Pi*ZZ liver stained with H&E (left panel) and Periodic Acid-Schiff with digestion (PAS with digestion, right panel) showing inclusions (“globules”) of AAT mutant Z polymerized protein visible within some hepatocytes (arrows).
Liver disease in Pi*ZZ AATD may have a variety of clinical presentations, including chronic hepatitis, cirrhosis, hepatocellular carcinoma or the rare occurrence of fulminant hepatic failure (Citation1, Citation3, Citation8). In the neonatal period, the typical presentation is the “neonatal hepatitis syndrome,” which includes cholestatic jaundice, pruritis, poor feeding, poor weight gain, hepatomegaly, and splenomegaly (Citation9, 10). Laboratory evaluation may reveal elevated total or conjugated bilirubin, elevated serum AST and ALT, hypoalbuminemia, or coagulopathy due to vitamin K deficiency or to liver synthetic dysfunction.
There are rare reports of severe vitamin K deficient coagulopathic hemorrhage as the presenting feature of AATD in infants, which may result from impaired vitamin K absorption during sub-clinical neonatal cholestasis. Liver biopsy findings may be highly variable in infants including giant cell transformation, lobular hepatitis, significant steatosis, fibrosis, hepatocellular necrosis, bile duct paucity or bile duct proliferation (Citation1, Citation8, Citation11).
Differentiation from other cholestatic liver diseases of infancy by liver biopsy alone is not reliable. Globular, eosinophilic inclusions in some but not all hepatocytes are usually seen under conventional H&E stain, which represent dilated ER membranes engorged with polymerized AAT mutant Z protein () (Citation12). Staining with periodic acid-Schiff (PAS) followed by digestion with diastase, a technique which stains glycoproteins red, is used to highlight the “globules” (“PAS-positive”) within hepatocytes on a neutral background. Significant accumulations of PAS-positive material are not usually seen in normal hepatocytes.
Examination of liver biopsies for PAS-positive globules should be done with caution, as similar structures have sometimes been described in other liver diseases. Furthermore, the globules are not present in all hepatocytes or can be small and “dustlike” in small infants. Globules may be absent in the neonatal liver. Some pathology laboratories apply the technique of AAT immunostain as a confirmatory test to assist in the labeling of the globules.
The best, prospective, unbiased data on the natural history of AATD is the study by Sveger and colleagues who screened 200,000 newborns in Sweden in the 1970's and identified 127 Pi*ZZ who are still followed as a study cohort (Citation3). These data show that life-threatening liver disease can occur in the first few months or years of life, but that 80% of Pi*ZZ patients presenting with neonatal cholestasis are healthy and free of chronic disease by age 18 years (Citation13).
The overall risk of life-threatening liver disease in childhood was about 5%. The Swedish study is the best unbiased, population based cohort data available, although there is concern that the outcomes reported might not be fully representative of a less homogenous genetic population, such as North America to the British Isles, which may carry a different array of modifier genes. A smaller newborn screening series from the US based in the state of Oregon, which has not been consistently followed up, also suggested a low rate of life threatening childhood liver disease, as has a Canadian cohort (Citation14).
In toddlers and older children, Pi*ZZ AAT deficiency may present as asymptomatic chronic hepatitis, failure to thrive, possibly with poor feeding, or as isolated hepatomegaly or splenomegaly. The occurrence of these various problems ranged from 15% to 50% in the Swedish cohort, although many of these patients may have escaped medical attention without newborn screening (Citation9). Many children are completely healthy, without evidence of liver injury, except for mild and usually clinically insignificant elevations of serum AST or ALT.
The liver biopsy findings in later childhood often become more classic with easy to identify, large globules in many but not all hepatocytes, steatosis, possible lobular inflammation, and possible fibrosis. Occasionally, children with previously unrecognized chronic liver disease and cirrhosis present with ascites, gastrointestinal bleeding, or hepatic failure. There has also been a common observation that some children with severe liver disease in the first few months or years of life may enter a “honeymoon period” with few signs or symptoms and normal growth, before entering a period of renewed progressive injury and decompensation as older children or teenagers. However, even Pi*ZZ children with established cirrhosis and portal hypertension may remain stable and grow normally for years to decades with minimal intervention (Citation8, Citation15–17).
Progressive liver disease in previously well young and middle aged Pi*ZZ adults appears to be uncommon, although this is largely based on anecdote. There is very little published data on adult AATD liver disease (Citation3, 4, Citation13, Citation18). Adults may develop chronic hepatitis, with or without cirrhosis, but the risk of clinically significant disease likely increases with advancing age (Citation4). The biochemical and histopathologic findings in Pi*ZZ adults may be similar to those of alcoholic liver disease, which may lead to diagnostic confusion if specific serum testing for AATD is not performed in patients undergoing evaluation for unexplained liver disease.
Liver biopsy findings in adults may include lobular inflammation, variable hepatocellular necrosis, fibrosis, cirrhosis, steatosis, and PAS-positive, diastase-resistant globules in some, but not all, hepatocytes (Citation8). These findings can be similar to those of alcoholic liver disease if the globular inclusions are misinterpreted. There also appears to be an increased risk of hepatocellular carcinoma in Pi*ZZ adults, although the magnitude of the risk is unclear (Citation4, Citation11, Citation19).
The risk of hepatocellular carcinoma may, in part, be independent of the development of hepatic cirrhosis. The rate of significant liver injury increases in older ages. Autopsy studies show that histologically significant, but possibly clinically undetected, liver injury and cirrhosis may be present in 40–50% of elderly Pi*ZZ adults (Citation4). As middle-aged emphysema is more effectively treated, or prevented altogether as a result of decreased smoking, it is possible that more older adults with Pi*ZZ liver disease will come to medical attention.
The diagnosis of AATD does not require liver biopsy. The gold standard for the diagnosis of AATD is the analysis of the “phenotype” of AAT protein in a patient's serum or the genotype analysis of genomic DNA. The phenotype gel analysis is technically demanding and is therefore best performed in an experienced reference laboratory. Some clinicians advise the use of a serum AAT level as a screening test and then perform the gold standard test if the result is outside the normal range (). This approach is common in some liver clinics. The results should be interpreted with caution, however, as AAT is an acute phase reactant and even a Pi*ZZ patient will have modest increases in serum level during times of systemic inflammation.
Table 1. α1AT Phenotypes and corresponding Typical α1AT serum levels
Although a Pi*ZZ patient would not be expected to ever produce a level in the normal range, this author has seen Pi*SZ patients with liver disease occasionally have AAT levels reported in the normal range during episodes of systemic inflammation. Care should also be taken not to obtain serum for a level or phenotype if the patient has recently had a plasma transfusion, as is sometimes used to treat patients with severe liver disease, as the result may reflect the status of the plasma donor and not the host patient. Liver biopsy during a liver disease evaluation can be an important tool to assess the degree of liver injury and is still regarded as the gold standard to determine the extent of hepatic fibrosis and to diagnose cirrhosis.
Heterozygotes and other genotypes
It is commonly accepted that individuals who are compound heterozygotes for the S and the Z alleles of AAT (Pi*SZ) may develop liver disease identical to Pi*ZZ patients, including PAS-positive, diastase-resistant globules. However, the magnitude of the risk of disease in Pi*SZ patients is not well established. The S allele is common in North American and Western European populations, especially in Spain and Portugal. Studies have shown that the AAT mutant S protein can heteropolymerize intracellularly only when it is co-expressed with the mutant Z protein, which may explain the occurrence of liver injury in Pi*SZ patients when liver disease is absent in Pi*SS individuals (Citation3, Citation20, Citation21).
Over 100 other rare mutations in the AAT gene have been described, some of which yield a gene product with a normal M migration on the phenotype test gel but when present in the heterozygous state with a Z allele can accumulate within the liver and have been associated with liver disease (Citation3, Citation22, Citation23). Two examples are the M Duarte and M malton alleles (Citation3, Citation22). Such patients are usually recognized by a profoundly low AAT level in peripheral blood that is not in keeping with the higher level expected by an “Pi*MZ” phenotype result. Serum deficiency states caused by null genes, or other unusual alleles which to not direct the synthesis of a protein product which accumulates within the liver are not associated with liver disease(Citation3).
Individuals who are heterozygous for AAT, carrying one normal M allele and one mutant Z allele (“Pi*MZ’’ or “MZ’’) are generally considered asymptomatic and healthy with regard to liver disease (Citation3). However, data from retrospective, referral center studies report a 2 to 5 fold over representation of Pi*MZ patients in groups with chronic liver diseases, such as cryptogenic cirrhosis, sometimes in association with concurrent viral hepatitis (Citation3). Limited but unselected population-based studies have thus far failed to confirm this increased risk (Citation15). The most widely accepted explanation is that the Pi*MZ heterozygous state likely represents a genetic modifier of other liver diseases.
There are anecdotal case reports of rare Pi*MZ adults developing liver disease, including the development of PAS-positive globules in hepatocytes, without other apparent risk factors for liver disease, although the possible genetic or environmental influences on the development of this injury remains controversial (Citation3, Citation24–26). Pi*MZ children appear to be completely healthy, and even in adults an Pi*MZ phenotype result is not readily accepted as the cause of otherwise unexplained liver disease without extensive further evaluation.
Management
There is no specific treatment for the liver disease associated with AATD. However, the general supportive measures used in many liver diseases are applied. Management focuses on preventing the complications of chronic liver disease, such as bleeding, ascites, pruritis, malnutrition, fat-soluble vitamin deficiency, infection, hepatocellular carcinoma and growth disturbances, or attenuating the repercussions if they do occur (Citation1, Citation3, Citation8). In pediatric liver clinics ursodeoxycholic acid is often prescribed, although the results of some small reports are inconclusive (Citation27).
Some authorities cite the anti-apoptotic effects of ursodeoxycholic acid documented in in vitro studies as being theoretically able to inhibit the pro-apoptotic effects of AAT mutant Z intracellular retention as justification. However, recent trials of ursodeoxycholic acid in other liver diseases have shown it to be associated with increased mortality when used in high doses, which has tempered some of the enthusiasm for this approach.
It is common practice for Pi*ZZ patients to be followed annually by a physician knowledgeable in liver disease, although many patients have normal health and can be monitored conservatively. Even patients with significant liver disease and fibrosis may remain stable for many years, although the reasons for this kind of stabilizing course of liver disease are unclear. Monitoring for progressive liver disease requires careful attention to many aspects of the patients’ health status. This includes repeated questioning for liver related symptoms such as pruritis and fatigue, among others, and repeated extensive physical examination to monitor for hepatosplenomegaly, or for other less obvious signs of liver disease.
Laboratory tests should not exclusively focus on serum transaminase levels, as some patients with cirrhosis can have normal or nearly normal ALT and AST. Bilirubin levels, coagulation, and fat soluble vitamin levels should occasionally be examined. The recommendations of the American Association for the Study of Liver Diseases (AASLD) guidelines for surveillance of liver cancer recommend liver ultrasound every 6 months for all liver disease patients with >2% risk per year of liver cancer. Although data are lacking to firmly apply this recommendation to Pi*ZZ patients, a conservative approach would be to obtain liver ultrasound every 6 months in any patient with cirrhosis, significant fibrosis or persistently large AST or ALT elevation. Some patients with significant degrees of liver injury, and even cirrhosis, can remain stable for many years with very little need for other interventions.
If life-threatening liver disease does develop, then liver transplantation is commonly employed with excellent success rates. Following liver transplantation, the serum phenotype and AAT levels will return to normal, Pi*MM, assuming the donor is Pi*MM. It is generally thought that lung deterioration will stop, but not be reversed after a liver transplant, although little prospective confirmation of this logical conclusion has been reported. Exogenous AAT replacement has no effect on the development of liver disease since liver injury is related to the accumulation of the AAT mutant Z protein within hepatocytes, not a lack of circulating anti-protease activity.
Lifestyle and environmental factors may also be important in the management of liver disease. Although there is very little data specific to AATD, avoiding obesity and fatty liver and limiting alcohol are likely to be beneficial. Patients with advanced liver disease should abstain from alcohol completely, while the recommendations are less clear for patients without advanced disease. Adult liver data in patients with hepatitis C but without progressive injury suggest that up to the equivalent of the 3 alcoholic drinks per week may be safe (see AASLD guidelines).
Studies in animal models of AATD liver disease show that non-steroidal anti-inflammatory drugs (NSAIDS) may be uniquely toxic to the Pi*ZZ liver. NSAIDS in model systems increase AAT mutant Z protein synthesis, increase the hepatic burden of accumulated mutant protein and potentiate liver injury (Citation28). Acetaminophen is not shown to have the same effect. Although there has been no examination of this issue in human studies, it seems prudent to limit the intake of NSAIDS in Pi*ZZ patients if possible, although this is controversial.
All AATD patients, regardless of the presence of lung disease or liver disease are urgently cautioned to avoid cigarette smoke and other inhalation exposures. Some studies suggest that exposure to even secondhand smoke and environmental air pollutants in childhood is an important risk factor for the development AATD associated adult emphysema (Citation3, Citation29-31). Therefore, Pi*ZZ children and their household contacts are urgently cautioned against smoking, even if they come to medical attention primarily because of liver disease. Children with Pi*ZZ AATD generally do not develop clinically detectable emphysema, although they may be at increased risk for childhood asthma (Citation32, 33). Pi*ZZ children are commonly referred to an adult pulmonologist at age 18 years for a baseline evaluation, unless asthma or other respiratory symptoms are present necessitating treatment in childhood.
Pathophysiology of homozygous Pi*ZZ liver disease
The evolution of the understanding of liver injury in this disease has been a lengthy process, and is still not yet fully understood. However, over the 50 years since the disease was first described there have been a few seminal observations that have driven the field and now provide new insights into potential therapies. First, was the original recognition by Sharp and colleagues that pediatric Pi*ZZ patients can develop liver disease.
Then, studies of patient tissues by Perlmutter and colleagues showed reduced intracellular clearance of mutant Z protein correlated to life-threatening liver disease, which gave the strongest support up to that time that accumulation of the mutant Z protein in the liver was the key trigger of liver injury (Citation34). At the same time, the polymerized conformation was discovered by Lomas, Carrell, and others, which focused the field on the key concepts of protein conformation and trafficking (Citation35).
More recently, the discovery by Teckman and Perlmutter that autophagy was a primary route of intracellular degradation for the mutant Z protein, when combined with these other concepts has led to multiple new therapeutic approaches (Citation36). Finally, documentation in the last decade by Teckman and Perlmutter, of the hepatocellular apoptosis and compensatory proliferation in the liver revealed how mutant Z protein accumulation was likely linked to cirrhosis and HCC (Citation2, Citation6). We will review how these discoveries have now led to our concept of the mechanism of AATD liver injury.
The pathophysiology of liver injury in a1AT deficiency results directly from the accumulation of the abnormal, AAT mutant Z protein within liver cells ()(Citation37). The lack of circulating anti-protease activity does not play a role in liver disease. Various studies have also provided data to support the hypothesis that accumulation within hepatocytes of AAT mutant Z protein specifically in the polymerized conformation is a key factor in liver injury (Citation5, Citation34, Citation38, 39). During biosynthesis the AAT mutant Z gene is appropriately transcribed, translated, and then the nascent mutant Z polypeptide chain is translocated into the ER lumen.
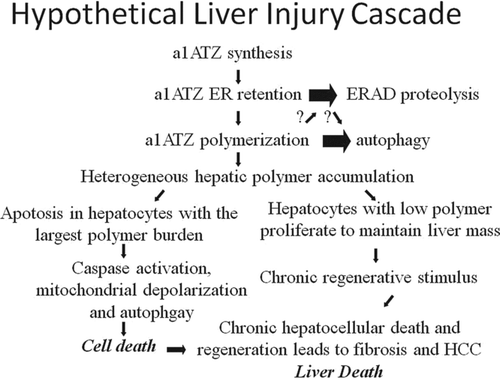
Figure 2. Hypothetical liver injury pathway in Pi*ZZ AATD. The AAT mutant Z protein is appropriately synthesized, but then retained in the ER of hepatocytes rather than being secreted. Quality control processes within the cells direct most of the mutant Z protein molecules into intracellular proteolysis pathways related to the proteasome (ERAD). However, some of the mutant Z protein molecules escape proteolysis and may attain a unique, polymerized conformation forming inclusions in the ER. Autophagic degradation is upregulated as a response to mutant Z polymer accumulation. For reasons that are not clear, a small population of hepatocytes develops especially large accumulations of polymerized mutant Z protein and undergo apoptosis. The hepatocytes with a smaller burden of mutant Z protein proliferate, possibly with the input of a liver stem cell population, to maintain the functional liver mass. This chronic process of injury, cell death, and compensatory proliferation is known to lead to end organ processes of fibrosis, cirrhosis, and HCC. Given the variable nature of clinical liver injury between individuals with the same genotype, and the usually slow disease progression, there are likely to be important environmental and genetic disease modifiers affecting the rate and magnitude of these processes.
However, in the ER the mutant Z protein molecule folds slowly and inefficiently into its final, secretion-competent conformation (Citation35, Citation40). A system of proteins within the ER, termed the “quality control” apparatus, recognizes these mutant Z molecules as abnormal and directs them to a series of proteolytic systems rather than allowing progression down the secretory pathway (Citation34, Citation36, Citation39, Citation41, Citation42, 43).
However, this process of quality control holds the mutant Z molecules in the ER lumen for a longer time than would normally be the case during secretion of the wild type M protein. For reasons that are not clear, but which might be related to this “lag” in degradation, some of the mutant Z molecules escape proteolysis and may attain a variety of abnormal conformations including a unique state in which multiple molecules aggregate to form large polymers (discussed further later) (Citation35, Citation38, Citation44). This polymer conformation is highly thermodynamically stable and links large groups of mutant Z molecules together with non-covalent bonds. These polymers have a long biological half life within cells.
Accumulations within hepatocytes of the polymerized mutant Z protein may be large enough to be seen under light microscopy and represent the “globules” observed in the Pi*ZZ liver (). The result of these processes is that only approximately 15% of AAT mutant Z protein molecules are secreted into the serum.
Further data on the importance of polymerization come from studies of the AAT mutant S molecule, which is retained intracellularly when expressed alone in cells. These data shown that the S protein does not form polymers unless mutant Z molecules are also present in the same cell (Citation20, 21). In that case there is formation of S and Z heteropolymers (Citation20). Patients who are homozygous Pi*SS do not develop liver disease, but Pi*SZ heteropolymerization would represent the physiology of an Pi*SZ compound heterozygous patient, who is at risk of liver disease.
Furthermore, studies in animal models have shown that liver injury increases if the intrahepatic burden of retained mutant Z polymer increases, and liver injury is reduced when the intrahepatic mutant Z accumulation is reduced (Citation28, Citation45-47). Taken together, these data support the hypothesis that polymer formation and accumulation is likely to be the key event in triggering liver injury. There has also been the suggestion that there could be a dosage affect, or possibly a threshold of globule formation for injury initiation, given the observation that the vast majority of Pi*MZ patients do not have hepatocellular globules of protein polymer or evidence of liver injury, while Pi*SZ and Pi*ZZ patients do.
Once the mutant Z protein is retained in the ER, the hepatocyte attempts to deal with this burden of misfolded protein via intracellular pathways for protein degradation. These include ubiquitin dependent and ubiquitin independent proteasomal pathways, and possibly other mechanisms (Citation42, 43, Citation48, 49). These pathways are sometimes referred to as “ER associated degradation” (ERAD), and are thought to be critical mechanisms for liver cells to “protect” themselves from the accumulation of abnormally folded proteins.
It is thought that these pathways are the primary route for degradation of AAT mutant Z protein in the non-polymerized conformation. These proteolytic pathways successfully process the vast majority of AAT mutant Z protein molecules retained within the ER, or the liver would quickly cease to function since the liver synthesizes 2 to 3 grams per day of AAT protein. Although many of the mechanistic steps in the degradation process, and their specific sequence, are still under investigation, previous work has shown that two molecules present in the ER, calnexin and ER manosidase I (ERmanI), are likely to be critical points of control.
Calnexin is a transmembrane ER chaperone which binds AAT mutant Z, becomes targeted for degradation by linkage to ubiquitin, and then is degraded as this trimolecular complex (AAT mutant Z-calnexin-ubiquitin) by the proteasome (Citation39). Studies in human fibroblast cell lines established from Pi*ZZ homozygous patients show that patients susceptible to liver disease have less efficient ER associated degradation of AAT mutant Z protein than Pi*ZZ patients without liver disease (Citation21, Citation34).
The reduced efficiency of degradation in the liver disease patients presumably leads to a greater steady state burden of mutant Z protein within liver cells and increased liver injury. Similarly, studies of the enzyme ERmanI suggest that it also may have a critical role in directing AAT mutant Z molecules to the proteasome for degradation. These data raise the possibility that allelic variations in calnexin, ERmanI, or in other proteins involved in the quality control or proteolytic systems might alter susceptibility to liver injury by changing the efficiency of degradation (Citation41, Citation50).
There has been a report that susceptibility to liver disease might also be related to allelic variations in the AAT gene itself, which would not otherwise be considered disease-associated mutations (Citation51, 52). Evidence also shows that the accumulation of the polymerized AAT mutant Z protein within cells induces an autophagic response. Autophagy is a highly conserved degradation system whereby unique vacuoles degrade abnormal proteins and larger structures such as senescent organelles. It is thought that autophagy is an important route for the degradation of AAT mutant Z polymers, and in experimental systems liver injury can be reduced by increased autophagic degradation of mutant Z polymer protein (Citation45, 46).
For many years it was recognized that mutant Z protein accumulation in the liver, and likely polymer accumulation drove liver injury, but it was difficult to reconcile these data with the observation that clinical liver injury in Pi*ZZ humans is usually a slow process over years to decades and that the mutant Z protein accumulation was heterogeneous. That is, not all hepatocytes contain the globules of accumulated mutant Z polymer.
Typically, hepatocytes, which on histology appear to be healthy, may contain large globules but may be immediately adjacent to hepatocytes with no globules. Experiments in human tissue and animal models have recently explained this by showing that a cellular injury cascade in the liver is triggered within the small population of hepatocytes with the largest AAT mutant Z polymerized protein accumulation (Citation6, Citation37, Citation53-56).
Hepatocytes with the largest AAT mutant Z protein accumulation, perhaps only a few percent of the total hepatocytes, have increased cleavage of the ER stress caspases involved in apoptotic signaling. These heavily burdened hepatocytes also have mitochondrial injury and increased signaling of apoptosis through mitochondria, including cytochrome c release (Citation53, 54). Mitochondria have also been implicated in autophagic signaling and in modulating some inflammatory pathways, which might contribute to hepatocellular damage (Citation36, Citation56).
What follows is a low, but higher than normal baseline rate of hepatocyte death in Pi*ZZ liver tissue compared to normal liver (Citation2). The cells with low polymer accumulation then proliferate to maintain the functional liver mass. Over time, the continued stress, death, and repair leads to liver fibrosis, cirrhosis, HCC, and chronic organ injury. This model of slow, but continued progressive injury fits the clinical pattern of chronic liver disease observed in many Pi*ZZ patients, which progresses over a time span of months to many years. Environmental and genetic modifiers of the trafficking, degradation, apoptotic, or regenerative processes would then by hypothesized to influence the progression of liver disease in an individual patient.
New discoveries and technologies suggest new therapies
The improved insights, discussed here, into the cellular processes related to liver injury in this disease has led to new hypotheses for approaches to treatment and new hope that a treatment for this liver disease will be developed. When considering approaches to treatment for AATD associated liver disease it is helpful to think in terms of the progression of the pathophysiology. First, the mutant gene directs synthesis of the mutant protein. A variety of gene therapy approaches have been examined for many of the single gene defects, with limited success in human trials, and also with significant safety concerns.
Mouse models of Pi*ZZ AATD have been successfully treated with adeno-associated viral vectors, or other vectors, containing siRNAs, ribozyme, and other mechanisms for inhibiting transcription or translation of the mutant gene. In one recent publication involving the mouse model, it was even possible to inhibit the mutant Z protein synthesis, while generating wild-type, M, AAT protein synthesis with an exogenous mRNA (Citation57).
It has also been recently announced that there are pharmaceutical companies with siRNA technology that can target the liver in AATD. Press releases and meeting abstract presentations suggest that knocking down AAT mutant Z mRNA in the liver has been accomplished in model systems with excellent safety data and that possible human trials of this technology are not far away. The hope in this strategy is that if mutant Z protein production in the liver could be shut down, that the liver would itself clear the accumulated Z polymer. However, patients would still require treatment for the lung disease associated with AATD
After transcription and translation, comes folding of the nascent polypeptide, trafficking and retention of the mutant Z protein in the ER. There has been, and continues to be, intense interest in small molecules which might alter the folding of the AAT mutant Z molecule to prevent retention, prevent polymerization, allow secretion, or promote degradation. If secretion could be increased and polymerization in the periphery prevented, then lung disease might also be treated by such a method. The so called chemical chaperone, 4-phenylbutyrate, was shown in in vitro studies and in animal models to increase secretion of AAT mutant Z.
However, human studies have shown no therapeutic effect as a clinical treatment, possibly because the drug levels required to improve secretion in animals could not be reached in human serum before side effects became intolerable (Citation58). Several rationally designed molecules have been tested in vitro and have been shown to inhibit polymerization, but so far they have not been developed enough to allow a clinical trial.
When the mutant Z protein is retained in the ER, the cell attempts to cope with this burden of misfolded protein by directing the protein molecules into intracellular degradation pathways. Again, several potential therapeutic drugs have been tested in mice which may reduce liver injury by accelerating proteolysis pathways. Two of the most interesting recent reports involve compounds that appear to induce increase autophagy in the liver, carbamazepine and rapamycin (Citation45, 46). The autophagic pathway is a highly conserved intracellular pathway involved in the degradation of protein aggregates, damaged organelles, and a variety of other cellular functions.
Autophagy is involved in the degradation of AAT mutant Z protein polymers, as discussed above. However, there were concerns raised about extending these studies to human trials. One was the known potential toxicity of rapamycin, and the other was that the positive effect of carbamazepine was seen in mice at 10 times the usual human dose. A human trial for Pi*ZZ individuals with end stage cirrhosis is underway, but only using normal dose ranges so the expectation of benefit in this trial is unclear.
Another strategy is to inhibit the downstream consequences of liver injury associated with mutant Z protein accumulation, such as apoptotic signaling, mitochondrial depolarization, or fibrosis. Drug strategies aimed at inhibiting points in these intrahepatic injury cascades, including the use of cyclosporine to inhibit mitochondrial injury and reduce liver damage, have shown promise in animal studies, but have not yet been extended to humans (Citation47).
A wide range of anti-fibrotic strategies, some of them targeting activated stellate cells are also under investigation in a wide range of liver diseases. Finally, there has also been continued interest in treatments at the level of the liver cell or the organ itself, such as hepatocyte transplantation as a method to “replace” Pi*ZZ liver cells with Pi*MM liver cells. However, a lack of supply of viable livers from which to isolate cells has delayed further trials until a reliable method of growing hepatocytes from an as yet unidentified source of stem cells, can be developed.
Conclusions
Homozygous Pi*ZZ AATD is a common metabolic liver disease which can affect adults and children. The clinical manifestations are highly variable, with many patients remaining healthy or exhibiting only mild biochemical abnormalities until late in life. Accumulation of the AAT mutant Z protein within hepatocytes activates an intracellular injury cascade of apoptotic liver cell death and compensatory hepatocellular proliferation leading to end organ injury. Genetic and environmental disease modifiers are thought to be improtant, but are still poorly described. There is no specific treatment for AATD associated liver disease, but there are treatment options involving supportive measures and liver transplant. New technologies aimed at stimulating proteolysis pathways, small molecule chaperones, gene therapy, RNA technologies, or cell transplantation may hold promise for the treatment of this diseases. Future research is likely to lead to studies of these new approaches, although the high degree of clinical variability will pose a challenge to the design of clinical trials.
Declaration of Interest Statement
The author has been the recipient of grant support from the Saint Louis University Liver Center, Alnylam Pharmaceuticals, the NIH (Dk ) and the Alpha-1 Foundation and has served in an advisory capacity to the Alpha-1 Foundation, the Alpha-1 Association and the Alpha-1 Project. The author serves on the Steering Committee of the Childhood Liver Disease Research and Education Network. The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Acknowledgments
The author recognizes Dr. David Perlmutter for his invaluable mentoring, leadership and devoted service to this author personally and to the field of alpha-1 antitrypsin.
References
- Nelson DR, Teckman J, Di Bisceglie AM, Brenner DA. Diagnosis and management of patients with a(1)-Antitrypsin (A1AT) Deficiency. Clin Gastroenterol Hepatol. 2011. Epub 2011/12/28.
- Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology 2004; 39(4):1048–1055.
- American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am J Respir Crit Care Med 2003; 168(7):818–900.
- Eriksson S. Alpha-1-antitrypsin deficiency: natural course and therapeutic strategies. In: Boyer JL, Blum, H.E., Maier, K.-P., Sauerbruch, T., Stalder, G.A., editor. Falk Symposium 115: Liver Cirrhosis and its Development. Dordrecht: Kluwer Academic Publishers; 2001. p. 307–315.
- Qu D, Teckman JH, Perlmutter DH. Review: Alpha 1-antitrypsin deficiency associated liver disease. J Gastroenterol Hepatol 1997; 12(5):404–416.
- Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007; 46(4):1228–1235. Epub 2007/09/22.
- Eriksson S. Discovery of alpha 1-antitrypsin deficiency. Lung 1990; 168 Suppl:523–529.
- Perlmutter DH. Alpha-1-antitrypsin deficiency: diagnosis and treatment. Clin Liver Dis 2004; 8(4):839–859, viii–ix.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294(24):1316–1321.
- Sveger T. alpha 1-antitrypsin deficiency in early childhood. Pediatrics 1978; 62(1):22–25.
- Eriksson S. Alpha 1-antitrypsin deficiency. J Hepatol 1999; 30 Suppl 1:34–39.
- An JK, Blomenkamp K, Lindblad D, Teckman JH. Quantitative isolation of alphalAT mutant Z protein polymers from human and mouse livers and the effect of heat. Hepatology 2005; 41(1):160–167.
- Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology 1995; 22(2):514–517.
- Cruz PE, Mueller C, Cossette TL, Golant A, Tang Q, Beattie SG, In vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab Invest 2007; 87(9):893–902. Epub 2007/06/27.
- Sveger T. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand 1988; 77(6):847–851.
- Mowat AP. Alpha 1-antitrypsin deficiency (PiZZ): features of liver involvement in childhood. Acta Paediatr Suppl 1994; 393:13–17.
- Pittschieler K, Massi G. Alpha 1 antitrypsin deficiency in two population groups in north Italy. Paediatr Padol 1988; 23(4):307–311.
- Eriksson S. A 30-year perspective on alpha 1-antitrypsin deficiency. Chest 1996; 110(6 Suppl):237S–42S.
- Eriksson S, Lindmark B, Olsson S. Lack of association between hemochromatosis and alpha-antitrypsin deficiency. Acta Med Scand 1986; 219(3):291–294.
- Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest 1999; 103(7):999–1006.
- Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem 1996; 271(22):13215–13220.
- Lomas DA, Elliott PR, Sidhar SK, Foreman RC, Finch JT, Cox DW, alpha 1-Antitrypsin Mmalton (Phe52-deleted) forms loop-sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J Biol Chem 1995; 270(28):16864–16870.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53–>Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268(21):15333–15335.
- Pittschieler K. Liver disease and heterozygous alpha-1-antitrypsin deficiency. Acta Paediatr Scand 1991; 80(3):323–327.
- Kaserbacher R, Propst T, Propst A, Graziadei I, Judmaier G, Vogel W. Association between heterozygous alpha 1-antitrypsin deficiency and genetic hemochromatosis. Hepatology 1993; 18(3):707–708.
- Propst T, Propst A, Dietze O, Judmaier G, Braunsteiner H, Vogel W. High prevalence of viral infection in adults with homozygous and heterozygous alpha 1-antitrypsin deficiency and chronic liver disease. Ann Intern Med 1992; 117(8):641–645.
- Lykavieris P, Ducot B, Lachaux A, Dabadie A, Broue P, Sarles J, Liver disease associated with ZZ alpha1-antitrypsin deficiency and ursodeoxycholic acid therapy in children. J Pediatr Gastroenterol Nutr 2008; 47(5):623–629. Epub 2008/10/29.
- Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology 2006; 44(4):976–982. Epub 2006/09/29.
- Piitulainen E, Sveger T. Respiratory symptoms and lung function in young adults with severe alpha(1)-antitrypsin deficiency (PiZZ). Thorax 2002; 57(8):705–708.
- Sveger T, Thelin T, McNeil TF. Young adults with alpha 1-antitrypsin deficiency identified neonatally: their health, knowledge about and adaptation to the high-risk condition. Acta Paediatr 1997; 86(1):37–40.
- Mayer AS, Stoller JK, Vedal S, Ruttenber AJ, Strand M, Sandhaus RA, Risk factors for symptom onset in PI*Z alpha-1 antitrypsin deficiency. Inter J Chron Obstruct Pulmon Dis 2006; 1(4):485–492. Epub 2007/11/30.
- Sveger T, Piitulainen E, Arborelius M, Jr. Lung function in adolescents with alpha 1-antitrypsin deficiency. Acta Paediatr 1994; 83(11):1170–1173.
- Eden E, Hammel J, Rouhani FN, Brantly ML, Barker AF, Buist AS, Asthma features in severe alpha1-antitrypsin deficiency: experience of the National Heart, Lung, and Blood Institute Registry. Chest 2003;123(3):765–771.
- Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci USA 1994; 91(19):9014–9018.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357(6379):605–607.
- Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol 2000; 279(5):G961–974.
- Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G Signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant alpha1-antitrypsin Z in the classical form of alpha1-antitrypsin deficiency. J Biol Chem 2007; 282(38):27769-80. Epub 2007/07/20.
- Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest 2002; 110(11):1585–1590.
- Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem 1996; 271(37):22791–22795.
- James EL, Bottomley SP. The mechanism of alpha 1-antitrypsin polymerization probed by fluorescence spectroscopy. Arch Biochem Biophys 1998; 356(2):296–300.
- Cabral CM, Choudhury P, Liu Y, Sifers RN. Processing by endoplasmic reticulum mannosidases partitions a secretion-impaired glycoprotein into distinct disposal pathways. J Biol Chem 2000; 275(32):25015–25022.
- Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, Perlmutter DH. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem 2001; 276(48):44865–44872.
- Sifers RN. Cell biology. Protein degradation unlocked. Science 2003; 299(5611):1330–1331.
- Dafforn TR, Mahadeva R, Elliott PR, Sivasothy P, Lomas DA. A kinetic mechanism for the polymerization of alpha1-antitrypsin. J Biol Chem 1999; 274(14):9548–9555.
- Kaushal S, Annamali M, Blomenkamp K, Rudnick D, Halloran D, Brunt EM, Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood) 2010; 235(6):700–709. Epub 2010/06/01.
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010; 329(5988):229–232. Epub 2010/06/05.
- Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol 2004; 286(5):G851–G862.
- Wu Y, Swulius MT, Moremen KW, Sifers RN. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci USA 2003; 100(14):8229–8234.
- Teckman JH, Gilmore R, Perlmutter DH. Role of ubiquitin in proteasomal degradation of mutant alpha(1)-antitrypsin Z in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol 2000; 278(1):G39–G48.
- Cabral CM, Liu Y, Moremen KW, Sifers RN. Organizational diversity among distinct glycoprotein endoplasmic reticulum-associated degradation programs. Mol Biol Cell 2002; 13(8):2639–2650.
- Chappell S, Hadzic N, Stockley R, Guetta-Baranes T, Morgan K, Kalsheker N. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology 2008; 47(1):127–132. Epub 2007/11/01.
- Pan S, Huang L, McPherson J, Muzny D, Rouhani F, Brantly M, Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology 2009; 50(1):275–281. Epub 2009/05/16.
- Lawless MW, Greene CM, Mulgrew A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol 2004; 172(9):5722–5726.
- Miller SD, Greene CM, McLean C, Lawless MW, Taggart CC, O'Neill SJ, Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of Bad. Hepatology 2007; 46(2):496–503. Epub 2007/06/15.
- Rudnick DA, Perlmutter DH. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology 2005; 42(3):514–521.
- Schmidt BZ, Perlmutter DH. Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol 2005; 289(3):G444–G455.
- Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther 2012. Epub 2012/01/19.
- Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr 2004; 39(1):34–37.