
Abstract
The search for COPD biomarkers has largely employed a targeted approach that focuses on plasma proteins involved in the systemic inflammatory response and in lung injury and repair. This proof of concept study was designed to test the idea that an open, unbiased, in-depth proteomics approach could identify novel, low abundance plasma proteins i.e., ng/mL concentration, which could serve as potential biomarkers. Differentially expressed proteins were identified in a discovery group with severe COPD (FEV1 <45% predicted; n = 10). Subjects with normal lung function matched for age, sex, ethnicity and smoking history served as controls (n = 10). Pooled plasma from each group was exhaustively immunodepleted of abundant proteins, d separated by 1-D gel electrophoresis and extensively fractionated prior to LC-tandem mass spectroscopy (GeLC-MS). Thirty one differentially expressed proteins were identified in the discovery group including markers of lung defense against oxidant stress, alveolar macrophage activation, and lung tissue injury and repair. Four of the 31 proteins (i.e., GRP78, soluble CD163, IL1AP and MSPT9) were measured in a separate verification group of 80 subjects with varying COPD severity by immunoassay. All 4 were significantly altered in COPD and 2 (GRP78 and soluble CD163) correlated with both FEV1 and the extent of emphysema. In-depth, plasma proteomic analysis identified a group of novel, differentially expressed, low abundance proteins that reflect known pathogenic mechanisms and the severity of lung remodeling in COPD. These proteins may also prove useful as COPD biomarkers.
Keywords :
Introduction
Chronic obstructive pulmonary disease (COPD), a disease characterized by lung inflammation and remodeling but which also has systemic effects (Citation1–3), is an increasingly important public health concern in the United States and worldwide (Citation4,5). In the United States, COPD is the third-leading cause of death (Citation6,7) Epidemiological studies indicate that the disease affects 24 million Americans of whom one half remain undiagnosed (Citation4,5).
Recently, considerable efforts have been made to identify biomarkers that can be used to identify pathogenetic mechanisms; serve as surrogate markers for treatment responses; and to distinguish COPD phenotypes (Citation8–12). Studies to date have generally utilized a targeted approach in which proteins of interest have been assayed based on their putative role in the disease using antibody based assays either as individual candidates i.e., ELISA or as commercially available multiplex arrays (Citation9–11,Citation13,14). In general, cytokines, chemokines, metallo-proteinases, tissue inhibitors of metallo-proteinases (TIMPs), etc., have been measured and found to differentiate COPD from controls (Citation11,Citation13,Citation15,16). In addition, highly abundant, acute phase reactants produced by the liver which reflect the systemic inflammatory component of the disease e.g., C-reactive protein (CRP), fibrinogen, haptoglobin, etc., differentiate COPD from controls (Citation14,Citation17–21).
The directed approach has several advantages including ease of measurement and the ability to screen large numbers of subjects i.e., high throughput. However, it suffers from the fact that proteins not previously suspected of involvement in the disease are not identified. As a result, it has been suggested recently that more “open,” non-biased approaches which examine the entire proteome using mass spectroscopy are required to elucidate novel differentially expressed proteins (Citation13,Citation2,Citation23).
Study of the plasma proteome by mass spectroscopy to identifydisease biomarkers is extremely challenging, however, because 99% of the total protein mass is accounted for by a small number of highly abundant proteins (i.e., albumin, acute phase reactants, coagulation factors, etc.) (Citation24,25). Moreover, the remaining 1% of relatively low abundance proteins among which organ-specific, disease-specific markers may be detected, span a concentration range of more than 7 orders of magnitude (Citation26–28).
To date, a limited number of studies of the serum or plasma proteome in COPD have been performed using mass spectroscopy (Citation21,Citation29–31). These have utilized sample fractionation methods which essentially identify only high abundance plasma proteins like acute phase reactants, coagulation factors, and complement components, etc. most of which are synthesized in the liver not the lung (Citation23). Accordingly, low abundance, disease-specific proteins which are likely to reflect the pathogenic mechanisms operative in the lung require alternative approaches.
More recent proteomic approaches to identify low abundance plasma proteins have utilized robust immunodepletion to remove the most abundant plasma proteins which exert a “masking” effect and extensive multi-modality sample fractionation prior to mass spectroscopy (MS) (Citation24,Citation26–28,Citation32). In particular, one-dimensional SDS-PAGE with extensive fractionation (10 fractions) prior to LC-MS/MS, a method termed GeLC-MS/MS, provides extensive proteome coverage with high depth of analysis (Citation32). In fact, GeLC-MS/MS identifies proteins in the picogram/mL concentration range and has been used successfully to identify low abundance proteins some of which serve as cancer biomarkers (Citation32–34).
Accordingly, this proof of concept study was designed to test the ability of an open, unbiased proteomics approach using robust immunodepletion followed by extensive fractionation (15 fractions) to identify novel, low abundance plasma proteins which reflect the remodeling processes in the lung in subjects with COPD. The lung remodeling process in COPD was assessed from the FEV1 and the chest CT scan.
Methods
Study population
A subset of subjects enrolled in the NIH COPDGene® project, a multi-center, genome-wide association study designed to elucidate the genetic basis for COPD, served as the study group. The plasma proteome is affected by cigarette smoking, age, sex and ethnicity (Citation16,Citation35–37). Accordingly the study population was selected so as to control these several confounding variables and thus, reduce the environmental and biological “noise,” which might obscure the COPD-related “signal.”
Specifically, ex-smokers (i.e., cessation for >2 years) of a single ethnicity (i.e., non-Hispanic Caucasians) and sex (male) with airflow obstruction (i.e., FEV1 < 80% predicted and FEV1/FVC < 0.70) were chosen to comprise the COPD group (n = 50 subjects). A matched population of ex-smokers of the same age, sex and ethnicity without airflow obstruction (i.e., FEV1 >80% predicted and FEV1/FVC >0.70) served as controls (n = 30 subjects). The severity of COPD was assessed by spirometry performed at the time of study enrollment and subjects classified according to the 2007 Global Initiative for Obstructive Lung Disease (GOLD) guidelines (Citation5). The severity of emphysema at the time of study entry was quantitated on chest CT scan as the number ofl lung voxels with an attenuation value of less than −950 houndsfield units using the Slicer software package (http://www.slicer.org) as described previously (Citation38).
All subjects were clinically stable at the time of study. This research protocol was approved by the institutional review board at each institution and all participants provided written informed consent.
Sample collection and quality control
Plasma samples (7–8 mL of whole blood) were collected at the time of enrollment in the COPDGene® project in a vacutainer blood collection system containing a protease inhibitor cocktail specifically made for proteomic studies (Beckton Dickenson, P100, Franklin Lakes, NJ). Blood was centrifuged at room temperature within 30 minutes of collection. The separated plasma was aliquoted into sterile freezer vials (500 μL each) and stored at −80°C until used.
Initial analysis performed in the discovery group (see Supplementary ) assessed the degree of hemolysis and proteolysis of the plasma sample. No detectable hemolysis was present in any sample as determined spectrometrically. No detectable proteolysis was observed as assessed from the intensity of the <10 kD protein bands on a 1-D gel indicated (Supplementary Figure S1).
Table 1. Study population (Mean ± 1 SE)
Immunodepletion of high-abundance proteins
Plasma samples in the discovery group were stringently immunodepleted to remove as many of the highly abundant proteins as possible with a tandem, antibody-affinity resin column approach [Seppro Human IgY14 resin and Human Supermix resin system, Sigma-Aldrich Inc., St. Louis, MO] (Citation24,25,Citation33). This approach removed the 14 most abundant proteins and ∼50 moderately abundant proteins. Following tandem antibody-affinity resin column immunodepletion, individual samples from the discovery groups were then pooled to form two samples in preparation for GeLC-MS/MS.
GeLC-MS/MS
GeLC-MS, a highly robust, label-free method which we have shown is capable of detecting low abundance proteins is described in detail in supplementary methods (Citation33,Citation39).
Western blot / ELISA analysis
Selected differentially expressed proteins identified by GelC- MS in the discovery group were measured by immunoassay (Western blotting or ELISA) using commercially available antibodies in all 80 subjects as described in detail in supplementary methods.
Statistics
The statistical significance of differences in identified proteins between study groups was assessed by ANOVA. Post hoc testing was performed by Neuman-Keuls or nonparametric Mann–Whitney U-tests. Receiver operating characteristics (ROC) were constructed for validated, differentially expressed proteins based on logistic regression, and the area under the ROC curve (AUC) was calculated (Citation40). The relationship of validated proteins to the FEV1 or chest CT score was assessed by univariate or multivariate linear regression.
Results
Characteristics of the study population
By design, subjects in all groups were Caucasian males of similar age and smoking history (n = 80 subjects; ). Also by design, the FEV1 and FEV1/FVC, differed across study groups (). For example, FEV1 was 29 ± 2% SE predicted; 64 ± 2% predicted; and 94 ± 2% predicted in GOLD 3-4, GOLD 2 and controls, respectively (p < 0.01 by ANOVA). In addition, the extent of emphysema differed significantly across the three study groups. The area of emphysema was 25 ± 3% SE; 16 ± 3%; and 3 ± 1% in GOLD 3-4, GOLD 2 and control subjects, respectively (p < 0.01 by ANOVA).
Body weight and BMI were significantly different across groups (p < 0.05 by ANOVA) and less in GOLD 2 and 3-4 groups than controls (p < 0.05 for both comparisons). The incidence of common co-morbidities such as diabetes, cancer, hypertension, coronary artery disease and rheumatoid arthritis was similar across COPD and control groups (p = NS) ().
GeLC-MS assessment of differentially expressed plasma proteins in the discovery group
GeLC-MS was performed on a discovery subgroup of 10 subjects with severe COPD i.e., FEV1 < 45% predicted (GOLD 3-4) and 10 controls i.e., FEV1 > 80% predicted and FEV1/FVC >0.70. Characteristics of the discovery sub-group are shown in Supplementary Table S1.
Over 712 unique plasma proteins were identified in the two groups with concentrations that varied by at least 7 orders of magnitude (550 picograms/ml to 2 milligram/ml) based on reported plasma concentrations (Citation24).
COPD plasma demonstrated 31 differentially expressed proteins compared with controls ( and Supplementary ). Six proteins were increased (1.95-fold to 4.15-fold), and 7 proteins were decreased (0.69-fold to 0.28-fold) in the COPD samples. Seven proteins were identified only in the COPD group, and 11 proteins were identified only in the control group ().
Table 2. Differentially expressed proteins in COPD determined by GeLC-MS/MS
The differentially expressed proteins in the COPD group included proteins previously identified by others including coagulation factors (e.g., fibrinogen); acute phase reactants (e.g., c-reactive protein); metalloproteinase inhibitors (i.e., TIMP1 and 2); and adhesion molecules (e.g., VCAM1) (Citation13,14,Citation19).
Of importance, a number of novel proteins were also identified in the COPD group. These proteins are involved in oxidant defense [e.g., glucose regulated protein of 78 kD (GRP78) and peroxiredoxin]; macrophage activation [e.g., soluble CD163 (sCD163), macrophage stimulating factor 9 (MSTP9) and, interleukin 1 receptor accessory protein (IL1AP]; anti-microbial defense [e.g., cathelicidin, dermacidin and MUC18]; and tissue inflammation and repair [e.g., proteoglycan 4, procollagen endopeptidase enhancer 1 (PCOC1), fetuin, S-100-A6 and CD115]. Of considerable interest, two of the identified proteins have no known function (e.g., lethal malignant brain tumor protein and GPR 25).
Validation of selected differentially expressed proteins
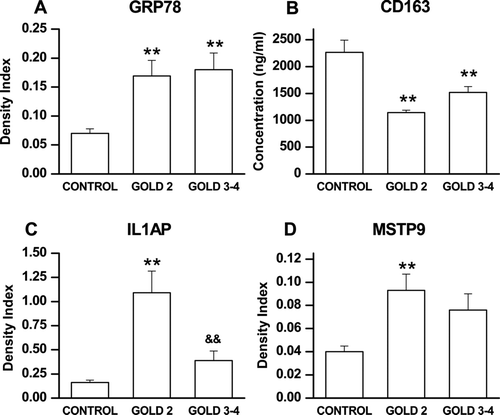
The expression of GRP78, IL1AP, sCD163 and MSPT9 was then examined by immunoassay in the entire group of 80 subjects (). These 4 proteins were chosen based on their potential importance in the pathogenesis of COPD. The immunoassay results for GRP78, MSTP9, sCD163, and IL1AP were in agreement with results on the pooled samples in the discovery group. That is, GRP78, IL1AP and MSTP9 levels were significantly increased in all COPD groups compared to controls (p < 0.01 by ANOVA for each comparison), while sCD163 levels were significantly decreased in all COPD groups (p < 0.01 by ANOVA).
Figure 1. Group mean ± SE data showing levels of GRP78, IL1AP, CD163 and MSTP9 in the 80 subject validation group as assessed by immunoassay. Panel A: GRP78 was significantly different across the 3 groups (p < 0.01 by ANOVA) and both GOLD 2 (p < 0.05) and GOLD 3-4 (p < 0.01) were significantly different than control by post-hoc testing. Panel B: sCD163 was significantly different across the 3 groups (p < 0.01 by ANOVA and GOLD 2 (p < 0.01) and GOLD 3-4 (p < 0.01) were both significantly different than control. Panel C: IL1AP was significantly different across the 3 groups (p < 0.01 by ANOVA) and both GOLD 2 (p < 0.01) and GOLD3-4 (p < 0.05) were significantly different than control. Moreover, GOLD2 differed from GOLD 3-4 (p < 0.01). Panel D: MSTP9 was significantly different across the 3 groups (p < 0.01 by ANOVA) and both GOLD 2 (p < 0.01) and GOLD 3-4 (p < 0.05) were significantly different thn control. Symbols indicate **p < 0.01 for comparison of GOLD 3-4 with control; *p < 0.05 for comparison with control; and &&p < 0.01 for comparison with GOLD 2.
Relationship of validated proteins to disease severity
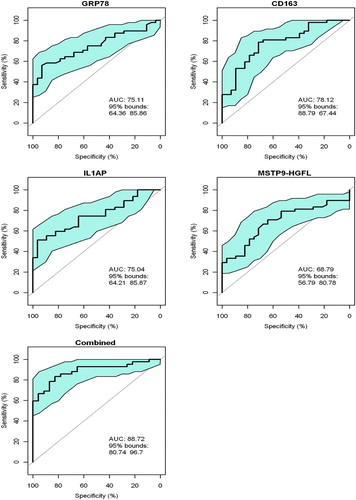
The ability of the four validated differentially expressed proteins, i.e., GRP78, IL1AP, MSTP9 and sCD163, to reflect the severity of COPD in the 80 subjects was assessed in two ways. First, we determined their ability to distinguish COPD per se from controls based on GOLD categorization as assessed by receiver operating curves (ROC) (). Specifically, ROCs for the separation between GOLD class 2, 3-4 from controls demonstrated mean AUCs of >0.74 for GRP78, IL1AP, or CD163 assessed individually. The AUC of MSTP9 individually was somewhat lower at 0.67 (95% CI 0.533 to 0.800). Of interest but not surprisingly, the combination of all 4 proteins performed better with an AUC of 0.89 (95% CI 0.807 to 0.967).
Figure 2. Receiver operating curves (ROCs) for GRP78, sCD163, IL1AP and MSTP9 for separation of COPD i.e., GOLD 2, 3–4 vs controls in all 80 subjects in the validation group. ROCs were computed for each individual protein and separately for the combination of all 4 proteins. The blue area in each ROC demarcates the upper and lower 95% confidence intervals around the mean (solid line). ROC curves for GRP78, IL1AP and MSTP9 were obtained from the ROCR library in R (reference) by providing it with disease category (1, 0) and marker value for each subject in ng/ml. Since sCD163 is negatively correlated with disease category, its values were negated prior to submission to the R procedure i.e., high values became large negative values. For the 4 markers in combination, values were submitted to logistic regression (R, glm procedure). The resulting log odds of disease as determined by the ROCR procedure was: Log(p/1-p) = 14.56 GRP78 − 0.0013 sCD163 + 3.46 IL1AP − 12.68 MSTP9 = y.
Second, we assessed the relationship of each of the 4 proteins to either the FEV1 or the percentage of emphysema determined by CT scan (). For FEV1% predicted, GRP78, sCD163, and MSTP9 each assessed individually correlated significantly (p < 0.02 for each one); GRP78 and sCD163 had the closest association (r = 0.40 and 0.31, respectively) (). In contrast, IL1AP did not correlate significantly with FEV1. Moreover, the combination of GRP78 and sCD163 performed better than each alone (r = 0.47, p < 0.001). Addition of IL1AP or MSTP9 individually or together did not significantly improve the correlation of GRP78 and sCD163 to the FEV1.
Table 3. Correlation coefficients for relationship of individual plasma biomarkers with FEV1 or emphysema
For the percentage of emphysema, GRP78 and sCD163 correlated significantly individually (r = 0.35 and 0.31, respectively; p < 0.01 for both) () and the combination performed even better (r = 0.40, p = 0.008). In general, however, the association of each of these proteins with emphysema was less than for the FEV1.
Discussion
In general, COPD biomarkers have been identified using a directed approach based on knowledge of lung physiology/pathophysiology and, in particular, the fact that immune/inflammatory responses are activated in the lung and blood (Citation9, Citation11, Citation12). In fact, existing plasma biomarkers for COPD are acute phase reactants (e.g., CRP, fibrinogen, etc.); a variety of Th1 and Th2 cytokines and chemokines; tissue growth factors; coagulation factors; proteases/protease inhibitors (e.g., MMP-9, SERPINA3, TIMP 1 and 2); or lung specific proteins (e.g., surfactant proteins A and D and PARC/CCL-18) (Citation10,11,Citation13,14,Citation17–19,Citation41,42).
In contrast, this study used an “open,” non-biased approach which scanned the plasma proteome of subjects with advanced COPD to identify low abundance proteins which may reflect lung damage and, hence, may potentially serve as novel COPD biomarkers. Considerable attention was paid to study design and methodology. First, to minimize biological or environmental “noise” related to age, gender, ethnicity and cigarette smoking history on the plasma proteome, a highly select group of subjects formed the discovery population. Specifically, the discovery group comprised Caucasian male, ex-smokers with advanced COPD (FEV1 <45% predicted; GOLD 3-4) while age-, sex- and ethnicity - matched, ex-smokers without airflow obstruction served as controls. Of note, the cigarette exposure history i.e., pack-years, was similar in the COPD and control subjects. The larger validation group differed only in that it included subjects with milder disease.
Co-morbidities may have confounding effects on the plasma proteome especially those characterized by an underlying inflammatory state. To assess this possibility, we compared the incidence of several common co-morbidities like diabetes, coronary artery disease, rheumatoid arthritis and hypertension. Of importance for the interpretation of our data, the COPD and control groups demonstrated a similar incidence of these co-morbidities suggesting that the presence of other common inflammatory and metabolic diseases did not explain our results.
Second, to minimize “noise” related to sample quality, i.e., protein degradation or hemolysis, special proteomics collection tubes (P100 tubes) were used. Third, since immunodepletion of highly abundant plasma proteins increases mass spectroscopy sensitivity and the ability to detect low abundance proteins, we used an extremely robust, double affinity column, which we have shown previously removes most high or moderately abundant proteins which comprise >90% of the mass of plasma proteins (Citation24). Next, 1 dimensional gel electrophoresis separation of the intact proteins followed by reversed-phase HPLC fractionation of the tryptic peptides generated from each gel slice was used to further enhance detection of unique proteins prior to mass spectrometry identification (Citation39).
In fact, the GeLC-MS/MS method allowed greater coverage of the plasma proteome in COPD than prior studies which utilized 2-D gel electrophoresis or strong cation exchange chromatography (Citation21,Citation29–31). Specifically, we detected over 700 unique proteins which included several differentially expressed proteins whose concentrations are in the ng/mL range e.g., GRP78, sCD163, MSTP9, IL1AP, TIMP1, ICAM1, proteoglycan 4, etc. (Citation43). In contrast, prior mass spectroscopy studies have observed differences in proteins whose concentration is in the mg/mL range, e.g., haptoglobin, fibrinogen, hemopexin, ceruloplasmin, complement components, etc. (Citation21,Citation29–31).
Of note, several of the 31 differentially expressed proteins in the COPD discovery group have been reported previously using either immunoassay or mass spectroscopy methods (Citation13,Citation15,Citation19,Citation21,Citation31). Specifically, we observed increases in COPD of acute phase response proteins e.g., CRP, fibrinogen; coagulation factors e.g., coagulation factor V; adhesion molecules e.g., VCAM-1; and decreases in molecules involved in protease-induced lung tissue injury and repair e.g., TIMP-1 and TIMP-2 (Citation13,Citation15,Citation19,Citation21,Citation31).
To our knowledge, however, 25 of the 31 differentially expressed proteins have not been reported previously as altered in COPD. Of these 25 novel proteins, 4 were chosen for immunoassay validation [i.e., GRP78, IL1AP, MSTP9 and sCD163].
Results obtained by immunoassay of all 80 subjects confirmed findings in the discovery group for all 4 proteins. That is, GRP78, IL1AP, MSTP9, sCD163 were all significantly different in the COPD groups compared to controls and all changed in the same direction as in the discovery group. The AUCs of ROC curves for distinguishing subjects with COPD from subjects without the disease using individual proteins ranged from 0.666 to 0.775 and increased to 0.887 (95% CI 0.807 to 0.967) using the combination of all 4 proteins.
That a combination of several markers performed better in predicting disease is not surprising given recent studies which demonstrate that groups of markers generally outperform single markers (Citation13,Citation15,Citation20,Citation44). Of interest, the AUC of these individual proteins to distinguish COPD from normals approximates that of putative biomarkers like SP-A, SP-D, MMP-9 and TIMP-1 and the combination of all 4 proteins exceeds that of the best single predictor, SP-A whose AUC was 0.842 [95% CI 0.785 to 0.8990] (Citation16). Finally, changes in the levels of GRP78 and sCD163 individually (and more so in combination) correlated significantly with the severity of the lung remodeling process in COPD as reflected in the FEV1 and the extent of emphysema. In fact, individual correlation coefficients for GRP78, sCD163 and MSTP9 against FEV1 (r > 0.30) were considerably greater than any one of the 84 protein markers of inflammation and tissue repair/remodeling assayed by multiplex array reported previously (Citation13).
The 4 proteins chosen for validation reflect a diversity of biological processes potentially involved in the pathogenesis of COPD including lung structural cell survival and the intensity of the lung inflammatory response (Citation3,Citation45). In particular, they play a role in oxidant stress, protein metabolism and alveolar macrophage activation.
Specifically, GRP78, glucose regulated protein of 78 kD/BIP a multi-functional member of the heat shock 70 chaperone family, is an endoplasmic reticulum (ER) chaperone which regulates responses to cell stress and cell protein homeostasis (Citation46). Moreover, GRP78 is a hallmark effector of the compensatory response to ER stress, a condition in which misfolded, non-functional proteins accumulate in the ER (Citation46) In fact, heightened ER stress promotes lung cell inflammation and induces lung cell apoptosis (Citation47). Increased expression of GRP78 as part of the unfolded protein response improves ER protein folding capacity and inhibits ER stress-induced apoptosis and lung inflammation (Citation46). Of particular interest, ER stress is present in the lungs of chronic cigarette smokers and in subjects with COPD (Citation48,49). Increases in GRP78 in the plasma in COPD in proportion to the severity of airway obstruction and emphysema, therefore, may be an indicator of the severity of ER stress in the lung in COPD.
On the other hand, sCD163, IL1AP and MSTP-9 all regulate macrophage responses to microorganisms and inflammatory cytokines. Their expression is affected by the level of macrophage activation. Specifically, CD163, which is expressed primarily by cells of the monocyte lineage such as alveolar macrophages, is a plasma membrane glycoprotein receptor and clearance mechanism for hemoglobin-haptoglobin complexes (Citation50). Thus, CD163 plays an important compensatory role in the setting of oxidative stress (Citation50). CD163 induces anti-inflammatory, anti-oxidant responses in macrophages (Citation51).
For example, hemoglobin-haptoglobin induced activation of CD163 up-regulates heme oxygenase -1 expression by monocytes/macrophages and induces release of carbon monoxide and IL-10 (Citation51). Soluble CD163 is released from its parent molecule, CD163, in response to pro-inflammatory stimuli and oxidant stress and, hence, is a marker of monocyte and tissue macrophage activation (Citation50). In contrast to CD163, relatively little is known about the biological functions of sCD163. Reduction in sCD163 levels in COPD subjects is a surprising finding in this study since the number of alveolar macrophages and their activation state is heightened in the COPD lung (Citation52).
The IL-1 receptor accessory protein [IL1AP], a member of the interleukin-1β receptor family, promotes IL-1β signaling when in its membrane bound form but enhances binding to the circulating decoy IL1R1 receptor in the soluble form (Citation53). IL1AP expression is actively regulated and reductions in membrane bound IL1AP inhibit NF-κB expression and hence the pro-inflammatory response to IL-1β (Citation54). In human respiratory cells, expression of inhibitors of IL-1β signaling such as IL1R1 decoy are stimulated in parallel with increases in IL-1β itself but are quantitatively greater (Citation55). IL-1β expression is enhanced in the COPD lung (Citation45,Citation56). Accordingly, increases in plasma soluble IL1AP in COPD likely reflect the intensity of IL-1β−induced inflammation in the lung.
Macrophage stimulatory protein (MSTP9), which is increased in COPD, is a serum protein belonging to the plasminogen-related growth factor family (Citation57). MSTP9 is primarily produced in the liver as a biologically inactive single-chain pro-MSP and is converted to its active form by several coagulation cascade enzymes. The specific receptor for MSTP9 is the RON (recepteur d'origine nantais) receptor tyrosine kinase - a member of the MET proto-oncogene family (Citation58). Activation of RON by MSPTP9 exerts dual functions on macrophages. Stimulatory activities include the induction of macrophage spreading, migration and phagocytosis. However, MSTP9 also inhibits lipopolysaccharide (LPS)-induced production of inflammatory mediators, including inducible nitric oxide, IL-12 and prostaglandins (Citation58). Thus, MSPTP9 is believed to regulate macrophage activities during bacterial infection (Citation59). Increases in MSTP9 may be a response to ongoing bacterial colonization or infection in the COPD lung.
This study tested the idea that an open, unbiased proteomics approach could identify novel, low abundance plasma proteins which could serve as potential biomarkers in COPD. In fact, we identified 25 differentially expressed proteins not previously recognized in COPD and validated 4. Nonetheless, the present study has several limitations which should be pointed out. First, the study group involved only male Caucasians, a limited subset of subjects with COPD. Whether the results obtained extend to females and subjects of other ethnicities requires further study. Second, the method of robust immunodepletion employed removed a large number of the most abundant plasma proteins whose mass comprises >90% of the plasma proteome (Citation24). Accordingly, robust immunodepletion likely eliminated a variety of previously described markers that are elevated in COPD like the iron binding proteins, ceruloplasmin, hemopexin and haptoglobin (Citation21). We also did not detect the many cytokines and chemokines which are increased in COPD (Citation13,Citation15). Our failure to detect these very low abundance proteins that are present in pg/mL amounts likely reflects the lower sensitivity of present day mass spectrometry than immunoassay or protein expression array (Citation28).
Finally, of 25 novel, differentially expressed proteins identified in the discovery group, 21 were not evaluated. These included two proteins of considerable interest since their function is unknown. These are lethal malignant brain tumor-like 3 protein (LMBL3), which was detected in COPD only, and probable G protein receptor 25 (GPR25), which was detected only in controls. The lack of available antibodies for many of these proteins prevented us from studying them further. These deserve further study.
Nonetheless, the validated proteins, GRP78, sCD163, IL1AP and MSTP9, are of interest both as indicators of the presence of COPD and, in the case of GRP78 and sCD163, the severity of lung and airway remodeling in this disease. Given the function of these proteins, they also may shed new light on the molecular mechanisms that underlie this disease.
Supplementary Material
Supplementary Methods
GeLC-MS/MS
Plasma samples from the 10 COPD and 10 control subjects in the discovery group were pooled for further analysis by mixing 50 μL of each individual's sample. The final volume of the pooled sample was 500 μL for each group. Each of the pooled COPD and control samples was diluted at a 1:2 ratio with Laemmli sample buffer (BioRad, Hercules, CA) containing 5% β-mercaptoethanol, heated for 10 minutes at 90°C and loaded onto a 10–14% polyacrylamide gel. Electrophoresis was performed using a mini Protean II system (BioRad) at 200 V for 45 minutes. Separation was confirmed by staining with SimplyBlue SafeStain (Invitrogen). Each sample lane was sliced into 20 sections, and each section further cut into ∼1 mm3 slices in preparation for tryptic digestion.
The desalted tryptic peptides were dried in a vacuum centrifuge and resolubilized in 30 μL of 0.1% (vol/vol) trifluoroacetic acid. The tryptic peptide sample was loaded onto a 2 μg capacity peptide trap (CapTrap™; Michrom Bioresources, Auburn, CA), separated by a C18 capillary column (15 cm 75 μm, Agilent) at 300 nL/min (delivered by an Agilent 1100 LC pump). A mobile-phase gradient was run using mobile phase A (1% acetonitrile/0.1% formic acid) and B (80% acetonitrile/0.1% formic acid) from 0 to 10 minutes with 0–15% B followed by 10–60 minutes with 15-60% B and 60–65 minutes with 60–100% B.
Nanoelectrospray ionization (ESI) tandem MS was performed using a Brukers HCT Ultra ion trap mass spectrometer. ESI was delivered using a distal-coating spray Silica tip (ID 20 μM, tip inner ID 10 μM, New Objective) at a spray voltage of -1300 V. Using automatic switching between MS and MS/MS modes, MS/MS fragmentation was performed on the two most abundant ions on each spectrum using collision–induced dissociation with active exclusion (excluded after two spectra, and released after 2 min). The complete system was fully controlled by HyStar 3.1 software.
Mass spectra (MS) processing was performed using Brukers Biotools (Version 2.3.0.0) with search and quantitation toolbox options. The generated de-isotoped peak list was submitted to an in-house Mascot server 2.2 and searched against the Swiss-Prot database (version 56.6 of 16-Dec-2008, 405506 sequences). Mascot search parameters were set as follows: Homo sapiens (20413 sequences); enzyme, trypsin with maximum 1 missed cleavage; fixed modification, cysteine carbamidomethylation; variable modification, methionine oxidation; 0.50 Da mass tolerance for precursor peptide ions; and 0.6 Da for MS/MS fragment ions. All peptide matches were filtered using an ion score cutoff of 10. The following two criteria were used to evaluate protein identification: one peptide with ion score ≥ 35, two or more peptides with at least one ion score ≥ 20 (p < 0.05 threshold) and the cumulative Mascot scores ≥ 35. For all proteins with cumulative MOWSE scores ≥ 20 and ≤ 35, the theoretical and experimental gel molecular weights had to be consistent. When these criteria were used to search against a reversed decoy Swiss-Prot database, no false positive match was obtained (false discovery < 0.5%). For added stringency, proteins with scores above 40 were used for comparisons between samples. Protein quantitation using label-free methods was performed by averaging the results of: 1) the relative intensities of extracted ion chromatograms determined by Mascot Distiller; 2) the number of peptide fragments of a given protein isolated; 3) sequence coverage; and 4) the exponentially modified Protein Abundance Index (emPAI).
Western Blotting/ELISA of Individual Samples
Western blotting was used to assess expression of 3 proteins increased in the COPD pooled sample i.e., glucose regulated protein 78 (GRP 78), interleukin 1 receptor accessory protein (IL1AP), and macrophage stimulating protein 9 (MSTP9). Proteins (30 μg) were separated by 10–14% gradient SDS-PAGE and transferred to a nitrocellulose membrane in a semi-dry blotting chamber (Biorad, Hercules, CA). Blots were blocked with 5% milk in Tris-buffer saline solution (pH 7.6) containing 0.05% Tween-20 (TBS/T), and probed with the following rabbit anti-human antibodies (Santa Cruz Biotechnology, Santa Cruz, CA): GRP78, IL1AP and MSTP9. Primary antibodies were used at a concentration of 0.4 μg/mL. Goat anti-human IgG HRP (Santa Cruz Biotechnology) was used as a loading control. Blots were incubated with primary antibody overnight at 4°C with gentle shaking and then incubated with a mouse, anti-rabbit HRP-conjugated secondary Ab (1:10000) (Biomeda Corp, Foster City, CA) for 1 hr at room temperature. Blots were exposed by a chemiluminescent method (Enhanced ECL Detection System, Amersham Biosciences) and scanned by FLA 5100 (FujiFilm, Edison, NJ). The density of protein bands was determined using NIH free-ware (ImageJ software).
Soluble CD163 (sCD163) was measured by ELISA using a commercially available 96-well plate immunoassay kit according to the manufacturers’ directions (Macro163, Trillium Diagnostics, Groningen, Netherlands). A 1:500 dilution of plasma was assayed in each well in a final volume of 100 μL in each well. Assays were run in duplicate. Optical density changes were read in a microtiter plate reader (BioRad, model 550)., ,
Figure S1. Individual 1D SDS PAGE gel of 10 COPD (rows #1 to #10) and 10 control subjects (rows #11 to #20) who comprise the discovery group. Gels show plasma samples in each subject prior to pooling.

Table S1 Supplementary .
Table S2. Supplementary Proteins differentially expressed in COPD
Declaration of Interest Statement
This research was supported in part by NIH grants: 5RC2 HL101713, 2R01 HL089897, RO1 HL 095432, and 2R01 HL089856.
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgments
Salim Merali and Carlos A. Barrero are equal co-authors. We would like to thank Monica Sowers for help with the organization of the proteomics data.
References
- Nussbaumer-Ochsner Y, Rabe KF. Systemic manifestations of COPD. Chest 2011; 139:165–173.
- Patel AR, Hurst JR. Extrapulmonary comorbidities in chronic obstructive pulmonary disease: state of the art. Expert Rev Respir Med 2011; 5: 647–662.
- Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007; 87:1047–1082.
- Punturieri A, Croxton TL, Weinmann GG, Kiley JP. Chronic obstructive pulmonary disease: a view from the NHLBI. Am J Respir Crit Care Med 2008; 178:441–443.
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176:532–555.
- Godtfredsen NS, Lam TH, Hansel TT, Leon ME, Gray N, Dresler C, COPD-related morbidity and mortality after smoking cessation: status of the evidence. Eur Respir J 2008; 32:844–853.
- Minino AM, Murphy SL. Death in the United States, 2010; NCHS Data Brief Jul(99):1–8.
- Ubhi BK, Riley JH, Shaw PA, Lomas DA, Tal-Singer R, MacNee W, Metabolic profiling detects biomarkers of protein degradation in COPD patients. Eur Respir J 2012; 40:345–355.
- Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J 2008; 31:869–873.
- Sin DD, Miller BE, Duvoix A, Man SF, Zhang X, Silverman EK, Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 183:1187–1192.
- Sin DD, Vestbo J. Biomarkers in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6:543–545.
- Tzortzaki EG, Lambiri I, Vlachaki E, Siafakas NM. Biomarkers in COPD. Curr Med Chem 2007; 14:1037–1048.
- Pinto-Plata V, Toso J, Lee K, Park D, Bilello J, Mullerova H, Profiling serum biomarkers in patients with COPD: associations with clinical parameters. Thorax 2007; 62:595–601.
- Pinto-Plata VM, Mullerova H, Toso JF, Feudjo-Tepie M, Soriano JB, Vessey RS, C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax 2006; 61:23–28.
- Devanarayan V, Scholand MB, Hoidal J, Leppert MF, Crackower MA, O'Neill GP, Identification of distinct plasma biomarker signatures in patients with rapid and slow declining forms of COPD. COPD 2010; 7:51–58.
- Ilumets H, Mazur W, Toljamo T, Louhelainen N, Nieminen P, Kobayashi H, Ageing and smoking contribute to plasma surfactant proteins and protease imbalance with correlations to airway obstruction. BMC Pulm Med 2011; 11:19.
- Agusti A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE, Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One 2012; 7(5):e37483.
- Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 185:1065–1072.
- Duvoix A, Dickens J, Haq I, Mannino D, Miller B, Tal-Singer R, Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax 2013; 68:670–676.
- Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG. Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:982–988.
- Verrills NM, Irwin JA, He XY, Wood LG, Powell H, Simpson JL, Identification of novel diagnostic biomarkers for asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 183:1633–1643.
- Auffray C, Adcock IM, Chung KF, Djukanovic R, Pison C, Sterk PJ. An integrative systems biology approach to understanding pulmonary diseases. Chest 2010; 137:1410–1416.
- Chen H, Wang D, Bai C, Wang X. Proteomics-based biomarkers in chronic obstructive pulmonary disease. J Proteome Res 2010; 9:2798–2808.
- Patel BB, Barrero CA, Braverman A, Kim PD, Jones KA, Chen DE, Assessment of two immunodepletion methods: off-target effects and variations in immunodepletion efficiency may confound plasma proteomics. J Proteome Res 2012; 11:5947–5958.
- Qian WJ, Kaleta DT, Petritis BO, Jiang H, Liu T, Zhang X, Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol Cell Proteomics 2008; 7:1963–1973.
- Faca V, Pitteri SJ, Newcomb L, Glukhova V, Phanstiel D, Krasnoselsky A, Contribution of protein fractionation to depth of analysis of the serum and plasma proteomes. J Proteome Res 2007; 6:3558–3565.
- Zhang Q, Faca V, Hanash S. Mining the plasma proteome for disease applicat-ions across seven logs of protein abundance. J Proteome Res 2011; 10:46–50.
- Qian WJ, Jacobs JM, Liu T, Camp DG, 2nd, Smith RD. Advances and challenges in liquid chromatography-mass spectrometry-based proteomics profiling for clinical applications. Mol Cell Proteomics 2006; 5:1727–1744.
- Bandow JE, Baker JD, Berth M, Painter C, Sepulveda OJ, Clark KA, Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies–COPD biomarker discovery study. Proteomics 2008; 8:3030–3041.
- Rana GS, York TP, Edmiston JS, Zedler BK, Pounds JG, Adkins JN, Proteomic biomarkers in plasma that differentiate rapid and slow decline in lung function in adult cigarette smokers with chronic obstructive pulmonary disease (COPD). Anal Bioanal Chem 2010; 397:1809–1819.
- York TP, van den Oord EJ, Langston TB, Edmiston JS, McKinney W, Webb BT, High-resolution mass spectrometry proteomics for the identification of candidate plasma protein biomarkers for chronic obstructive pulmonary disease. Biomarkers 2010; 15:367–377.
- Fang Y, Robinson DP, Foster LJ. Quantitative analysis of proteome coverage and recovery rates for upstream fractionation methods in proteomics. J Proteome Res 2010; 9:1902–1912.
- Ke E, Patel BB, Liu T, Li XM, Haluszka O, Hoffman JP, Proteomic analyses of pancreatic cyst fluids. Pancreas 2009; 38:e33–42.
- Xie LQ, Zhao C, Cai SJ, Xu Y, Huang LY, Bian JS, Novel proteomic strategy reveal combined alpha1 antitrypsin and cathepsin D as biomarkers for colorectal cancer early screening. J Proteome Res 2010; 9:4701–4709.
- Bortner JD, Jr., Richie JP, Jr., Das A, Liao J, Umstead TM, Stanley A, Proteomic profiling of human plasma by iTRAQ reveals down-regulation of ITI-HC3 and VDBP by cigarette smoking. J Proteome Res 2011; 10:1151–1159.
- de Torres JP, Casanova C, Pinto-Plata V, Varo N, Restituto P, Cordoba-Lanus E, Gender differences in plasma biomarker levels in a cohort of COPD patients: a pilot study. PLoS One. 6(1):e16021.
- Kim CX, Bailey KR, Klee GG, Ellington AA, Liu G, Mosley TH Jr, Sex and ethnic differences in 47 candidate proteomic markers of cardiovascular disease: the Mayo Clinic proteomic markers of arteriosclerosis study. PLoS One. 5(2):e9065.
- Han MK, Kazerooni EA, Lynch DA, Liu LX, Murray S, Curtis JL, Chronic obstructive pulmonary disease exacerbations in the COPDGene study: associated radiologic phenotypes. Radiology 2010; 261:274–282.
- Han Z, Liang CG, Cheng Y, Duan X, Zhong Z, Potireddy S, Oocyte spindle proteomics analysis leading to rescue of chromosome congression defects in cloned embryos. J Proteome Res 2010; 9:6025–6032.
- Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics 2005; 21:3940–3941.
- Dahl M, Vestbo J, Zacho J, Lange P, Tybjaerg-Hansen A, Nordestgaard BG. C reactive protein and chronic obstructive pulmonary disease: a Mendelian randomisation approach. Thorax 2011; 66:197–204.
- Hurst JR, Donaldson GC, Perera WR, Wilkinson TM, Bilello JA, Hagan GW, Use of plasma biomarkers at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 174:867–874.
- Haab BB, Geierstanger BH, Michailidis G, Vitzthum F, Forrester S, Okon R, Immunoassay and antibody microarray analysis of the HUPO Plasma Proteome Project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics 2005; 5:3278–3291.
- Zethelius B, Berglund L, Sundstrom J, Ingelsson E, Basu S, Larsson A, Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med 2008; 358:2107–2116.
- Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J 2008; 31:1334–1356.
- Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev 2006; 86:1133–1149.
- Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 2011; 108:10562–10567.
- Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol 2008; 38:541–550.
- Malhotra D, Thimmulappa R, Vij N, Navas-Acien A, Sussan T, Merali S, Heightened endoplasmic reticulum stress in COPD lungs: The role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med 2009; 180:1196–1207.
- Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol 2010; 47:1650–1660.
- Philippidis P, Mason JC, Evans BJ, Nadra I, Taylor KM, Haskard DO, Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res 2004; 94:119–126.
- Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD 2004; 1:59–70.
- Subramaniam S, Stansberg C, Cunningham C. The interleukin 1 receptor family. Dev Comp Immunol 2004; 28:415–428.
- Chen R, Li M, Zhang Y, Zhou Q, Shu HB. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1beta-induced NF-kappaB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc Natl Acad Sci USA 2012; 109:14128–14133.
- Yang Y, Bin W, Aksoy MO, Kelsen SG. Regulation of interleukin-1beta and interleukin-1beta inhibitor release by human airway epithelial cells. Eur Respir J 2004; 24:360–366.
- Barnes PJ SS, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22:672–688.
- Wang MH, Zhou YQ, Chen YQ. Macrophage-stimulating protein and RON receptor tyrosine kinase: potential regulators of macrophage inflammatory activities. Scand J Immunol 2002; 56:545–553.
- Morrison AC, Wilson CB, Ray M, Correll PH. Macrophage-stimulating protein, the ligand for the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J Immunol 2004; 172:1825–1832.
- Ray M, Yu S, Sharda DR, Wilson CB, Liu Q, Kaushal N, Inhibition of TLR4-induced IkappaB kinase activity by the RON receptor tyrosine kinase and its ligand, macrophage-stimulating protein. J Immunol 2010; 185:7309–7316.