Abstract
Calprotectin comprises more than 45% of the cytosolic content of neutrophil granulocytes. Because pathogenesis, disease activity and disease progression in COPD are believed to be partly dependent of neutrophil driven inflammation we decided to investigate whether plasma level of calprotectin (p-calprotectin) was associated with all-cause mortality in patients with COPD. We measured p-calprotectin in blood samples from 460 patients with moderate to very severe COPD in stable phase. Patients were stratified into three groups according to p-calprotectin level. Outcome measure was all-cause mortality. Analyses were adjusted for factors known to influence mortality using a Cox regression analysis. We found a time dependent correlation between p-calprotectin levels and mortality during the first 5 years of follow-up. Increasing levels of p-calprotectin were associated with concomitant increases in mortality from HR 1.56 (CI 95%: 1.03 –2.38) at calprotectin between 100 –200 ng/ml to HR 2.02 (CI 95%: 1.27-3.19) at calprotectin >200 ng/ml. P-calprotectin could be a useful marker of all-cause mortality in patients suffering from moderate to very severe COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is associated with significant morbidity and mortality and is a leading cause of death in the world (Citation1–3). COPD is defined by airflow limitation, which is not fully reversible. The disease is usually progressive, with a decline in pulmonary function as a consequence (Citation4). The pathogenesis underlying the disease is complex, but is in part attributed to ongoing neutrophil granulocyte mediated inflammatory processes in the airways (Citation5), probably in response to noxious gases e.g. tobacco smoking (Citation6). Progression of COPD has been shown to be correlated to neutrophil granulocyte levels in the smaller airways (Citation7). In addition to inflammatory activity in the airways, increased systemic inflammation has also been recognized as part of the disease activity, and this is reflected by an increased number of neutrophil granulocytes in the circulation, and an increase in circulating inflammatory mediators (Citation8, 9). Thus, both systemic and localized inflammation implicates neutrophil granulocytes in the pathogenesis of the diseases (Citation10).
COPD is characterized by acute episodes of worsening symptoms termed “acute exacerbations.” The number of such episodes is associated with the rate of lung function decline (Citation11). It is believed that exacerbations are caused by inflammation in response to infections in up to 80% of the cases (Citation12). Somewhat surprisingly, biomarkers of inflammation have been very poor at predicting progression of the disease in terms of exacerbations (Citation13), and until recently, significant predictors of mortality have been restricted to low BMI (Citation14), low FEV1 (Citation15), quality of life (Citation16), and dyspnea (Citation17). Several of these predictors have been compiled into refined indices like the BODE index (Citation18). In the last few years some progress have been made into the usage of biomarkers like CRP (Citation19), fibrinogen (Citation20), and serum PARC/CCL 18 (Citation21). Some of these were included in a panel of biomarkers, which found significant correlation between biomarker levels and mortality (Citation22). However not all of the studies have been consistent in their findings (Citation23), the reproducibility of several biomarkers have been found to be poor (Citation24) and for several biomarkers, there has been found no effect of anti-inflammatory drug therapy on biomarker levels (Citation25).
Calgranulins are a group of calcium-binding proteins that have been shown to have anti-infective and anti-inflammatory properties. The group consists of three different proteins termed S100A8, S100A9 and S100A12. S100A8 and S100A9 forms a heterodimer termed calprotectin (Citation26, 27). This heterodimer is expressed in neutrophil granulocytes where it contributes to more than 45% of the cytosolic content, and in monocytes where it comprises less than 1% (Citation28). Secretion from neutrophil granulocytes has been proposed to be dependent on release from either cell disruption /cell death (Citation29) or via active non-classical secretion (Citation30). Calprotectin signaling is believed to be an upstream activator of (Tumor Necrosis Factor alpha) TNF-α, dependent on signaling through Toll-like Receptor 4 (TLR-4) (Citation31). The proposed anti-infective and anti-inflammatory properties of calgranulins involves a variety of functions e.g. chelation of divalent cations (Citation32), scavenging of tissue damaging reactive oxygen species (Citation33, 34), chemotaxis (Citation35) and direct antimicrobial function (Citation36, 37). Calgranulins involvement in inflammatory activity has been demonstrated in a wide range of acute and chronic inflammatory diseases, e.g. acute pancreatitis (Citation38), pulmonary tuberculosis (Citation39) and inflammatory bowel disease (Citation40). In rheumatoid arthritis serum levels have been found to be correlated to disease activity (Citation41).
Studies have also implicated calprotectin in respiratory health. Sputum proteomics have shown elevated levels of calgranulins in several inflammatory/suppurative respiratory diseases (Citation42). This is interesting as inhibition of S100A8 and S100A9 in animal models of pneumonia effectively suppresses neutrophil granulocytes and macrophage migration into the alveolar compartment of the lung (Citation43). The clinical impact of these findings has been studied in patients suffering from cystic fibrosis (Citation44). Gray and colleagues demonstrated that calprotectin levels predicted time to next exacerbation, that serum levels decreased in patients treated with antibiotics and that calprotectin taken at baseline and after treatment with antibiotics levels, was inversely correlated with FEV1.
Because both low-grade systemic inflammation and lower respiratory tract inflammation are considered pivotal in the pathogenesis and progression COPD, we hypothesize that patients with rapid disease progression have higher plasma levels of calprotectin, which in turn would lead to a higher rate of mortality.
Methods
Study population
575 COPD patients were enrolled (May 2001 to April 2004) in a randomized clinical trial studying the effect of Azithromycin 500 mg, 3 days per month in a 36-month period. Primary outcome was changes in post-bronchodilator FEV1. Secondary outcomes included number of hospital admissions, number of hospital days, mortality, quality of life, use of medication, prevalence of respiratory pathogens and prevalence of macrolide resistance. The trials registered at http://clinicaltrials.com - identifier NCT00132860 (accessed 21 July 2012). Inclusion and exclusion criteria are displayed in Box 1. Ethical permission for the study was obtained from The Regional Scientific Ethical Committee for Southern Denmark approval number 19990031.
Plasma samples
First, 460 baseline plasma samples were available from the original study for analysis. Samples were taken before first dose of study medication was taken. The samples were centrifuged, aliquoted, frozen and stored at –80°C until analysis. This was done in EDTA tubes, which inhibits release of the intracellular stores of calprotectin from neutrophils (Citation45). Plasma level of calprotectin was measured using a commercially available ELISA kit (Hycult Biotech, Uden, NL). The measurement was done at the M 7641 department, Rigshospitalet, Copenhagen, Denmark. All measurements were performed in duplicate.
Statistics
Patients were divided in three groups according to plasma calprotectin levels <100 ng/ml, 100–200 ng/ml, and >200 ng/ml, which was the primary variable of interest. The rationale for choosing the lower cut-off level was data from two recently published studies that included healthy individuals, and used the same method for measuring p-calprotectin (Citation46, 47). In the study by Ole Hartvig Mortensen et al. participants in the healthy control group rarely had plasma levels above 100 ng/ml (personal communication). The upper cut-off level was arbitrarily defined as twice the lower cut-off level. Because of the limited knowledge of distribution of p-calprotectin levels in COPD patient populations we also decided to do the analyses dividing patients in tertiles.
For all three groups, person-years of follow-up was computed from inclusion in the study (May 2001 to April 2004) until date of death, emigration, lost to follow-up, or we had passed January 31st 2011, which was considered the end of our study, whichever came first. Patient date of death was registered in Danish central CPR registry and was collected up until January 31st 2011. Follow-up were 99.1% complete at 10 years.
Kruskal-Wallis equality-of-populations rank test or the Chi-squared test were used to compare distribution of baseline variables between different levels of p-calprotectin as appropriate. A p-value < 0.05 was considered significant.
We used Cox regression to calculate hazard rates (HR) and 95% confidence intervals (CI). Our primary variable was p-calprotectin level. Before analysis we decided to include COPD stage, Charlson score (< 3 or higher), age (as a continuous variable), treatment group (Azithromycin vs. placebo) and gender as possible confounders. In addition to this a potential panel of confounders were tested in univariate analyses as both continuous and categorical variables and included in the final model if they were significant at a level of 0.25 or below. For this reason neutrophil count as a categorical variable was included in the final model, while active smoking status at baseline, C-reactive protein (CRP), pack-years and BMI (<20) were not.
The number of events, 236 mortalities, was well within the 10 events per variable, as suggested by Peduzzi et al. (Citation48). Variables were tested for interaction using likelihood ratio test statistics and there were found no significant interactions. Continual confounders were tested for linearity and this assumption was met in all cases. Proportional hazard assumptions were tested individually for each confounder. We used log-log plots and observed vs. expected plots for categorical confounders and observed vs. expected plots for continuous confounders. These tests showed a violation of the proportional hazard assumption on our main variable after 1700 days. No violation was displayed on the remaining covariates. Because of this we restricted our multivariate regression model to the time ≤ 1700 days in the final Cox regression model.
Results are presented as median with 95% confidence interval (CI) or interquartile range or rates as appropriate. All statistical analyses were carried out using STATA 11.1 (Stata Corp LP, College Station, TX, USA).
Results
The baseline characteristics of the patients stratified by p-calprotectin levels are summarized in . Our study population was characterized by having advanced COPD, a relatively high BMI and by a high median age. Distribution of baseline characteristics showed significantly higher neutrophil granulocyte counts with higher levels of plasma calprotectin. In addition there were relatively more men and fewer smokers in groups with higher plasma levels of calprotectin. Lower levels of calprotectin were associated with lower BMI.
Table 1. Distribution of variables at baseline stratified by calprotectin
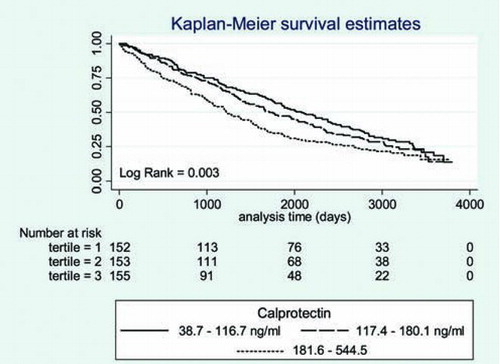
We computed a Kaplan–Meier curve to illustrate the time to death in the three different levels of calprotectin (). There was a significant association between higher p-calprotectin levels and increased mortality. Similar results were found when plotting calprotectin distributed by tertiles (). The 50% cumulative survival in the three different groups was 2234 days for calprotectin <100 ng/ml, 1651 days for calprotectin levels between 100–200 ng/ml and 1201 days for levels > 200 ng/ml. The Log-rank test was significant for both analyses.
Figure 1. Survival according to the level of calprotectin. Result of the log rank test is displayed in the lower left corner.
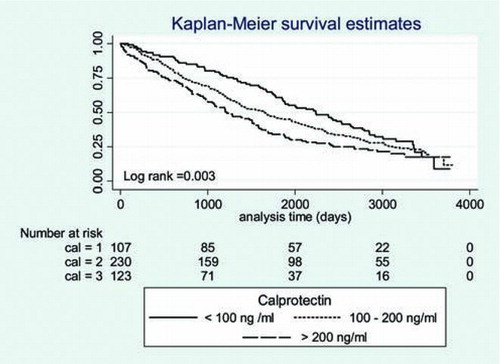
Figure 2. Survival according to the level of calprotectin stratified on tertiles. Result of the log Rank test for trend is displayed bottom left corner.
shows the results of multivariate Cox analysis up to 1700 days of follow-up. The model shows that high level of p-calprotectin is an independent risk factor for all-cause mortality. The risk of death rises from calprotectin 100–200 ng/ml [HR 1.56 (CI 95%: 1.03–2.38)] to [HR 2.02 (CI 95%: 1.27–3.19)] at levels above 200 ng/ml. This effect was comparable to that observed for severe COPD [HR 1.42 (CI 95%: 1.08–1.89)] and very severe COPD [HR 2.04 (CI 95%: 1.48–2.83)]. In addition to p-calprotectin levels and COPD severity, age [HR 1.04 (CI 95%: 1.03–1.06)] and elevated neutrophil granulocyte count [HR 1.30 (CI 95%: 1.02–1.65)] were also associated with increased hazard.
Table 2. Multivariate regression analysis of potential confounders –A multivariate Cox regression was performed to the time period before 1700 days
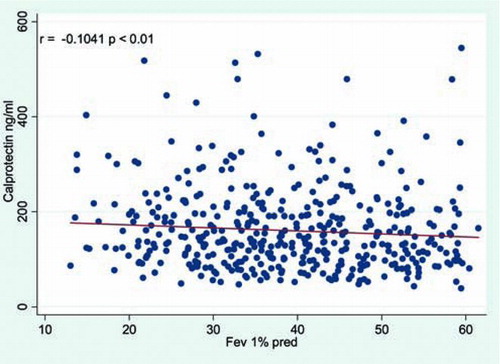
Finally a scatterplot () examining for a potential correlation between FEV1 pred.% and calprotectin was computed. This plot shows a significant, inverse relationship between baseline p-calprotectin levels and FEV1% pred.
Figure 3. Scatterplot showing p-calprotectin against FEV 1% predicted. Spearman correlation coefficient and the calculated p-value are displayed upper left corner.
Discussion
In patients diagnosed with moderate-to-very severe COPD, we found a significant association between baseline plasma levels of calprotectin and mortality during the first 5 years of follow-up. In patients with plasma levels above 200 ng/ml, the risk of dying within the initial 5 years of follow-up was more than twice as high, as that seen among patients with p-calprotectin levels below 100 ng/ml. At baseline there was a significant association between higher plasma levels of calprotectin and lower levels of pulmonary function. Although this does not prove causality, it could indicate a possible role of chronic neutrophil granulocyte mediated airway inflammation in disease progression.
We did not include the period after 1700 days in our multivariate Cox regression model because the model showed a violation of the proportional hazard assumption after this time. Therefore our data are only valid for the time period up to 1700 days. It is not surprising that a biological inflammatory marker such as p-calprotectin is unable to predict outcome in a probably highly selected group of survivors almost 5 years later.
We adjusted our analyses for two factors that are known to be associated with mortality in COPD i.e. FEV1% predicted (Citation15) and low BMI (Citation14). We found a correlation between FEV1% predicted and mortality, but were unable to demonstrate a correlation with low BMI (< 20), possibly because there were too few individuals with low BMI in our population (see ).
Although other studies have shown CRP plasma levels to be a marker of increased mortality in patients suffering from COPD we did not find such an association (Citation19, Citation49). This may reflect differences in methodology and patient populations. In contrast to these studies we did not use high sensitivity CRP assays, and our study population had overall more advanced COPD. In a study with a patient population similar to ours, the investigators did not find an association between CRP levels and mortality (Citation23), indicating that CRP may not be useful as a mortality marker in patients with advanced CRP. The usage of CRP in the monitoring of disease activity has also been questioned. In a recent study the reproducibility of CRP showed a high degree of variation limiting its scope as a marker of disease activity (Citation24).
Calprotectin has been shown to be a useful marker of inflammation in inflammatory bowel disease (fecal levels of calprotectin), and rheumatoid arthritis (serum levels), and is correlated to the inflammatory activity of these diseases (Citation41, Citation50). Likewise, a study conducted on patients with cystic fibrosis found that serum calprotectin lowered when patients were treated with antibiotics for an acute infectious episode (Citation44). The study also demonstrated that serum calprotectin levels measured after antibiotic treatment predicted time to next infectious episode. This is interesting as studies in mouse models have shown that inhibition of calprotectin impairs the recruitment of neutrophil granulocytes to the smaller airways (Citation43). Since calprotectin is almost completely derived from activated and dying neutrophil granulocytes, it is likely that plasma level of calprotectin reflects neutrophil granulocyte activity, and in the case of COPD most likely activity in the airway tissue and subsequent overspill into the systemic circulation.
We found an inverse correlation between FEV1% predicted and calprotectin levels (). This does not prove a causal relationship, but if future studies can show the same association between high levels of p-calprotectin and the rate of decline in pulmonary function this would strengthen the hypothesis that factors resulting in higher p-calprotectin levels are associated with more rapid airway destruction in COPD. In the same context it would also be of interest to determine whether calprotectin can be used to predict time to next exacerbation and exacerbation severity.
Neutrophil granulocyte count above the upper limits of our reference levels was associated with increased mortality. This is interesting as the same association was recently reported in another study (Citation22). In addition to this, there was a significant association between plasma levels of calprotectin and neutrophil granulocytes count (see ). This is not surprising as the major contributor of calprotectin in plasma is neutrophil granulocytes. We did in our data however adjust for neutrophil granulocyte count, and after this p-calprotectin level remained an independent and significant predictor of death. Calprotectin comprises a large proportion of the protein content in the cytosol of neutrophil granulocytes (Citation28).
Earlier studies have shown that only neutrophil granulocytes with disrupted membranes, dead neutrophil granulocytes or monocytes activated through protein kinase c dependent pathways release calprotectin (Citation29, 30). Thus, calprotectin levels most likely reflect the number of neutrophil granulocytes currently participating in inflammatory activity, and are not just markers of total numbers of neutrophil granulocytes.
It remains to be seen whether p-calprotectin is a marker of airway inflammation in COPD, but our results suggest that p-calprotectin levels can be used to identify a COPD population with shortened survival. Furthemore, p-calprotectin measurements may help to identify COPD phenotypes dominated by a high level of basal inflammation, a potential target for pharmacological intervention. We currently lack useful biomarkers that can predict progression of the disease and especially biomarkers that can be used to evaluate response to treatment, something that is currently sought after (Citation51).
Our study was fairly small, when compared to similar studies studying the effect of biomarkers and their effect on mortality (Citation19–21). This is somewhat balanced out by the very high number of events (fatalities) during the follow-up period in which 236 of our 460 patients died within the first 1700 days of the study. In addition to this the follow-up period was long and we had almost complete follow-up on our patients (99.4%).In addition to this the patient group was well characterized.
The study included patients suffering from moderate-to-severe COPD with multiple comorbidities, which was reflected by the very high mortality rate. The results of our study may not be the same in other populations with COPD. Another limitation of our study is that we only measured plasma calprotectin levels at one time point. It is possible that more frequent measurement will give a more complete picture of the interaction between p-calprotectin levels, disease activity and death.
To properly assess the baseline inflammatory state of these patients, it is imperative that no acute inflammatory event took place right before the blood samples were drawn. In our case we believe that this condition is met to some degree, by the inclusion criteria of our cohort, in the sense that no patient who had received antibiotics up until one week before inclusion were included, in addition to this, patients who had been hospitalized within the last month before inclusion was also excluded (Box 1).
Conclusions
In summary calprotectin levels above 100 ng/ml is associated with increased mortality in patients with moderate-to-very severe COPD in stable phase during the first 1700 days of observation. This coincided with a significant relationship between p-calprotectin levels and degree of airway obstruction suggesting a potential role for p-calprotectin in the monitoring of patients with COPD.
Declaration of Interests Statement
The study was supported by the Research Council of Southern Denmark, Overlægerådets Legatråd, the Danish Lung Association and the Danish Council for Independent Research (Grant no. 22-04-0636).
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
We are grateful for the continual support of the clinical staff of our departments. In particular we would like to thank technician Hanne Villumsen at department M 7641, Rigshospitalet for measuring p-calprotectin analysis.
References
- Lash TL, Johansen MB, Christensen S, Baron JA, Rothman KJ, Hansen JG, Hospitalization rates and survival associated with COPD: a nationwide Danish cohort study. Lung 2011; 189(1):27–35. Epub 2010/12/21.
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364(9434):613–620. Epub 2004/08/18.
- Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006; 367(9524):1747–1757. Epub 2006/05/30.
- Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. The European respiratory journal : official journal of the European Soc Clin Respir Physiol 2004; 23(6):932–946. Epub 2004/06/29.
- Stockley RA. Neutrophils and the pathogenesis of COPD. Chest 2002; 121(5 Suppl):151S–155S. Epub 2002/05/16.
- Cornwell WD, Kim V, Song C, Rogers TJ. Pathogenesis of inflammation and repair in advanced COPD. Semin Respir Crit Care Med 2010; 31(3):257–266. Epub 2010/05/25.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, The nature of small-airway obstruction in chronic obstructive pulmonary disease. New Engl J Med 2004; 350(26):2645–2653. Epub 2004/06/25.
- Moermans C, Heinen V, Nguyen M, Henket M, Sele J, Manise M, Local and systemic cellular inflammation and cytokine release in chronic obstructive pulmonary disease. Cytokine 2011; 56(2):298–304. Epub 2011/09/02.
- Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: A result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax 2010; 65(10):930–936. Epub 2010/07/16.
- Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2007; 119(5):1065–1071. Epub 2007/02/03.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002; 57(10):847–52. Epub 2002/09/27.
- Sethi S. Infectious etiology of acute exacerbations of chronic bronchitis. Chest 2000; 117(5 Suppl 2):380S–385S. Epub 2000/06/10.
- Hurst JR, Donaldson GC, Perera WR, Wilkinson TM, Bilello JA, Hagan GW, Use of plasma biomarkers at exacerbation of chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2006; 174(8):867–874. Epub 2006/06/27.
- Landbo C, Prescott E, Lange P, Vestbo J, Almdal TP. Prognostic value of nutritional status in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1999; 160(6):1856–1861. Epub 1999/12/10.
- Anthonisen NR, Wright EC, Hodgkin JE. Prognosis in chronic obstructive pulmonary disease. Amer Rev Respir Dis 1986; 133(1):14–20. Epub 1986/01/01.
- Fan VS, Curtis JR, Tu SP, McDonell MB, Fihn SD. Using quality of life to predict hospitalization and mortality in patients with obstructive lung diseases. Chest 2002; 122(2):429–436. Epub 2002/08/13.
- Nishimura K, Izumi T, Tsukino M, Oga T. Dyspnea is a better predictor of 5-year survival than airway obstruction in patients with COPD. Chest 2002; 121(5): 1434–1440. Epub 2002/05/15.
- Celli BR, Cote CG, Marin JM, Casanova C, Montes de Oca M, Mendez RA, The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. New Engl J Med 2004; 350(10):1005–1012. Epub 2004/03/05.
- Man SF, Connett JE, Anthonisen NR, Wise RA, Tashkin DP, Sin DD. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax 2006; 61(10):849–53. Epub 2006/06/02.
- Mannino DM, Valvi D, Mullerova H, Tal-Singer R. Fibrinogen, COPD and Mortality in a Nationally Representative U.S. Cohort. COPD August 2012; 9(4):359–366. Epub 2012/04/12.
- Sin DD, Miller BE, Duvoix A, Man SF, Zhang X, Silverman EK, Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2011; 183(9):1187–1192. Epub 2011/01/11.
- Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2012; 185(10):1065–1072. Epub 2012/03/20.
- de Torres JP, Pinto-Plata V, Casanova C, Mullerova H, Cordoba-Lanus E, Muros de Fuentes M, C-reactive protein levels and survival in patients with moderate to very severe COPD. Chest 2008; 133(6):1336–1343. Epub 2008/03/15.
- Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res 2011; 12(1):146. Epub 2011/11/08.
- Dentener MA, Creutzberg EC, Pennings HJ, Rijkers GT, Mercken E, Wouters EF. Effect of infliximab on local and systemic inflammation in chronic obstructive pulmonary disease: a pilot study. Respir Inter Rev thor Dis 2008; 76(3):275–282. Epub 2008/02/16.
- Striz I, Trebichavsky I. Calprotectin - a pleiotropic molecule in acute and chronic inflammation. Physiol Res/ Acad Scient Bohemoslovaca 2004; 53(3):245–253. Epub 2004/06/24.
- Hsu K, Champaiboon C, Guenther BD, Sorenson BS, Khammanivong A, Ross KF, Anti-infective protective properties of S100 calgranulins. Anti-Inflam Anti-Allergy Agents Med Chem 2009; 8(4):290–305. Epub 2010/06/05.
- Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem 1991; 266(12):7706–7713. Epub 1991/04/25.
- Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leuk Biol 2001; 70(1):130–134. Epub 2001/07/04.
- Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem 1997; v272(14):9496–9502. Epub 1997/04/04.
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 2007; 13(9):1042–1049. Epub 2007/09/04.
- Clohessy PA, Golden BE. Calprotectin-mediated zinc chelation as a biostatic mechanism in host defence. Scand J Immunol 1995; 42(5):551–6. Epub 1995/11/01.
- Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem 1999; 274(13):8561–8569. Epub 1999/03/20.
- Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem 2001; 276(36):33393–33401. Epub 2001/07/11.
- Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 2003; 170(6):3233–3242. Epub 2003/03/11.
- Sohnle PG, Collins-Lech C, Wiessner JH. The zinc-reversible antimicrobial activity of neutrophil lysates and abscess fluid supernatants. J Infect Dis 1991; 164(1):137–142. Epub 1991/07/01.
- Sohnle PG, Collins-Lech C, Wiessner JH. Antimicrobial activity of an abundant calcium-binding protein in the cytoplasm of human neutrophils. J Infect Dis 1991; 163(1):187–192. Epub 1991/01/01.
- Carroccio A, Rocco P, Rabitti PG, Di Prima L, Forte GB, Cefalu AB, Plasma calprotectin levels in patients suffering from acute pancreatitis. Digest Dis Sci 2006; 51(10):1749–1753. Epub 2006/09/12.
- Pechkovsky DV, Zalutskaya OM, Ivanov GI, Misuno NI. Calprotectin (MRP8/14 protein complex) release during mycobacterial infection in vitro and in vivo. FEMS Immunol Med Microbiol 2000; 29(1):27–33. Epub 2000/09/01.
- Manolakis AC, Kapsoritakis AN, Tiaka EK, Potamianos SP. Calprotectin, calgranulin C, and other members of the s100 protein family in inflammatory bowel disease. Digestive diseases and sciences. 2011; 56(6):1601–1611. Epub 2011/01/05.
- Andres Cerezo L, Mann H, Pecha O, Plestilova L, Pavelka K, Vencovsky J, Decreases in serum levels of S100A8/9 (calprotectin) correlate with improvements in total swollen joint count in patients with recent-onset rheumatoid arthritis. Arthr Res Ther 2011; 13(4): R122. Epub 2011/07/28.
- Gray RD, MacGregor G, Noble D, Imrie M, Dewar M, Boyd AC, Sputum proteomics in inflammatory and suppurative respiratory diseases. Amer J Respir Crit Care Med 2008; 178(5):444–452. Epub 2008/06/21.
- Raquil MA, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol 2008; 180(5):3366–3374. Epub 2008/02/23.
- Gray RD, Imrie M, Boyd AC, Porteous D, Innes JA, Greening AP. Sputum and serum calprotectin are useful biomarkers during CF exacerbation. J Cyst Fibr Euro Cyst Fibr Soc 2010; 9(3):193–198. Epub 2010/03/20.
- Dale I. Plasma levels of the calcium-binding L1 leukocyte protein: standardization of blood collection and evaluation of reference intervals in healthy controls. Scand J Clin Lab Invest 1990; 50(8):837–841. Epub 1990/12/01.
- Mortensen OH, Nielsen AR, Erikstrup C, Plomgaard P, Fischer CP, Krogh-Madsen R, Calprotectin—A novel marker of obesity. PloS One 2009; 4(10): e7419. Epub 2009/10/14.
- Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 2009; 17(11):2014–2018. Epub 2009/04/25.
- Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis. II. Accuracy and precision of regression estimates. J Clinical Epidemiol 1995; 48(12):1503–1510. Epub 1995/12/01.
- Dahl M, Vestbo J, Lange P, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. C-reactive protein as a predictor of prognosis in chronic obstructive pulmonary disease. Amer J Resp Crit Care Med 2007; 175(3):250–255. Epub 2006/10/21.
- Ricanek P, Brackmann S, Perminow G, Lyckander LG, Sponheim J, Holme O, Evaluation of disease activity in IBD at the time of diagnosis by the use of clinical, biochemical, and fecal markers. Scand J Gastroenterol 2011; 46(9):1081–1091. Epub 2011/05/31.
- Vestbo J, Rennard S. Chronic obstructive pulmonary disease biomarker(s) for disease activity needed—urgently. Amer J Respir Crit Care Med 2010; 182(7):863–864. Epub 2010/10/05.