
Abstract
Cigarette smoke has been shown to cause chronic inflammation of the lungs, eventually leading to chronic obstructive pulmonary disease (COPD). Additionally, recent studies have suggested that mesenchymal stem cells (MSCs) can mediate local inflammatory responses in the lungs. Thus, the aim of the present study was to test the effects of rat MSCs (rMSCs) on inflammation of the lungs and destructive pulmonary function induced by cigarette smoke in rats. Rats were exposed to cigarette smoke for 7 weeks. rMSCs were cultured in vitro and infused intratracheally into cigarette smoke-exposed rats. The total and differential cell counts in the bronchoalveolar lavage fluid (BALF), histological changes, pro-inflammatory cytokines, transforming growth factor-β1 (TGF-β1) expression, and pulmonary function were evaluated. Additionally, human peripheral blood mononuclear cells and human MSCs were cocultured in vitro to detect cytokines and TGF-β1 levels. We found that rMSC administration resulted in downregulation of pro-inflammatory cytokines in the lungs while increasing TGF-β1 expression, reducing total inflammatory cell numbers in the BALF, and improving pulmonary histopathology and airflow obstruction. Coculture revealed that human MSCs mediated an anti-inflammatory effect partly via upregulation of TGF-β1. These findings suggested that MSCs may have therapeutic potential in cigarette smoke-induced inflammation and airflow obstruction, partly via upregulation of TGF-β1.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disease characterized by an abnormal inflammatory response of the lungs to noxious particles and gases (Citation1). Cigarette smoke (CS) is the major risk factor for the development of COPD, and smoking has been shown to lead to airway inflammation with an increase of inflammatory cells from both the innate and adaptive immune system (Citation2–4). Upregulation of inflammatory processes leads to irreversible events such as apoptosis of epithelial cells, proteolysis of the terminal air-space and lung extracellular matrix components. Therefore, the development of novel, effective therapeutic approaches based on reducing chronic inflammation and airflow limitation is critical.
Mesenchymal stem cells (MSCs) are multipotent stem cells capable of differentiating into mesenchymal and nonmesenchymal lineages, including airway and alveolar epithelial cells (Citation5–7). Experimental studies have provided evidence indicating that mesenchymal stem cells (MSCs) may be useful for the treatment of a variety of clinical disorders, including sepsis, acute lung injury (ALI) (Citation8,9). MSCs protect lung tissue through suppression of proinflammatory cytokines, and through triggering production of reparative growth factors (Citation10–14).
In murine models of acute lung injury (ALI), MSCs suppress local lung inflammation by paracrine protective factors including angiopoietin-1, vascular endothelial growth factor, keratinocyte growth factor (KGF), and hepatocyte growth factor (Citation15,16). Recent study in double-blind, placebo controlled study using allogeneic hMSCs in patients with moderate-severe COPD has demonstrated an early, significant decrease in levels of circulating C-reactive protein (CRP) in patients treated with MSCs who had elevated CRP levels at study entry (Citation17,18). Although the mechanism of MSCs anti-inflammatory effect in chronic pulmonary and systemic inflammation of COPD is still unknown.
The transforming growth factor (TGF)-β superfamily comprises more than 40 members, which are essential during organ development, a process often recapitulated in chronic diseases. Recently, researchers have become interested in the role of TGF-β signaling in the pathogenesis of COPD. Airway remodeling is one of the most important mechanisms in the pathogenesis of COPD and is triggered by chronic inflammation mediated by angiopoietin-1 (Ang-1), interleukin-8 (IL-8) and transforming growth factor-β1 (TGF-β1)(Citation18). Königshoff et al. described the 2 main features of COPD as small airway disease (SAD), which includes airway inflammation and remodeling and emphysema which is characterized by airspace enlargement (Citation19). Although increased TGF-β signaling clearly triggers the development of SAD and decreased TGF-β signaling in emphysema may lead to increased expression of matrix metalloproteinases (MMPs) and subsequent extracellular matrix degradation, which may contribute to existing genetic or acquired susceptibility to emphysema (Citation19–22).
Although the recent studies have shown that MSCs can restore alveolar architecture and emphysema induced by papain (Citation23), elastase (Citation24), and CS (Citation25), the effects of MSCs on pulmonary inflammation induced by subacute CS exposure have not been elucidated. Here, we hypothesized that administration of rat MSCs (rMSCs) may reduce lung inflammation and destructive pulmonary function resulting from CS exposure in rats. Furthermore, we sought to explore the possible paracrine mechanisms mediating this process, including TGF-β1 signaling, by coculturing human peripheral blood mononuclear cells (PBMCs) and human MSCs (hMSCs) in vitro.
Materials and methods
MSC culture and characterization
rMSCs were isolated from the bone marrow of tibias and femurs from male Sprague-Dawley rats (Chinese Academy of Sciences, Shanghai, China) weighing 140–160 g, as previously reported, with slight modifications (Citation16, Citation26). The experimental protocol was approved by the Committee of Animal Care and Use of Shanghai Jiaotong University, according to National Institutes of Health guidelines. Briefly, adherent bone marrow cells were incubated for a week until colony-forming units (CFUs) were established.
The rMSCs of these CFUs were subcultured in Dulbecco's modified Eagle medium (DMEM)/F12 containing 10% fetal bovine serum (FBS). Passage 2 rMSCs were subjected to adipogenic, osteogenic, and chondrogenic differentiation assays, flow cytometry for the cell surface markers CD11b/c, CD45 (Invitrogen, Carlsbad, CA, USA), CD29 (eBioscience, San Diego, CA, USA), and CD90 (Biolegend, San Diego, CA, USA). Similar to rMSC cultures, hMSCs were isolated from bone marrow aspirates taken from the iliac crest of a healthy adult volunteer with informed consent and subsequently cultured in DMEM/F12 containing 20% FBS. hMSCs were stained with antibodies targeting CD11b/c, CD19, CD34, CD44, CD90, and CD105 (Becton, Dickinson, Franklin, NJ, USA). Passage 2 rMSCs and hMSCs were used in in vivo and in vitro experiments, respectively.
CS exposure and experimental protocol
Sprague–Dawley (SD) rats weighing 250–280 g each were divided into 3 groups: the sham-exposed group (rats were exposed to air), smoke-exposed group, and smoke-exposed group with rMSC treatment. The rats were placed in 6-L perspex chambers (5 rats/chamber) and exposed to CS generated from commercial cigarettes (“Da Qianmen,” Shanghai, China, 1.25 mg nicotine and 12 mg tar oil per cigarette) with fresh air being pumped in for the remaining time. CS was collected by burning 18 cigarettes per day, 5 days a week for 7 weeks. On the eighth week, rats in the rMSC treatment group were anesthetized and infused with 6 × 106 MSCs via the trachea, while rats in the smoke-only group were treated with 0.15 mL PBS. At the 12th week, 4 weeks after the MSCs infusion, pulmonary function was evaluated and samples of serum, BALF, and lung tissues were collected.
Evaluation of pulmonary function
At the endpoint of the experiment, pulmonary function was evaluated using an invasive pulmonary function device (Buxco Research Systems, Wilmington, NC, USA), as previously described with slight modifications (Citation27). Briefly, rats were anesthetized, trachea-intubated, and placed (whole body) in a forced pulmonary maneuver system. An average breathing frequency of 50 breaths per min was imposed. The following parameters were recorded by the software: functional residual capacity (FRC), total lung capacity (TLC), forced vital capacity (FVC), forced expiratory flows (FEVs), such as FEV in 100 ms (FEV100) and 200 ms (FEV200), maximal expiratory flow at 25% vital capacity (FEF25), maximal expiratory flow at 50% vital capacity (FEF50), and maximal expiratory flow at 75% vital capacity (FEF75).
Inflammatory cell counts in bronchoalveolar lavage
The left lungs were lavaged 3 times with 2 mL ice-cold PBS each time, and the recovery ratio was above 90%. The total and differential cell counts in the bronchoalveolar lavage fluid (BALF) were determined by globulimeter and Wright's staining, respectively.
Measurement of pro-inflammatory cytokines by enzyme-linked immunosorbent assay (ELISA)
The bronchi were extracted from the right lung and the lungs were homogenized in RIPA buffer (Beyotime, Shanghai, China). Interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, monocytes chemotactic protein (MCP)-1, and TGF-β1 levels in lung homogenates (pg/mg protein) and IL-1β, IL-6, and TNF-α in the serum (pg/mL) were measured with ELISA kits (eBioscience Inc. and Genetimes Inc., Shanghai, China). All procedures were performed in accordance with the manufacturer's instructions. The cytokine concentration in the lungs was normalized to the total protein concentration of the homogenate as measured by bicinchoninic acid protein assay (Pierce, Rockford, USA).
Assessment of lipid peroxidation products in lung homogenates
The levels of the lipid peroxidation product malondialdehyde (MDA) in the lungs were estimated using the thiobarbituric acid reaction (Beyotime, China) in accordance with the manufacturer's instructions, as previously described (Citation28). The concentration of MDA (nM/mg protein) was calculated by standard curve and normalized to the total protein concentration.
Pulmonary histopathology
The right inferior lobe lungs were harvested and fixed in 10% neutral formalin for 48 h. The lung tissues were then embedded in paraffin and cut into 4-μm-thick sections. Hematoxylin-eosin staining was performed using standard protocols for histological analysis.
Coculture of cigarette smoke extract (CSE)-stimulated human PBMCs with hMSCs
Human PBMCs were isolated from 50 mL peripheral venous blood of a healthy volunteer by a density gradient centrifugation method. Briefly, heparinized blood (20 U/mL) was layered onto lymphoprep (Axis-Shield, Norway) and centrifuged at 2500 rpm for 30 min. The layer of mononuclear cells was aspirated, washed 2 times with PBS, and suspended in RPMI-1640 medium for further experiments. In order to confirm whether MSCs have anti-inflammatory properties due to paracrine mechanisms, 6-well Transwell chambers (0.4-μm pore, Costar, Corning, NY, USA) were used, with PBMCs (1.5 × 106) suspended in 1.5 mL medium in the lower chambers and hMSCs (2 × 105) suspended in 0.5 mL medium in the upper chambers. The optimal ratio of PBMCs to MSCs was identified by a preliminary experiment. CSE was prepared at a concentration of 1 cigarette/40 mL in serum-free DMEM. This medium was defined as 100% CSE and was used after adjusting the pH to 7.4 and filtering the extract through a 0.22-μm filter.
First, we confirmed that 5% CSE did not affect the viability of PBMCs or hMSCs. Subsequently, cells suspended in serum-free RPMI-1640 were subjected to the following conditions for 12 h: PBMCs (1.5 × 106) were cultured alone with or without 5% CSE, human recombinant TGF-β1 (40 ng/mL, Peprotech, Rocky Point, NJ, USA) was added to cultured PBMCs exposed to 5% CSE, PBMCs exposed to 5% CSE were cocultured with hMSCs, or anti-TGF-β1 neutralizing antibody (500 ng/mL, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added into the MSC-PBMC coculture system. The conditioned medium was collected for the evaluation of TGF-β1 and TNF-α (pg/mL) by ELISA, using the same protocol as described above without normalization. Tgf-β1 mRNA levels in cultured PBMCs exposed to 5% CSE and the mRNA levels of Tgf-β1 and Tnf-α in PBMCs and hMSCs in the coculture system were analyzed by real-time quantitative PCR using SYBR Green PCR Mix (Takara, Dalian, China).
PCR was performed using the following conditions: initial incubation at 95°C for 30 seconds, followed by 40 cycles of 95°C for 5 seconds and 64°C for 34 seconds. The mRNA abundances of various cytokines were quantified as the ∆Ct values. The following primers were used: TGF-β1, 5′-AGCGACTCGCCAGAGTG GTTA-3′, 5′-GCAGTGTGTTATCCCTGCTGTCA-3′; TNF-α, 5′-GTGACAAGCCTGTAGCCCATGTT-3′, 5′-TTATCTCTCAGCTCCACGCCATT-3′; GAPDH, 5′-GCACCGTCAAGGCTGAGAAC-3′, 5′-TGGTGAA GACGCCAGTGGA-3′.
Statistical analyses
Data are shown as the mean ± SD and were analyzed using SPSS11.5 software. Analysis between the different groups was performed using one-way analysis of variance followed by the LSD significant difference test in the case of equal variances and Dunnet's post hoc test in the case of unequal variances. A value of p < 0.05 was considered statistically significant.
Results
Identification of cultured rMSCs and hMSCs
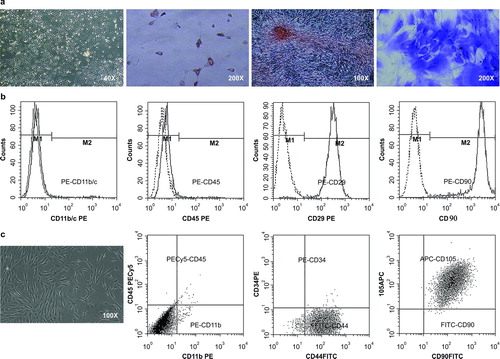
After 2 weeks of culture, primary fibroblast-like rMSCs () and hMSCs () were observed. Differentiation assays demonstrated that rMSCs had the ability to form adipocytes, osteoblasts, and chondrocytes (). Flow cytometry analysis showed that rMSCs did not express CD11b/c or CD45, but did express CD29 and CD90 (). hMSCs did not express CD11b/c, CD19, or CD34, but did express CD44, CD90, and CD105 (). These results indicated that cultured rMSCs and hMSCs exhibited phenotypes consistent with international standards of MSCs (Citation29).
Figure 1. Characterization of MSCs. (a) Primary fibroblast-like rMSCs was observed. The multipotential differentiation of rMSCs was verified via Oil Red staining for adipocytes, Alizarin S staining for osteoblasts, and toluidine blue staining for chondrocytes. (b) The rMSCs did not express CD11b or CD45, but expressed CD29 and CD90, as measured by flow cytometry. (c) Primary fibroblast-like hMSCs were observed. The hMSCs did not express CD11b/c, CD19, or CD34, but expressed CD44, CD90, and CD105.
Administration of rMSCs reduced the expression of pro-inflammatory cytokines and MDA and increased the expression of TGF-β1 in the lungs
In order to evaluate the anti-inflammatory effects of rMSCs, we measured levels of pro-inflammatory mediators in lung homogenates and serum. Subacute CS exposure upregulated the levels of IL-1β, IL-6, and TNF-α in the lungs, and infusion of rMSCs significantly reduced the levels of these cytokines ().
Figure 2. rMSC infusion downregulated pro-inflammatory cytokines levels, while increasing TGF-β1 expression. The levels of IL-1β, IL-6, MCP-1, TNF-α, and TGFβ-1 in lung homogenates and serum were measured by ELISA. Data are shown as the mean ± SD (n = 6). *p < 0.05, **p < 0.01 vs. sham exposed group; #p < 0.05, ##p < 0.01 vs. smoke-exposed group.
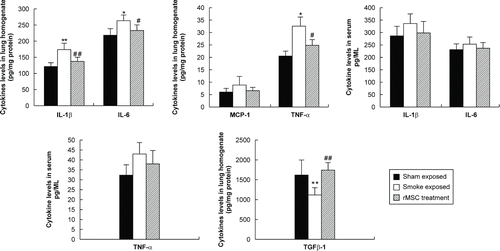
Similarly, in lung homogenates, MCP-1 showed a tendency to increase in smoke-exposed rats and exhibited a reduction after the administration of rMSCs; however, the differences were not statistically significant (). Given the low level of MCP-1 in the lungs, we evaluated only IL-1β, IL-6, and TNF-α levels in the serum. We found that the levels of these cytokines were higher in smoke-exposed rats than in sham-exposed rats and tended to be lower after infusion of rMSCs; however, again, the differences were not statistically significant ().
We hypothesize that 7 weeks of exposure to CS may be too short, not causing the induction of obvious systemic inflammation. In addition to measurement of the above cytokines, we also measured TGF-β1 levels in lung homogenates. Smoke-exposed rats had significantly lower TGF-β1 levels than sham-exposed rats, whereas rats treated with rMSCs had significantly higher TGF-β1 levels than smoke-exposed rats ().
Because CS-induced oxidative stress has been strongly implicated in the inflammatory response (Citation3, Citation30), MDA levels in different groups were evaluated. In response to CS, significantly higher MDA levels were observed in smoke-exposed lungs as compared with sham-exposed lungs (1.5 ± 0.3 vs. 0.8 ± 0.2 nM/mg protein, respectively), while infusion of rMSCs resulted in a dramatic decrease in MDA in the lungs (1.0 ± 0.2 nM/mg protein).
Infusion of rMSCs reduced smoke-induced inflammation in the BALF and in histopathological samples
In order to further confirm the anti-inflammation effects of rMSCs, total and differentiated cell counts in the BALF and pathological changes in the lungs were analyzed. Compared with sham-exposed rats, total inflammatory cells and percentages of neutrophils and lymphocytes in the BALF were significantly increased in smoke-exposed rats. In contrast, infusion of rMSCs significantly reduced the total numbers of inflammatory cells (). Histological analysis revealed infiltration of inflammatory cells in the airways and lungs in smoke-exposed rats, while administration of rMSCs reduced these changes, resulting in the observation of fewer inflammatory cells in peribronchial and alveolar structures ().
Figure 3. rMSC infusion reduced inflammation of peribronchial and alveolar structures. Pulmonary histopathological changes were observed by hematoxylin and eosin staining. The arrows indicate airway and lung inflammation in smoke-exposed rats and reduced inflammatory cells in peribronchial and alveolar structures after rMSC administration.
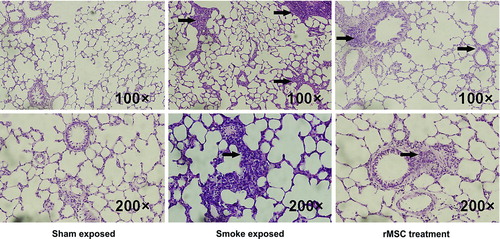
Table 1. The total and differential cell counts of leukocytes in BALF (1 × 108/L)
Infusion of rMSCs reduced airflow obstruction
shows the results of pulmonary function tests. A statistically significant increase in FVC and TLC was found in smoke-exposed rats as compared with sham-exposed rats. There were no differences in FEV100, FEV200, or FEF25 among experimental rats. However, markers for airflow obstruction, i.e., FEV100/FVC, FEV200/FVC, FEF50, and FEF75, exhibited a statistically significant reduction in smoke-exposed rats. Importantly, administration of rMSCs administration significantly reduced airflow obstruction, as demonstrated by statistically higher FEV100/FVC, FEV200/FVC, FEF50, and FEF75, with nonstatistically significant reductions in FRC and FRC/TLC.
Table 2. rMSC administration reduced airway obstruction
Coculture with hMSCs reduced TNF-α production from PBMCs via TGF-β1
Our in vivo results revealed that rMSC transplantation reduced pro-inflammatory cytokines and local inflammation in the lungs, with a concomitant increase in TGF-β1 expression. In addition, the anti-inflammatory capacity of TGF-β1 has been previously suggested in a report demonstrating that TGF-β1-null mice died of overwhelming inflammation within 1 month of birth (Citation31). To further confirm whether MSCs exerted an anti-inflammatory role via paracrine TGF-β1 signaling, in vitro coculture experiments were performed. We cocultured PBMCs and hMSCs with CSE stimulation using Transwell system, which physically separated the 2 cell types and thereby inhibited cell-cell contact.
We observed that CSE stimulation reduced TGF-β1 levels in cell conditioned media of cultured PBMCs (p < 0.01); in contrast, TGF-β1 increased dramatically in cell suspensions of CSE-stimulated PBMCs cocultured with hMSCs (). Next, we evaluated the expression of the pro-inflammatory factor TNF-α in cell supernatants. CSE stimulation inhibited the release of TNF-α from PBMCs. Recombinant TGF-β1 further suppressed the production of TNF-α by CSE-stimulated PBMCs (). Furthermore, a dramatic reduction in TNF-α was detected when hMSCs were added; in contrast, the inhibitory effect on TNF-α in the coculture system was partly blocked by the addition of an anti-TGFβ-1 neutralizing antibody (). The above results indicated that cocultured hMSCs suppressed TNF-α production from PBMCs partly via TGF-β1 ().
Figure 4. Evaluation of pro-inflammatory cytokine levels in cell supernatants. Human PBMCs were cultured alone without 5% CSE (control group) or with 5% CSE stimulation (5% CSE group). Recombinant TGFβ-1 (40 ng/mL) was added into cultured PBMCs exposed to 5% CSE(TGFβ-1 group). In addition, PBMCs exposed to 5% CSE were cocultured with hMSCs using a transwell system (hMSCs group) and an anti-TGFβ-1 neutralizing antibody (500 ng/mL) was added into the coculture system (anti-TGFβ-1 group). The cell supernatant was collected to evaluate TNF-α levels (pg/mL) by ELISA. *p < 0.05, **p < 0.01 vs. cultured PBMCs without CSE; ##p < 0.01 vs. cultured PBMCs with CSE; ++p < 0.01 vs. cocultured PBMCs with hMSCs without anti-TGFβ-1 antibody.
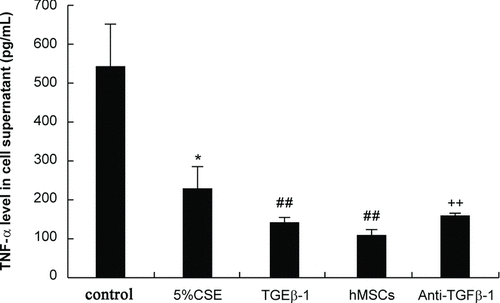
Table 3. TGF-β1 levels in different groups
Discussion
In general, MSCs secrete soluble factors that are thought to play a significant role in tissue repair rather than transdiff erentiation (Citation32,33). In the present study, MSCs were administered in the treatment of CS-exposed rats. The results demonstrated that MSCs have therapeutic potential in cigarette smoke-induced inflammation and airflow obstruction. Up-regulation of TGF-β1 by MSCs administration may play an important role in ameliorating chronic inflammation.
Exposure to CS has been shown to cause progressive chronic inflammation of airway and lung parenchyma, characterized by increased numbers of neutrophils, activated macrophages, and T-lymphocytes. In the inflammatory response, CS-induced pro-inflammatory cytokines and chemokines, such as TNF-α, IL-1β, MIP-1α, IL-6, c-x-c motif chemokine ligand (CXCL) 8 (IL-8), and MCP-1 by airway epithelial cells and alveolar macrophages elicits the recruitment of neutrophils and inflammatory monocytes to the lungs (Citation34–36). Therefore, the main focus of our work was to determine whether rMSCs could reduce CS-induced sub-acute inflammation. We found that rMSCs had the capacity to suppress IL-1β, IL-6, and TNF-α expression, with a tendency to reduce MCP-1 expression, in the lungs of smoke-exposed rats. In our animal model, we observed that serum IL-1β, IL-6, and TNF-α were increased after CS exposure and that infusion of rMSCs reduced the levels of these cytokines, although these differences were not statistically significance.
Furthermore, we observed that rMSC transplantation reduced the number of total inflammatory cells in the BALF and decreased infiltration of inflammatory cells in peribronchial and alveolar structures, indicating that rMSCs improved CS-induced local inflammation in the lungs. A similar study showed that systemic administration of MSCs weakened the systemic inflammatory response (decreased IL-1β, IL-6, MIP-1α, and IL-8 in serum) of endotoxin-induced ALI in mice, but these pro-inflammatory cytokines were not statistically reduced in the BALF after MSC administration (Citation16). Different approaches to the administration of MSCs may result in disparate effects on local and systemic inflammation.
The excessive exogenous oxidative stress derived from the increased burden of inhaled oxidants, such as CS, together with the increased endogenous reactive oxygen species generated by several inflammatory cells, immune cells, and structural airways cells, have been implicated in the progression of CS-induced diseases and can be reflected by increased markers of oxidative stress, including H2O2, MDA, and 4-hydroxy-2-nonenal in the sputum, airways, and blood (Citation37, 38). Therefore, when evaluating MDA levels in lung homogenates, we found that rMSC infusion significantly reduced the increased MDA levels induced by CS.
Among inflammatory and extracellular matrix regulatory cytokines, transforming growth factor-beta (TGF-β) stands central, as it possesses both important immunomodulatory and fibrogenic activities, and should be considered a key for understanding inflammation and remodeling processes. TGF-β is involved in the pathogenesis of several diseases, including COPD (Citation39, 40). The expression of TGF-β1, type I TGF-β1 receptor (Citation41), and type II TGF-β1 receptor (Citation42) in the lungs was significantly decreased in emphysema. The balanced TGF-β signaling is not only important for regulation of extracellular matrix turnover, but also for alveolar cell homeostasis. In line with the above reports, we observed that CS exposure reduced TGF-β1 levels in the lungs, and importantly, rMSCs administration significantly upregulated TGF-β1 levels.
Notably, TGF-β1-null mice have an excessive inflammatory response and develop multifocal inflammatory diseases that are ultimately lethal, involving massive infiltration of lymphocytes and macrophages accompanied with increased TNF-α and MIP-1 levels primarily in the lungs (Citation43). Mice deficient in β6 integrin, failing to activate latent TGF-β within lung, also develop macrophage-rich inflammation (Citation44). Therefore, we proposed that increased TGF-β1 may contribute to the decline in pro-inflammatory cytokines and subsequent improvement of inflammation in rMSC-treated lungs. To verify this hypothesis, we cocultured PBMCs and hMSCs, added recombinant TGF-β1 to cultures of PBMCs alone, and added anti-TGF-β1 neutralizing antibodies to the coculture system.
The results showed that coculture with hMSCs inhibited TNF-α release from PBMCs in part via TGF-β1 signaling. Interestingly, CSE stimulation unexpectedly inhibited TNF-α production from PBMCs in the current study, which is inconsistent with a report demonstrating that CSE promoted type II pneumocytes to release TNF-α (Citation45). This disparity may be due to variations in the nature of cells, culture/stimulation conditions, and duration of stimulation, and the complex regulation events involved need to be studied further before conclusions are drawn.
We also focused on pulmonary function loss and airflow limitation, which is relevant to inflammation. The pulmonary function of subacute smoke-exposed rats was evaluated. Administration of rMSCs significantly increased markers for airflow obstruction during expiration (FEV100/FVC, FEV200/FVC, FEF50, and FEF75), while FVC, TLC, FRC, and FEF25 parameters did not differ significantly. These results indicated that administration of MSCs improved pulmonary function in CS-exposed rats.
Importantly, it should be emphasized that the diverse mechanisms involved do not operate separately, but are strongly interrelated. For example, alveolar macrophages, epithelial cells, and recruited inflammatory cells are the principal sources of endogenous oxidants (Citation34). Excessive oxidative stress amplifies airway inflammation, induces lung structural cell death, and contributes to proteinase and anti-proteinase imbalance (Citation46). TGF-β1 can inhibit MMP9 and MMP12 expression in alveolar macrophages and monocytes, whereas the inhibitory signaling of TGF-β1 leads to increased MMP12 expression and subsequent extracellular matrix degradation, contributing to the development of emphysema (Citation41). Here, we speculated that reduced inflammation and oxidative stress and upregulated TGF-β1 exerted beneficial effects on each other and contributed to the rMSC-mediated reduction in airflow obstruction.
Speculations
The current study has shown that the administration of rMSCs downregulated IL-1β and IL-6, with a trend toward reductions in MCP-1 and TNF-α, alleviated oxidative stress and lung inflammation, and upregulated TGF-β1 levels, thereby reducing airflow obstruction. In vitro coculture studies provided further evidence that hMSCs suppressed TNF-α release from PBMCs, depending, at least in part, on the upregulation of TGF-β1. Thus, our data demonstrated that MSCs can sense the local microenvironment and upregulate the production of soluble factors and growth factors, such as TGF-β1, resulting in anti-inflammatory effects, which may facilitate the use of MSCs in the treatment of chronic inflammatory diseases such as COPD.
Acknowledgments
Authors Song and Guan contributed equally to this work. We would like to thank the staff of the Molecular and Biology Laboratory of Xinhua Hospital for their assistance.
Declaration of Interest Statement
The authors indicate that there are no potential conflicts of interest to declare. The authors are responsible for the content and writing of this paper.
This work was supported by grants from the Support Program of Shanghai Science and Technology Committee (No. 12431900701), Youth Project of Shanghai Municipal Public Health Bureau (No. 2009Y024) and State Natural Sciences Foundation Project (No. 81000014).
References
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364:613–620.
- Rabe KF, Hurd S, Anzueto A, Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176:532–555.
- Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 2012; 122(8):2749–2755.
- Nikota JK, Stämpfli MR. Cigarette smoke-induced inflammation and respiratory host defense: Insights from animal models. Pulm Pharmacol Ther 2012; 25(4):257–262.
- Abreu SC, Antunes MA, Pelosi P, Mechanisms of cellular therapy in respiratory diseases. Intensive Care Med 2011; 37(9):1421–1431.
- Sinclair K, Yerkovich ST, Chambers DC. Mesenchymal stem cells and the lung. Respirology 2013; 18(3):397–411.
- Aliotta JM, Passero M, Meharg J, Stem cells and pulmonary metamorphosis: New concepts in repair and regeneration. J Cell Physiol 2005; 204:725–741.
- Németh K, Leelahavanichkul A, Yuen PS, Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 2009; 15(1):42–49.
- Gupta N, Su X, Popov B, Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol 2007; 179:1855–1863.
- D'Agostino B, Sullo N, Siniscalco D, Mesenchymal stem cell therapy for the treatment of chronic obstructive pulmonary disease. Expert Opin Biol Ther 2010; 10(5):681–687.
- Katsha AM, Ohkouchi S, Xin H, Paracrine factors of multipotent stromal cells ameliorate lung injury in an elastase-induced emphysema model. Mol Ther 2011; 19(1):196–203.
- Iyer SS, Co C, Rojas M. Mesenchymal stem cells and inflammatory lung diseases. Panminerva Med 2009; 51(1):5–16.
- Lee JW, Gupta N, Serikov V, Potential application of mesenchymal stem cells in acute lung injury. Expert Opin Biol Ther 2009; 9(10):1259–1270.
- Kean TJ, Lin P, Caplan AI, MSCs: Delivery Routes and Engraftment, Cell-Targeting Strategies, and Immune Modulation. Stem Cells Int 2013; 2013:732–742.
- Matthay MA, Thompson BT, Read EJ, Therapeutic potential of mesenchymal stem cells for severe acute lung injury. Chest 2010; 138(4):965–972.
- Xu J, Woods CR, Mora AL, Prevention of endotoxin-induced systemic response by bone marrow-derived mesenchymal stem cells in mice. Am J Physiol Lung Cell Mol Physiol 2007; 293:L131–141.
- Weiss DJ, Casaburi R, Flannery R, A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest 2013; 143(6):1590–1598.
- Gao J, Zhan B. The effects of Ang-1, IL-8 and TGF-β1 on the pathogenesis of COPD. Mol Med Rep 2012; 6(5):1155–1159.
- Königshoff M, Kneidinger N, Eickelberg O. TGF-beta signaling in COPD: deciphering genetic and cellular susceptibilities for future therapeutic regimen. Swiss Med Wkly 2009; 139:554–563.
- Morty RE, Königshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6(7):607–613.
- Roberts AB. Medicine: Smoke signals for lung disease. Nature 2003; 422:130–131.
- Morris DG, Huang X, Kaminski N, Loss of integrin alpha(v)beta6-mediated TGFbeta activation causes Mmp12-dependent emphysema. Nature 2003; 422:169–173.
- Zhen G, Xue Z, Zhao J, Mesenchymal stem cell transplantation increases expression of vascular endothelial growth factor in papain-induced emphysematous lungs and inhibits apoptosis of lung cells. Cytotherapy 2010; 12:605–614.
- Katsha AM, Ohkouchi S, Xin H, Paracrine factors of multipotent stromal cells ameliorate lung injury in an elastase-induced emphysema model. Mol Ther 2011; 19:196–203.
- Huh JW, Kim SY, Lee JH, Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am J Physiol Lung Cell Mol Physiol 2011; 301:L255–266.
- Guan XJ, Song L, Han FF, Mesenchymal stem cells protect cigarette smoke-damaged lung and pulmonary funtion partly via VEGF-VEGF receptors. J Cell Biochem 2013; 114(2):323–335.
- Vanoirbeek JA, Rinaldi M, De Vooght V, Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol 2010; 42:96–104.
- Yao H, Edirisinghe I, Rajendrasozhan S, Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol 2008; 294:L1174–1186.
- Dominici M, Le Blanc K, Mueller I, Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8:315–317.
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011; 378(9795):1015–1026.
- Shapiro SD. Transgenic and gene-targeted mice as models for chronic obstructive pulmonary disease. Eur Respir J 2007; 29:375–378.
- Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells 2010; 28:585–596.
- Lee JW, Gupta N, Serikov V, Potential application of mesenchymal stem cells in acute lung injury. Expert Opin Biol Ther 2009; 9(10):1259–1270.
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22:672–688.
- Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007; 87:1047–1082.
- Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax 2010; 65:930–936.
- Lakhdar R, Denden S, Kassab A, Update in chronic obstructive pulmonary disease: role of antioxidant and metabolizing gene polymorphisms. Exp Lung Res 2011; 37:364–375.
- Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis 2011; 6:413–421.
- Yang YC, Zhang N, Van Crombruggen K, Transforming growth factor-beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy 2012; 67(10):1193–1202.
- Budd DC, Holmes AM. Targeting TGFβ superfamily ligand accessory proteins as novel therapeutics for chronic lung disorders. Pharmacol Ther 2012; 135(3):279–291.
- Zandvoort A, Postma DS, Jonker MR, Altered expression of the Smad signaling pathway: implications for COPD pathogenesis. Eur Respir J 2006; 28:533–541.
- Baraldo S, Bazzan E, Turato G, Decreased expression of TGF-β type II receptor in bronchial glands of smokers with COPD. Thorax 2005; 60:998–1002.
- Shull MM, Ormsby I, Kier AB, Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992; 359:693–699.
- Morris DG, Huang X, Kaminski N, Loss of integrin αvβ6-mediated TGF-β activation causes Mmp12-dependent emphysema. Nature 2003; 422:169–173.
- Lixuan Z, Jingcheng D, Wenqin Y, Jianhua H, Baojun L, Xiaotao F. Baicalin attenuates inflammation by inhibiting NK-κB activation in cigarette smoke induced inflammatory models. Pulm Pharmacol Ther 2010; 23:411–419.
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011; 378:1015–1026.