
Abstract
Background: Exacerbation of COPD is a major risk factor for bad prognosis of COPD. A few plasma proteins have been discovered to associate with hospital admission due to exacerbation up to date. We tried to find new plasma biomarkers to predict the exacerbation of COPD. Methods: We examined the plasma of normal control (n = 8) and COPD stable (n = 8) and exacerbation (n = 8) using 2-Dimentional Electrophoresis. The differentially expressed protein spots were identified by MALDI-TOF. ELISA were performed for quantitative measurement of RARα in plasma from normal control (n = 37) and COPD (n = 35). Results: 17 proteins were differentially expressed in plasma between stable and exacerbation state in the subjects with COPD. Identification using MALDI-TOF showed that retinoic acid receptor alpha, ninein, isoform CRA_a, alpha-1 antitrypsin, fibrinogen gamma, tyrosyl-DNA phosphodiesterase 2, and T cell receptor delta chain were increased in exacerbation of COPD, while fibrin beta, Crystal Structure Of An Autoimmune Complex Between A Human Igm RF* And Igg1 Fc, transferrin, serpin peptidase inhibitor member 6, complement factor B preproprotein, Chain B, Crig Bound To C3c, and WD repeat-containing protein 1 isoform 1 were decreased. Quantitative measurement showed that RARα plasma levels significantly increased in exacerbation state compared to stable state of COPD (n = 14). In the plasma of stable state, the COPD subjects (n = 14) having more than 0.4 time/yr of admission had very high levels of RAR alpha protein and those (n = 11) having less than 0.4 times/yr of admission had the intermediate levels compared to those having no exacerbation (n = 10). ROC analysis of RAR alpha levels to frequency of admission showed an area under the curve of 0.844. A cut-off of 0.154 ng/ml of RAR alpha predicted hospital admission with a sensitivity of 71.4% and a specificity of 92.8%. Conclusion: The proteomic analysis of plasma indicates that alteration of several proteins may be associated with admission of COPD. Among them, plasma RAR alpha level may predict hospital admission with a sensitivity of 71.4% and a specificity of 92.8%.
Introduction
Chronic obstructive pulmonary disease (COPD) is a persistent progressive disease of the lower respiratory tract. According to World Health Organization estimates in 2004, 64 million people had COPD, resulting in 3 million deaths that represented 5% of all global deaths (Citation1). COPD is now a major cause of disability, and is the third leading cause of death in the U.S. (Citation2). Total deaths from COPD are predicted to increase by more than 30% in the next 10 years, becoming the third leading cause of death worldwide by 2030 without any interventions to cut risk factors, particularly exposure to tobacco smoke (Citation3).
Frequency of acute exacerbations, history of previous hospital admission and intervention in hospital intensive care units (ICUs) are regarded as important clinical manifestations related to mortality among patients with COPD (Citation4, 5). Thus, it is crucial to identify patients at risk of acute exacerbation and subsequent hospital admission and to implement preventative measures aimed at avoiding these complications.
Forced expiratory volume in one second (FEV1) is associated with mortality in patients with COPD (Citation5). The grades of FEV1, severity of symptoms and frequency of exacerbation in the preceding year showed an increase in risk of exacerbation, hospitalization and death (Citation6, 7). Additional clinical findings such as old age, co-morbidity (Citation8), a high degree of functional breathlessness and a low body mass index (BMI) (Citation9) were significantly associated with the risk of frequent exacerbations and hospital admissions.
As a composition of clinical parameters (BMI, obstruction, dyspnea, and exercise capacity; BODE), the BODE index predicts the risk of mortality in patients with COPD (Citation9). However, these clinical parameters may not provide sufficiently precise estimates in individual patients. This ambiguity has prompted the search for other indicators to predict hospital admission and mortality of COPD.
In peripheral blood, C-reactive protein (CRP), fibrinogen, α1-antitrypsin, eotaxin, interleukin (IL)-4, IL-8, monocyte chemotactic protein-1 (MCP-1), vascular endothelial growth factor (VEGF), tumor necrosis factor receptor II (TNFRII) and the soluble form of vascular cell adhesion protein I (VCAM-1) are reportedly related to the rapid decline of symptoms and lung functions and with the increased risk of future hospitalizations (Citation10–12). Although no single biomarker has been widely accepted, these are useful clinical predictors for the course of COPD (Citation13).
Searching for other COPD-specific proteins that will shed light on early detection, prognosis and treatment of COPD is important. Large-scale, high-throughput, and whole-proteome studies are needed to understand the contribution of the proteome to COPD. By comparing protein expression in plasma from patients with stable COPD and those with COPD exacerbation, we adopted differential-display proteomics to discover novel protein markers predictive exacerbation-related hospitalization of COPD patients.
Materials and methods
Subjects
The characteristics of normal control subjects (n = 37) and patients with COPD (n = 35) are summarized in . All subjects were Korean. Informed written consent forms were obtained from the participants in the study, and the protocol was approved by the local Ethics Committee of the hospital (SCHBC_IRB_2010-007). All patients were diagnosed and treated by physicians as defined by the Global Initiative for COPD (GOLD) guidelines (Citation14).
Table 1. Clinical characteristics of the study subjects
Briefly, clinical diagnosis of COPD was considered in subjects who were smokers or ex-smokers (>10 pack-years), who complained of dyspnea, chronic cough, or sputum production. In the present study, subjects with COPD were included when the initial post-bronchodilator FEV1/FVC was less than 0.70 and FEV1 was less than 70% of the predicted value. All subjects were followed up with symptom and lung-function tests for at least 3 years at regular 2-month intervals.
A stable COPD state was defined as those with no requirement for increased treatment above maintenance therapy for 30 days without any evidence of chest infection. Indications for hospital admission due to acute COPD exacerbation were as follows: marked increase in intensity of dyspnea, signs of hypoxia, appearance of peripheral edema, and infection signs including purulent sputum, fever, leukocytosis with elevated erythrocyte sedimentation rate (ESR) and CRP.
Etiologic agents causing exacerbation were identified in sputum by bacterial culture and respiratory virus polymerase chain reaction (PCR). Patients previously diagnosed with bronchial asthma, other lung diseases such as bronchiectasis, tuberculosis and malignancies, or other immune-compromising illnesses were excluded. Normal subjects were recruited from the general population who answered negatively to a screening questionnaire for respiratory symptoms, and had FEV1 >75% of the predicted value, FEV1/FVC >70%, and normal findings on a simple chest radiogram. Peripheral venous blood from all study subjects was collected in a tube containing ethylenediamine tetraacetic acid (EDTA, 0.020 g/10-mL blood) and plasma was immediately separated from whole blood by centrifugation for 15 min at 2000 rpm and aliquots were stored without protease at −80°C. Samples from patients with acute COPD exacerbation were collected immediately after diagnosis of exacerbation before clinical intervention.
Preparation of plasma and two-dimensional gel electrophoresis (2DE)
A total of 24 plasma samples from 8 normal controls, and stable state and exacerbation state of 8 COPD subjects were resolved by 2DE. Their clinical characteristics are summarized in . Immobiline DryStrips (24 cm, pH 3–10, pH 3–7; Amersham Biosciences, Uppsala, Sweden) were used for isoelectric focusing (IEF) on an IPGphor system (Amersham Biosciences) using 1 mg of protein as described previously (Citation15). After IEF, the proteins were separated on 5–18.5% SDS‑polyacrylamide gels, using the Ettan Dalt II system (AP Biotech) At the end of each run, the gel was stained with Coomassie brilliant blue G-250. Digitized images of the stained gels were analyzed using the ImageMaster 2D (ver. 4.0, Amersham Pharmacia Biotech). The Coomassie-stained spots were quantified on the basis of the normalized volume, i.e., the spot volume divided by the sum of all spot volumes.
Intra-gel digestion and mass spectrometric analysis
Differentially expressed protein spots were excised from gels, cut into smaller pieces, digested with trypsin (Promega) and subjected to matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) (Citation16). For MALDI-TOF mass spectrometry (MS) analysis, tryptic peptides were concentrated on POROS® R2 columns (Applied Biosystems). After successive column washes with 40% methanol, 100% acetonitrile, and 50 mM ammonium bicarbonate, the samples were applied to the column and eluted in 2 ml of cyano-4-hydroxycinnamic acid. MALDI-TOF spectra were obtained using a Voyager DE PROMALDI-TOF spectrometer (Applied Biosystems). Protein database searches were performed with the MSFit program (http://prospector.ucsf.edu/ucsfhtml3.4/msfit.htm) using monoisotopic peaks. A mass tolerance within 50 ppm was allowed for the first analysis; the system was subsequently recalibrated at 20 ppm using the lists of proteins obtained from the initial analysis. The spectra were internally calibrated using trypsin autolysis products. Peptide matching and protein searches against the Swiss-Prot and NCBI databases were performed using Mascot (http://www.matrixscience.com/) and Pro Found (http://prowl.rockefeller.edu/).
Measurement of plasma RAR-α levels by ELISA
Plasma samples from 37 normal controls and 35 patients with stable state COPD were subjected to sandwich enzyme-linked immunosorbent assay (ELISA) (USCN Life Science Inc., Wuhan, China), and 14 subjects with COPD exacerbation underwent additional follow-up RARα measurements. The lower detection limit was 0.063 ng/ml. Values below this limit were assumed to be zero for the purposes of statistical analysis.
Statistical analysis
Statistical analyses were performed using SPSS, version 13.0. The Mann–Whitney U-test (two independent samples) and Wilcoxon signed-rank sum test (two related samples) were applied to the differences in densities of the spots on 2DE and RAR-α concentrations. Discrimination was assessed using the area under the receiver operating characteristic (ROC) curve to evaluate how well the model distinguished patients admitted to the hospital in the previous three years from those who were not admitted. All data are expressed as mean values and SD, and significance was defined as a value of p < 0.05.
Results
Clinical characteristics of the study subjects
Clinical characteristics are summarized in . We selected male subjects with COPD and normal controls. All subjects with COPD were smokers or ex-smokers (>10 pack-years) while 10% of normal controls were non-smokers. All of the normal control subjects had post-bronchodilator FEV1/FVC values of >0.7, whereas all subjects with COPD had values of <0.7. BMI was slightly lower in the COPD group than that in the normal control group, but the difference was minimal (p > 0.05). 2DE analysis of plasma from subjects with COPD (n = 8) showed markedly decreased FEV1 in those with COPD exacerbation compared to those with stable COPD (p < 0.001).
A total of 35 subjects with COPD were stratified based on hospital admission due to exacerbation during the follow-up period. Of these, 21 subjects admitted and had significantly lower FEV1, FVC and FEV1/FVC ratio compared to those having COPD without admission (n = Citation14). Among the 21 subjects with COPD, 14 ones with COPD exacerbation were subject(ed) to follow-up plasma samples drawn upon admission. The causes of acute exacerbation were bacterial infection (n = 10) and viral infection (n = 4). The causative agents were not documented in the other cases of exacerbation (n = 7).
2-D electrophoresis and plasma protein identification by MALDI-TOF in patients with stable COPD and COPD exacerbation
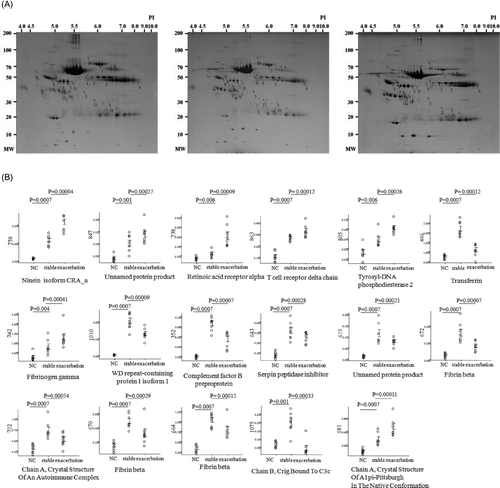
After Coomassie blue staining, protein spots from the normal control subjects (median number, 561; range, 430–730), patients with stable state COPD (median, 555; range, 452–653) and subjects with COPD exacerbation (median, 566; range, 477–696) were detected in 1-mg plasma protein. In plasma from eight subjects with stable COPD versus those with exacerbations, 17 proteins showed significant differences in relative intensity (). The relative intensities (%) of the spots are expressed in ; the locations of the identified protein spots are shown in the master image. Among them, the intensities of 7 spots were significantly increased and 10 were decreased in patients with COPD exacerbation compared to those with stable COPD ().
Figure 1. Photographs of two-dimensional electrophoresis (2-DE) separation of plasma obtained from eight normal controls and stable and exacerbation state of eight COPD subjects. The plasma proteins (1 mg) were focused on a pH 4–10 gradient strip and then separated on an 7.5–20% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis, stained, and visualized as described in the Methods section. Protein spots identified by MALDI-TOF (arrows) are marked by their spot numbers.
Identification of proteins by MALDI-TOF
The above-mentioned 17 spots were excised from the gels, incubated with trypsin for in-gel digestion, and identified by MALDI-TOF. The results are summarized in supplementary as well as . The proteins having highest matching score among the all candidate ones are presented in .
Table 2. List of differentially expressed proteins identified by MALDI-TOF
Retinoic acid receptor alpha (RAR-α, spot 13), ninein (GSK3B interacting protein) isoform CRA_a (spot 5), alpha-1 antitrypsin (A1pi, spot 8), fibrinogen gamma (spot 1), tyrosyl-DNA phosphodiesterase 2 (spot 14), and T cell receptor delta chain (spot 15) were increased in the plasma of subjects with COPD exacerbation, whereas fibrin beta (spots 2, 3, 4), crystal structure of an autoimmune complex between a human IgM RF* and IgG1 Fc (COAC, spot 6), transferrin (spot 7), Serpin peptidase inhibitor member 6 (Serpin B6, Spot 9), complement factor B preproprotein (spot 10), chain B, Crig bound to C3c (spot 11), and WD repeat-containing protein 1 isoform 1 (spot 12) were decreased. Fibrinogen gamma and fibrin beta are coagulation proteins; ninein (GSK3B interacting protein) is a cytoskeleton protein; COAC and transferrin are immune response proteins; complement factor B Chain B, Crig bound to C3c, A1pi, and Serpin peptidase inhibitor 6 are inflammatory proteins; And WD repeat containing protein 1, RAR-α, tyrosyl-DNA phosphodiesterase 2, and T cell receptor delta chain are signal transduction modifiers.
Changes in RAR-α levels in plasma from patients with stable COPD compared to those with COPD exacerbation
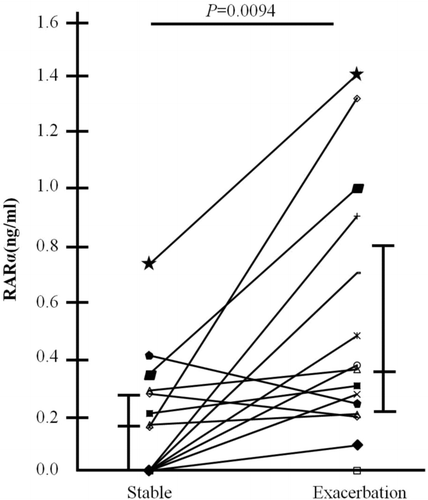
To validate the changes in retinoic acid receptor alpha (RAR-α) protein levels in the exacerbation from stable state, RARα was measured in plasma of patients with stable and exacerbated COPD (n = 15, ). Among them, 12 subjects showed increased RAR-α levels when exacerbated compared to stable COPD. The mean RAR-α concentration was elevated twofold in the exacerbation state as compared with that in the stable state.
Figure 2. Changes of retinoic acid receptor alpha level in plasma from stable to exacerbation state of the patients with COPD (n = 15).
Relation of RAR-α levels at stable state with frequency of hospitalization
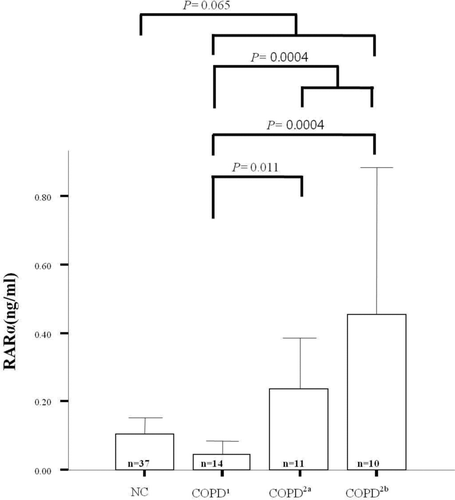
To determine whether RAR-α protein levels in the stable state were associated with the frequency of hospital admission due to exacerbations, RAR-α levels in normal controls (n = 37) and patients with COPD (n = 35) were compared (). Patients with COPD were stratified by the frequency of hospital exacerbation. Admission-free subjects with COPD (n = 14) had comparable levels of RAR-α to those of normal controls (p > 0.05). The subjects with COPD who were admitted to hospital (n = 21) had significantly higher levels of RAR-α compared to admission-free COPD patients (p = 0.0004). Subjects with COPD who were admitted to hospital more than 0.4 times /yr (n = 10) had very high levels of RAR-α protein, while those admitted fewer than 0.4 times /yr (n = 11) had intermediate levels.
Figure 3. Plasma retinoic acid receptor alpha level in nomal controls and stable state of the patients with COPD. (COPD: represents no exacerbation, COPD2a: represents the exacerbation frequency of less than 0.4/year, COPD2b: represents the exacerbation frequency of more than 0.4/year).
Receiver operating curve (ROC) analysis of RAR-α protein levels as a predictor of exacerbation in COPD
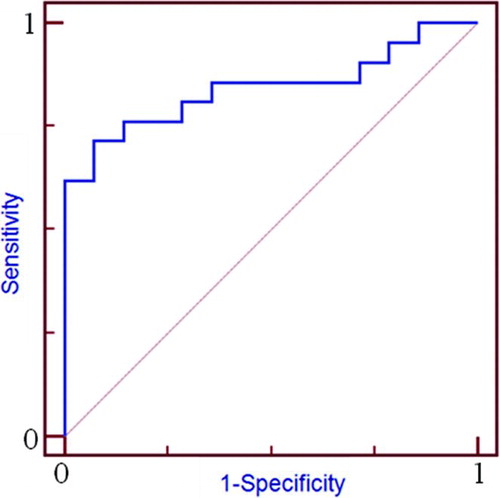
ROCs were created for RAR-α levels in subjects with stable COPD (n = 35, ). Using the plasma level variance estimates, we calculated 95% confidence intervals (CI). ROC analysis of RAR-α gave an area under the curve of 0.844 (). A cut-off of 0.154 ng/ml RAR-α predicted hospital admission with a sensitivity of 71.4% and a specificity of 92.8%.
Figure 4. Receiver operating curve (ROC) analysis of RAR alpha protein levels as a predictor of exacerbation in COPD. A cut-off of 0.154 ng/ml RAR-α predicted hospital admission with a sensitivity of 71.4% and a specificity of 92.8% (area under the curve of 0.844).
Discussion
In the present study, 17 protein spots were found to be differentially expressed in the plasma of subjects with COPD exacerbation compared to those with stable COPD. Among them, we investigated RAR-α because retinoic acid (RA), a ligand for RAR-α, is associated with lung development and repair (Citation17). Using plasma from normal controls and COPD subjects, the concentrations of RAR-α were twofold higher in patients with COPD exacerbation compared with those with stable COPD. Furthermore, the levels in subjects with stable COPD were correlated with the frequency of hospital admission. These data indicate that the plasma concentration of RAR-α is a promising marker for predicting hospital admission for COPD. To our knowledge, this is the first study to relate RAR-α levels to hospital admission due to COPD exacerbation.
RA is required throughout life for the maintenance of lung alveoli. Experimental rats lacking dietary retinol lose alveoli and show features of emphysema (Citation18). All-trans retinol (ATRA), the transported form of vitamin A, can be reversibly converted into retinyl esters, which serve as the storage form in mainly the liver, but also in the lungs (Citation19). In an experimental study, ATRA down-regulated MMP-9 and up-regulated TIMP-1 expression in alveolar macrophages (Citation20).
Furthermore, administration of ATRA to patients with emphysema reduced plasma MMP-9 and MMP-9 enzyme activity (Citation21). Thus, ATRA is believed to restore the protease–anti-protease imbalance in patients with COPD. Notably, smoking increases plasma levels of acid retinoids and retinol, both of which can replenish pulmonary retinoid levels (Citation22), whereas oxidative stress originating from smoke exposure reduces the expression of RAR (Citation23). In the present study, to exclude the effect of smoking, we matched 90% of the normal controls with ex-smokers and smokers because all subjects with COPD were either ex- or current smokers.
Because there is an increase in apoptotic alveolar epithelial and endothelial cells in the lungs in COPD patients, it is likely that apoptosis of structural cells in the lung is an important upstream event in the pathogenesis of COPD. (Citation24). The process of apoptosis is not counterbalanced by an increase in the proliferation of these structural cells, which leads to the destruction of lung tissue and the development of emphysema (Citation25). Because apoptosis can occur outside the lungs, including in skeletal muscle (Citation26) and circulating T cells (Citation27), intra-cytoplasmic proteins such as RAR can be detected in peripheral blood. The source of elevated plasma RAR in patients with COPD exacerbation remains unclear. However, considering the involvement of retinoic acids with lung development and the increased apoptosis of intra- and extra-lung tissues in COPD patients, the increased RAR levels may originate from the apoptotic cells, which is accentuated in patients with COPD exacerbation.
A systemic release of apoptosis-specific proteins as markers for increased cellular turnover is accompanied by progression of COPD. Serum content of the apoptotic end-products caspase-cleaved cytokeratin-18 and histone-associated DNA fragments increases in the peripheral blood of patients with COPD (Citation28). In the present study, ninein was significantly increased in patients with COPD exacerbation. Interestingly, ninein gene levels increase in the small airway epithelium of smoking patients with COPD (Citation29). Ninein is one of the proteins important for centrosomal function and anchoring the microtubules in epithelial cells. The airway epithelium is the first line of defense against cigarette smoke, and the airway epithelium of smokers demonstrates marked changes in gene expression compared to nonsmokers (Citation30, 31).
Considering that COPD exacerbation starts in the airway epithelium and lung parenchyma, the molecules released from the epithelium may be increased in the peripheral blood of the subjects with COPD exacerbation. WD repeat-containing protein 1 is a cytosolic protein involved in the disassembly of actin filaments. The assembly and maintenance of the microtubule-dependent cilia and flagella structures depends on intraflagellar transport (IFT). Anterograde IFT serves as a means to deliver proteins within cilia, retrograde IFT is used to recycle the IFT machinery back to the ciliary base (Citation32). Ciliary beating is depressed in the nasal cilia from subjects with COPD (Citation33). Although the molecular mechanism is not yet clear, low levels of WD repeat-containing protein 1 might be related to COPD exacerbation via decreased cilia movement. Tyrosyl-DNA phosphodiesterase 1 repairs DNA damage that is induced by topoisomerases I and II (Citation34). Likewise, the elevation of these proteins might be derived from increased apoptosis in patients with COPD.
In the present study, T cell receptor delta chain was increased in patients with COPD and was elevated even further in subjects with COPD exacerbation. T-lymphocytes are key inflammatory effector and regulatory cells that participate in the inflammatory response in COPD.TCR-γδ lymphocytes account for only ∼5% of the total T-cell subpopulation. However, they play a key role in the mucosal homeostasis and reparative response to tissue damage (Citation35). The percentage of circulating and BAL γδ T-lymphocytes is higher in smokers with normal lung function than in nonsmokers; this response is blunted in patients with COPD (Citation36). However, T-cell receptor delta chain levels were increased in the peripheral blood of patients in the present study, which suggests differences in the function of T-cell receptor delta chain in patients with COPD.
In this study, A1pi (alpha-1-antitrypsin, SERPINA1) was increased while Serpin B6 was decreased in COPD exacerbation compared to stable COPD. Serpins are a group of proteins with similar structures that were first identified as protease inhibitors. A1pi forms complexes predominantly with elastase, but also with trypsin, chymotrypsin, thrombin, and bacterial proteases (Citation37). In the A1pi deficiency, neutrophil elastase breaks down elastin, resulting in emphysema. Elevation of A1pi has been reported in both stable and exacerbated COPD (Citation37), which was confirmed in the present study.
Serpin B6 is a serine proteinase inhibitor present in the cytosolic fraction of myelomonocytes, capillary endothelial cells, platelets, and epithelial cells. Serpin B6 binds to endogenous membrane-associated serine proteinase such as cathepsin G (Citation38). When vacuole integrity is breached during phagocytosis, Serpin B6 rapidly inactivates any proteinases released into the interior of the cell. Control of intracellular cathepsin G may be particularly important, because it activates the proapoptotic proteinase, caspase-7 (Citation39). Thus, the lower levels of Serpin B6 reported in the present study may lead to increased apoptosis of lung cells or systemic organs. (Citation40).
In the coagulation pathway, fibrinogen gamma was elevated, whereas fibrin beta was decreased in subjects with COPD exacerbation. These are acute-phase glycoproteins, synthesized primarily in the liver and converted by thrombin into fibrin during blood coagulation. There is strong evidence of an association between fibrinogen levels and the presence of COPD and mortality (Citation12). Fibrinogen is a dimer consisting of three polypeptide chains, termed alpha, beta and gamma (Citation41). While all three chains are essential for the normal function of fibrinogen due to their intertwined three-dimensional structure, only the gamma chain contains a number of sites that interact with clotting factors, growth factors, and integrins (Citation42). In the present study, spots 2, 3, and 4 were identified as fibrin beta. Spots 3 and 4 are likely post-translational modifications of spot 2, because they have pIs of 5.95 and 6.25 whereas spot 2 has a pI of 6.25. All were significantly decreased in plasma from patients with COPD exacerbation compared to those with stable COPD. Why exacerbation was associated with a decreased amount of fibrinogen beta is not clear.
Transferrin, an iron-binding blood plasma glycoprotein that controls the level of free iron in biological fluids, is associated with the innate immune system. Decreased transferrin concentrations in the lower respiratory tract may decrease defenses against oxidant injury and bacterial infection in patients with respiratory failure due to COPD. Visceral protein stores are assessed using blood parameters such as albumin, prealbumin, or transferrin. Protein calorie malnutrition is likely to be present in patients with COPD and acute respiratory failure (Citation43). In the present study, BMI was significantly lower in patients with COPD, and was decreased slightly in those with COPD exacerbation, suggesting impaired nutritional status in patients with COPD.
Inflammation is one of the key processes in the pathogenesis of COPD, characterized by increased recruitment of inflammatory cells and up-regulation and down-regulation of inflammatory mediators in the bronchoalveolar lavage fluid, sputum, and lung tissues from patients with COPD (Citation44). Systemic inflammatory hyporeactions occurred during acute exacerbation, even though it remains unclear whether the hyporeaction results in or from acute exacerbation.
Plasma CRP concentration, serum IL-6, and copeptin in the presence of a major exacerbation symptom, is useful for confirmation of COPD exacerbation. (Citation45–47), although changes in these mediators were not identified in the present study. This discrepancy may originate from the use of different types of COPD exacerbation between our study and other studies. The definition of exacerbations in COPD remains controversial (Citation48). When reliant solely on symptoms or lung function, criteria are too subjective, either on the part of the patient or the physician.
In the present study, we enrolled subjects with COPD exacerbation who were admitted to the hospital. This suggests that the subjects in the present study experienced more severe COPD exacerbations than those in the other studies (Citation16, Citation47). A few studies using differential proteomics including 2DE and mass spectrometry have been published in COPD. Terracciano et al. (Citation49) found the alteration of Human alpha-defensins (human neutrophil peptide (HNP1, HNP2, HNP3) and three C-terminal amidated peptides, one of which is phosphorylated on serine in sputum of COPD compared to normal controls.
In lung tissues, Lee et al. (Citation50) found that MMP-13, transcription factor activator protein-4, peroxiredoxin-6, nonesterified fatty acid-interacting nuclear protein NIP30, hepatoma-derived growth factor, regulator of G protein signaling 16, thioredoxin-like protein 2, and histamine-releasing factor were elevated in the subjects with COPD compared to non-smoker normal controls. A panel of plasma biomarkers (a2-macroglobulin, haptoglobin, ceruloplasmin, and hemopexin) was found to discriminate the patients with COPD and normal controls (Citation51).
Recently, Pastor et al. (Citation52) identified a total of 16 oxidative stress regulatory proteins including CAT, PRDX1, PRDX2, and PRDX5 to express differentially in BAL samples from COPD patients as compared with control group. The spots identified in these studies were not detected in the present study except fibrin, fibrinogen and complement factor B. The reason for the low frequency of overlapping in the discovered proteins between the studies may be originated from the different types of samples between them. The limitation of the present study is a lack of replication data in different cohort samples. 2DE using plasma without removal of high abundant proteins is the other limitation. The presence of highly abundant plasma proteins, including albumin, IgG, antitrypsin, IgA, transferrin, and haptoglobin, would be a technical limitation of the present study, impairing the ability to identify proteins whose spots in 2DE images overlapped with these highly abundant proteins.
In summary, using 2DE and MALDI-TOF, we identified 17 proteins in plasma that were differentially expressed in patients with stable and exacerbated COPD. These identified proteins are promising candidate plasma biomarkers for prediction of COPD exacerbation. Quantitative measurement of one candidate biomarker, RAR-α, was performed using plasma from normal controls and subjects with COPD. In plasma from patients with stable COPD, RAR-α protein levels were related to the presence and frequency of hospital admission due to COPD exacerbation. A cut-off of 0.154 ng/ml of RAR-α showed a sensitivity of 71.4% and a specificity of 92.8% for hospital admission due to exacerbation. Our findings provide new insights into protein markers related to exacerbation of COPD.
Table s1. Supplementary Analysis of Each spot protein according to score
Declaration of Interests Statement
This work was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health, Welfare and Family Affairs, Republic of Korea (A090548) and the plasma samples were generously provided by a Collaborative Biobank of Korea in Soonchunhyang University Bucheon Hospital.
The study was supported by Soonchunhyang University Research Fund. For a certificate, see: http://www.textcheck.com/certificate/ ox3Vhg.
Acknowledgments
Jeong-Seok Heo and Jong-Sook Park equally contributed equally as the first author.
The authors thank the editors from textcheck.com, both native speakers of English, for their editing for grammar and typographic errors.
References
- CD Mathers, DM Fat, JT Boerma. The Global Burden of Disease: 2004 Update. Geneva: World Health Organization; 2008.
- DW Brown, JB Croft, PhD, KJ Greenlund, PhD, WH Giles, MD. Deaths from Chronic Obstructive Pulmonary Disease-United States, (2000–2005). Centers for Disease Control and Prevention. Atlanta, USA 2008 / 57(45); 1229–1232.
- Mathers CD, Loncar D. Projection of global mortality and burden of diseases from 2002 to 2030. PloS Med 2006; 3:e442.
- Burrows B, Earle RH. Course and prognosis of chronic obstructive lung disease: A prospective study of 200 patients. N Engl J Med 1969; 280: 397–404.
- B Celli, GL Snider, J Heffner. Definitions, epidemiology, pathophysiology, diagnosis, and staging. Am J Respir Crit Care Med 1995; 152: Suppl: S78–S83.
- Hurst JR, Vestbo J, Anzueto A, : Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–38.
- Decramer M, Celli B, Kesten S, Lystig T, Mehra S, Tashkin DP. Effect of tiotropium on outcomes in patients with moderate chronic obstructive pulmonary disease (UPLIFT): a prespecified subgroup analysis of a randomized controlled trial. Lancet 2009; 374:1171–1178.
- Gudmundsson G, Gislason T, Lindberg E, Hallin R, UlrikCS, Brøndum E, Nieminen MM, Aine T, Bakke P, Janson C. Mortality in COPD patients discharged from hospital: the role of treatment and co-morbidity. Respir Res 2006; 7:109
- Celli BR, Cote CG, Marin JM, The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in Chronic Obstructive Pulmonary Disease. N Engl J Med 2004; 350:1005–1012.
- .10. Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 2004; 59:574–580.
- Pinto-Plata V, Toso J, Lee K, Parks D, Bilello J, Mullerova H, De Souza MM, Vessey R, Celli B. Profiling serum biomarkers in patients with COPD: Associations with clinical parameters. Thorax 2007; 62:595–601.
- Dahl M, Tybjaerg-Hansen A, Vestbo J, Elevated plasma fibrinogen associated with reduced pulmonary function and increased risk of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164:1008–1125.
- Koutsokera A, Stolz D, Loukides S, Kostikas K. Systemic biomarkers in exacerbations of COPD: The evolving clinical challenge. CHEST 2012; 141(2):396–405.
- Global Initiative for Chronic Obstructive Lung Disease –GOLD [home page on the Internet]. Bethesda: Global Initiative for Chronic Obstructive Lung Disease [cited 2012 Mar 13]. Global strategy for the diagnosis, management and prevention of COPD - Revised 2011. [Adobe Acrobat document, 90p.]. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2011_Feb21.pdf
- Rhim T, Choi YS, Nam BY, Uh ST, Park JS, Kim YH, Paik YK, Park CS. Plasma protein profiles in early asthmatic responses to inhalation allergen challenge. Allergy 2009; 64(1):47–54.
- Shevchenko A, Wilm M, Vorm O, Mann M, Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 1996; 68:850–858.
- Maden M, Hind M. Retinoic acid in alveolar development, maintenance and regeneration. Philos Trans Roy Soc Lond B: Biol Sci 2004; 359:799.
- McGowan S, Jackson SK, Jenkins-Moore M, Dai HH, Chambon P, Snyder JM. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am J Respir Cell Mol Biol 2000; 23:162–167.
- Vogel S, Gamble MV, Blaner WS. Biosynthesis, absorption, metabolism and transport of retinoids. In: Retinoids. The Biochemical and Molecular Basis of Vitamin A and Retinoid Action. Nau H, Blaner WS, eds.; Berlin: Springer-Verlag, 1999.
- Frankenberger M, Hauck RW, Frankenberger B, Haussinger K, Maier KL, Heyder J, Ziegler-Heitbrock HW. All trans-retinoic acid selectively down-regulates matrix metalloproteinase-9 (MMP-9) and up-regulates tissue inhibitor of metalloproteinase-1 (TIMP-1) in human bronchoalveolar lavage cells. Mol Med 2001; 7:263.
- Mao JT, Tashkin DP, Belloni PN, Baileyhealy I, Baratelli F, Roth MD. All-trans retinoic acid modulates the balance of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in patients with emphysema. Chest 2003; 124:1724–1732.
- Ross AC. Retinoid production and catabolism: role of diet in regulating retinol esterification and retinoic acid oxidation. J Nutr 2003; 133: 291S–196S.
- Wang XD, Liu C, Bronson RT, Smith DE, Krinsky NI, Russell M. Retinoid signaling and activator protein-1 expression in ferrets given beta-carotene supplements and exposed to tobacco smoke. J Natl Cancer Inst 1999; 91:60–66.
- Gordon C, Gudi K, Krause A, Sackrowitz R, Harvey B-G, Strulovici-Barel Y, Mezey JG, Crystal RG. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med 2011; 184:224–232.
- Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 2006; 7:53
- Barreiro E, Ferrer D, Sanchez F, Minguella J, Marin-Corral J, Martinez-Llorens J, Lloreta J, Gea J. Inflammatory cells and apoptosis in respiratory and limb muscles of patients with COPD. J Appl Physiol 2011 Sep; 111(3):808–817.
- Lim SC, Ju JY, Chi SY, Ban HJ, Kwon YS, Oh IJ, Kim KS, Kim YI, Kim YC. Apoptosis of T lymphocytes isolated from peripheral blood of patients with acute exacerbation of chronic obstructive pulmonary disease. Yonsei Med J 2011; 52(4):581–587.
- Hacker S, Lambers C, Pollreisz A, Increased soluble serum markers caspase-cleaved cytokeratin-18, histones, and ST2 indicate apoptotic turnover and chronic immune response in COPD J Clin Lab Anal 2009; 23: 372–379.
- Tilley AE, O'Connor TP, Hackett NR, Strulovici-Barel Y, Salit J, Biologic phenotyping of the human small airway epithelial response to cigarette smoking. PLoS ONE 2011; 6(7):e22798.
- Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA 2004; 101:10143–10148.
- Pierrou S, Broberg P, O'Donnell RA, Pawlowski K, Virtala R, Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175:577–586.
- Blacque OE, Li C, Inglis PN, Esmail MA, Ou G, Mah AK, Baillie DL, Scholey JM, Leroux MR. The WD Repeat-containing Protein IFTA-1 Is required for retrograde intraflagellar transport. Mol Biol Cell 2006; 17(12):5053–5062.
- Yaghi A, Zaman A, Cox G, Dolovich MB. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir Med 2012; 106(8):1139–1147.
- Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J Biol Chem 2012; 287(16):12848–12857. Epub 2012 Feb 27.
- Jameson J, Witherden D, Havran WL. T-cell effector mechanisms: gammadelta and CD1d-restricted subsets. Curr Opin Immunol 2003; 15:349–353.
- Pons J, Sauleda J, Ferrer JM, Barceló B, Fuster A, Regueiro V, Julià MR, Agustí AG. Blunted gamma delta T-lymphocyte response in chronic obstructive pulmonary disease. Eur Respir J 2005; 25(3):441–446.
- Cox DW. Alpha-1 antitrypsin deficiency. In: Scriver CR, Beaudet AL, Sly WS, , eds. The Metabolic and Molecular Bases of Inherited Disease. 8th Ed.; Vol. 4. New York: McGraw-Hill, 2001: 5559–5584.
- Scott FL, Hirst CE, Sun J, Bird CH, Bottomley SP, Bird PI. The intracellular serpin proteinase Inhibitor 6 Is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, Cathepsin G. Blood 1999; 93:2089–2097.
- Zhou Q, Salvesen GS. Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem J 1997; 324:361.
- Scott FL, Coughlin PB, Bird C, Cerruti L, Hayman JA, Bird P. Proteinase inhibitor 6 cannot be secreted, which suggests it is a new type of cellular serpin. J Biol Chem 1996; 271:1605.
- Farrell DH. Pathophysiologic roles of the fibrinogen gamma chain. Curr Opin Hematol 2004; 11:151–155.
- Mosesson MW. Fibrinogen gamma chain functions. J Thromb Haemost 2003; 1:231–238.
- Schols A, Mostert R, Soeters P, Greve LH, Wouters EF. Inventory of nutritional status in patients with COPD. Chest 1989 Aug; 96(2):247–249.
- MacNee W. Pathogenesis of Chronic Obstructive Pulmonary Disease. Proc Am Thorac Soc 2005; 2:258–266.
- Perera WR, Donaldson GC, Wedzicha JA, Inflammatory changes, recovery and recurrence at COPD exacerbation. Eur Respir J 2007; 29:527–534.
- Hurst JR, Donaldson GC, Wedzicha A, Use of plasma biomarkers at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 174:867–874.
- Seligman R, Papassotiriou J, Morgenthaler NG, Meisner M, Teixeira PJZ. Copeptin, a novel prognostic biomarker in ventilator-associated pneumonia. CHEST 2007; 131:1058–1067.
- Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest 2000; 117:398S–401S.
- Terracciano R, Preiano M, Peptidome profiling of induced sputum by mesoporous silica beads and MALDI-TOF MS for non-invasive biomarker discovery of chronic inflammatory lung diseases. Proteomics 2011, 11, 3402–3414.
- Lee EJ, In KH, Proteomic analysis in lung tissue of smokers and COPD patients. CHEST 2009; 135:344–352.
- Verrills NM, Irwin JA, He XY, Identification of novel diagnostic biomarkers for asthma and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2011; 183:1633–1643.
- Pastor MD, Nogal A, Identification of oxidative stress related proteins as biomarkers for lung cancer and Chronic Obstructive Pulmonary Disease in bronchoalveolar lavage. Int J Mol Sci 2013; 14:3440–3455.