
Abstract
Despite extensive effort, only a few chronic obstructive pulmonary disease (COPD)-associated genes have been suggested, indicating that there must be additional risk-associated loci. Here we aimed to identify additional COPD-associated SNPs and to explore the potential relationship between COPD subgroups and the SNPs in the Korean population. We performed a genome-wide association study (GWAS) with 990 Korean individuals; 102 COPD cases and 544 controls for GWAS using Affymetrix SNP array 5.0, and 173 COPD cases and 171 controls for replication. After validating the candidate single nucleotide polymorphisms (SNP), we performed subgroup analysis by disease phenotype. Through GWAS, we identified a novel SNP in the phosphodiesterase-4D (PDE4D) gene [rs16878037 (C>T), p = 1.66 ◊ 10−6] that was significantly associated with COPD. This signal in PDE4D was successfully replicated in the independent set (p = 0.041). When we combined the discovery and replication data, the association signal became more significant (p = 5.69 ◊ 10−7). In the COPD subgroup analysis, the T allele of rs16878037 was significantly more frequent in COPD patients without severe diffusion capacity impairment (mild mixed and obstruction-dominant group) than in patients with severe impairment (severe mixed and emphysema-dominant groups). This result supports that PDE4D polymorphisms might be involved in the susceptibility to COPD especially in non-emphysematous individuals and that they could also affect the responsiveness of the PDE4 inhibitor treatment.
Introduction
Chronic obstructive pulmonary disease (COPD) is a condition characterized by persistent airway obstruction caused by chronic inflammation in the airway and lung. COPD is a major cause of death and morbidity worldwide and its socioeconomic burden is markedly increasing; indeed it is expected to be the third-leading cause of death in 2020 (Citation1). Cigarette smoking is the most important environmental risk factor for developing COPD. However, only a small fraction of smokers (10%–15%) develops symptomatic COPD, suggesting that genetic components must be associated with COPD (Citation2). A genetic contribution to COPD is supported by the higher concordance rate of COPD in monozygotic versus dizygotic twins and the high heritability of this disease (Citation3).
Indeed, twin and family studies have suggested consistently that COPD results from complex interactions between genetic and environmental factors (Citation4, 5). Accordingly, it has been widely accepted that ethnic differences can influence susceptibility to COPD (Citation6). In Homma et al.'s report, the significant association of phosphodiesterase-4D (PDE4D) gene with COPD was identified in Japanese, but not in Egyptians (Citation7). They suggested that the association of PDE4D polymorphism might be population specific, although COPD is a multifactorial disease with environmental and genetic factors involved in its aetiology.
Despite extensive effort, the only proven genetic risk factor for COPD is mutation of the SERPINA1 gene that causes severe α1-antitrypsin deficiency (Citation8, 9). Although the association has not been widely confirmed yet, the association of PDE4D polymorphisms with COPD and bronchial asthma has been reported in Japanese and Europeans (Citation7, Citation10). PDE4 phosphodiesterase enzyme family inactivates cyclic adenosine monophosphate (cAMP) and cyclic guanine monophosphate (cGMP) via hydrolysis into 5′-AMP and 5′-GMP. (Citation11).
cAMP has a variety of functions as a second messenger in controlling cell functions such as metabolism, differentiation, proliferation and inflammation (Citation8). Increases in intracellular cAMP levels result in activation of protein kinase A and enhanced protein phosphorylation, which in turn leads to inhibitory effects on many inflammatory and immunomodulatory cells (Citation12). PDE4 seems to be especially significant in the pathogenesis of COPD because it is a cAMP inactivating enzyme localized in airway smooth muscle and epithelial cells, nerves as well as inflammatory cells (Citation12, 13). Some PDE4 subtypes were reported to be overexpressed in lung inflammatory cells including alveolar macrophages from tobacco-smoking subjects with COPD compared with cells from controls (Citation14).
With the advent of genome-wide association studies (GWAS), several new candidate genes such as CHRNA3/5, HHIP, GSTO2, FAM13A, and IREB2 have been suggested to be associated with COPD (15–18), which can give us new insight into the pathophysiology of airway obstruction.
In this study, we performed a GWAS to identify COPD-associated SNPs and to explore their potential relationships with COPD subtypes and the SNPs specific to the Korean population. For this purpose, we performed the whole-genome SNP genotyping for 102 patients with COPD and 544 normal controls. To verify the findings in the GWAS discovery, we performed an independent replication test for the significant SNPs with a larger set of subjects (173 cases and 171 controls). After validation, we identified one COPD-associated SNP and observed the association of the SNP with COPD subgroups.
Methods
Study population
For discovery stage of GWAS, 102 COPD patients and 544 healthy individuals were recruited. The 102 COPD patients (97 males and 5 females; mean age, 65.8 ± 10.3 years) were collected from Yeouido St Mary's Hospital. Inclusion criteria of COPD patient ensured that participants had moderate to severe COPD according to 2011 Global Initiative for Chronic Obstructive Lung Disease (GOLD) guideline (Citation19), and smoking history over 10 pack years. The 544 healthy individuals (532 males and 12 females; mean age, 52.5 ± 9.2 years) reporting no COPD history were randomly selected from the Korean Genome Epidemiology Study (KoGES) as controls (Citation20).
For the replication of GWAS signals, 173 COPD patients (168 males and 5 females; mean age, 66.0 ± 7.1 years) and the 171 healthy controls (170 males and 1 female; mean age, 57.0 ± 11.7 years) who were determined to have no history of COPD or other respiratory disease were recruited from the Korean Obstructive Lung Disease (KOLD) cohort. Inclusion criteria are the same as for the GWAS. Details of the COPD criteria in this study are as follows: post bronchodilator FEV1/FVC < 0.7 and more than 10 pack-years of smoking history as well as no definite abnormality on chest radiographs.
For subgroup analysis, we combined the discovery and the replication stage COPD patients (n = 275). Among them, we selected 249 patients with relevant subgroup information. They were grouped into four phenotypic groups based on lung diffusion capacity (DLco) as a marker of the severity of emphysema and forced expiratory volume in the first second (FEV1) as a marker of the severity of airflow obstruction: mild obstruction with mild emphysema (mild mixed; DLco ≥ 50% and FEV1 < 50% of the normal predicted value, n = 109), severe obstruction with mild emphysema (obstruction dominant; DLco ≥ 50% and FEV1 < 50%, n = 71), severe obstruction with severe emphysema (severe mixed; DLco < 50% and FEV1 < 50%, n = 47) and mild obstruction with severe emphysema (emphysema dominant; DLco < 50% and FEV1 ≥ 50%, n = 22). The cut-off values for FEV1 were determined according to the 2011 GOLD guidelines (Citation19), and the cut-off values for DLco were chosen by a pulmonology specialist. As a control of subgroup analysis, we combined the discovery and replication stage controls (n = 715). This study was performed under the ethical review and approval of the Institutional Review Board of the Catholic University Medical College of Korea (CUMC09033).
SNP genotyping and quality control for GWAS
We genotyped 646 samples with Affymetrix Genome-Wide Human SNP array 5.0. Single nucleotide polymorphism (SNP) genotyping was performed using the Birdseed algorithm (Citation21). To remove false associations due to genotyping errors, we performed SNP quality control as follows: SNP call rate >95%, minor allele frequency >5% and Hardy-Weinberg equilibrium P >0.001. After filtration, we obtained 498,187SNPs from the initial 503,590 SNPs to test further association.
Association study
Genome-wide association analysis was performed using the software package PLINK (http://pngu.mgh.harvard.edu/∼purcell/plink/). Logistic regression was applied to test for association after adjusting age and sex. The p values were obtained from the four different models of additive, dominant, recessive and genotypic.
Replication study
Genotyping of the most significantly associated SNP (rs16878037 in the PDE4D gene) was performed by TaqMan analysis (ID: C_33833296_10, Applied Biosystems, Foster, CA, USA) as described elsewhere by using ViiA7 Real-time PCR system (Applied Biosystems) (Citation22).
Statistical analysis
The Hardy–Weinberg equilibrium was estimated using Chi-square tests. In both replication and subgroup analyses, logistic regression was performed using age and gender as covariates with the software package PLINK and SPSS (Version 17, SPSS Inc., Chicago, IL, USA). p < 0.05 was considered statistically significant.
Results
GWAS for COPD-susceptible loci
We performed GWAS with the whole-genome SNP genotyping data for 102 patients with COPD and 544 normal controls. To verify whether the process of SNP quality control effectively removed false associations, we drew a Quantile–Quantile plot based on the P values from a logistic regression analysis (Supplemental Figure S1). Because we did not observe any notable divergence from the null hypothesis in the plot with the genomic inflation factor (λGC) = 1, we performed a GWAS with these filtered SNPs (A).
Figure 1. The plots of GWAS and regional association of PDE4D. A. Manhattan plot of -log10 (p-values) from logistic regression applied to test for association after adjusting age and sex for 102 COPD cases and 544 controls. Arrow head indicates the PDE4D region. B. Regional association plot of the SNPs near PDE4D (upper plot) and LD pattern of rs16878035 and rs16878037 (lower plot). In the association plot, SNPs are represented with diamond dots and the the most strongly associated SNPs (rs16878035 and rs16878037) were marked with the largest diamond. The strength of LD relationship (r2) between the most associated SNP and the other SNPs is presented with red colour intensities. The background recombination rate curve is drawn based on the JPT+CHB HapMap data.
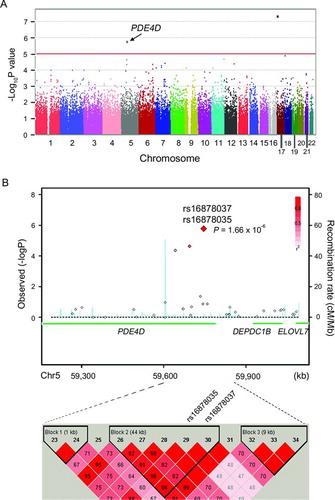
Three SNPs were found to satisfy the significance threshold of p < 10−5 (unadjusted; ). Because it is thought that a single significant SNP tends to be a false positive if it is the solely significant SNP around the region, we did not choose the most significant SNP in the POLR2A gene as the candidate target for replication (Supplemental Figure S2). Both the second and third significant SNPs were in the ≠phosphodiesterase-4D (PDE4D) gene [rs16878037 (C > T), p = 1.66 × 10−6; rs16878035 (G > T), p = 1.66 × 10−6] (B). When we observed the neighbouring SNPs, moderately significant SNPs (p < 10−4) were clustered around them and both were in the same haplotype block (B).
Table 1. Results of genome-wide association study for COPD in Korean population
Therefore, we decided to use PDE4D as a candidate for replication of the association with COPD. In the replication study, we genotyped only the rs16878037 because the nearby rs16878035 is in the same haplotype block. To increase the statistical power, we performed independent replication with a larger set of COPD cases than GWAS discovery (173 cases and 171 controls). As a result, association with the rs16878037 polymorphism was successfully replicated in the independent set (p = 0.041, OR = 11.78, 95% CI 1.1–125.9) (). When we combined the discovery and replication data, the rs16878037 signal became more significant (p = 5.69 × 10-7; OR = 45.69).
Table 2. Results of replication study between PDE4D polymorphisms (rs16878037) and COPD susceptibility in Korean population
Association of the PDE4D polymorphism with COPD phenotypes
Most patients had moderate-to-severe COPD according to the 2011 GOLD guidelines (): mild mixed group (43.8%, 109 cases), obstruction-dominant group (28.5%, 71 cases), severe mixed group (18.9%, 47 cases) and emphysema-dominant group (8.8%, 22 cases). When we compared the genotype frequency of PDE4D rs16878037 C>T between the COPD subgroups and healthy controls, the TT genotype was significantly more frequent than CC genotype in the mild mixed, obstruction-dominant and severe mixed groups than in healthy controls (p = 5.01 × 10−5, 0.001 and 0.013, respectively; ).
Table 3. Association of rs16878037 between control and COPD subgroups
However, although the distribution of TT genotype was significantly different between subgroups and controls, the allele frequency was not high enough to conclude a difference for the TT genotype, especially in the severe mixed group. Therefore, we also compared the T allele frequency between the subgroups and controls. Cohering with the genotype frequency, the T allele of PDE4D rs16878037 C > T was significantly more frequent in the mild mixed and obstruction-dominant groups than in controls (P = 6.79 × 10−5, 6.03 × 10−4, respectively). In contrast, the T allele was significantly less frequent in the severe mixed group (p = 0.01) or was not observed at all in the emphysema-dominant group ().
The mild mixed and obstruction-dominant groups showing significant differences in genotype and allele frequency compared with the control group were those patients with DLco ≥50%. Therefore, we merged them as a group with patients showing DLco ≥50%, and the remaining patients were grouped as having DLco < 50%. When we compared the genotype frequency between these two groups, the CT genotype was significantly more frequent in the patients with DLco ≥50% (p = 0.004) than among patients with DLco <50% (). The TT genotype was also more prevalent in the patients with DLco ≥50% compared with patients with DLco <50% (3.9% vs. 1.4%, respectively), although this was not statistically significant because of the small genotype counts. When we evaluated the combined genotype of CT+TT, they showed more significant difference between the DLco ≥50% and DLco < 50% patient groups than did the TT or CT genotypes alone (p = 0.003).
Table 4. Genotype and allele frequency of the PDE4D (rs16878037) according to impairment of diffusion capacity (DLco)
Discussion
In this study, through GWAS and independent replication, we identified that a SNP in PDE4D gene was associated with a risk of COPD in non-emphysematous Koreans. Association of PDE4D gene polymorphism with COPD has been previously suggested through target-based study in Japanese population (Citation7), but never been supported by GWAS. Although we could not explore the functional consequences of the PDE4D polymorphism in this study, we speculate that the risk allele of rs16878037 accelerates PDE4D enzyme activity, leading to the breakdown of intracellular cAMP. It could aggravate inflammation in the lungs and airways. Consequently, the ability to suppress lung inflammation against harmful substances including tobacco smoke might be low among individuals harbouring the risk allele of the candidate PDE4D polymorphism, so that their susceptibility to COPD would be high. PDE4D gene was also suggested to be linked to bronchial asthma in European subjects by GWAS analysis (Citation10).
Interestingly, in our COPD subgroup analysis, the risk allele of rs16878037 was significantly more frequent in non-emphysematous, obstruct-dominant COPD patients than in emphysema-dominant patients. Airflow limitation in subjects with COPD is mainly caused by inflammation-induced structural changes in the small airways (obstructive bronchiolitis) or destruction of the lung parenchyma caused by imbalance in the activities of proteases and anti-proteases (emphysema) (Citation19). The structural changes caused by inflammation induce fibrosis or luminal plugs in the small airways. In ≠contrast, airway obstruction in emphysema is provoked by loss of alveolar attachments to the small airways and this reduces lung elastic recoil (Citation23).
Thus, COPD is considered not to be just a single disease, but a cluster of several diseases sharing common clinical features of irreversible airflow limitation (Citation24). In this study, consistent with our hypothesis, the COPD subgroups with obstructive bronchitis dominating, but not with a serious reduction in DLco values, the mild mixed and the obstruction-dominant group, had a significantly higher frequency of the risk allele of PDE4D rs16878037 C > T compared with the groups showing destruction of the lung parenchyma, the severe mixed and emphysema-dominant groups.
In the emphysema-dominant subgroup, the risk allele (T allele) was not observed and all of them had the CC type. These results imply that this PDE4D SNP might be involved in the development of inflammation-induced small airway disease rather than in lung parenchymal destruction. Furthermore, such results imply that subjects with a chronic bronchitis type of COPD and an emphysematous type of COPD have different genetic backgrounds as well as pathophysiological mechanisms.
A recently approved second-generation PDE4 inhibitor, roflumilast, has beneficial effects in treating inflammatory airway diseases such as asthma and COPD (Citation25, 26). This drug is the first COPD phenotype-specific therapeutic agent, so it has stimulated the drive to provide a personalized treatment for patients with COPD. Interestingly, it is reported that roflumilast is highly effective for reducing acute exacerbation among patients with chronic bronchitis type COPD, but its efficacy has not been verified in patients with emphysema (Citation25, Citation27). Considering the importance of a reliable marker to predict the treatment response of the phenotype-based specific therapy, our result would be useful to predict the responsiveness of roflumilast treatment.
There were several limitations in this study. First, it was possible to miss some other potentially significant polymorphisms in our GWAS strategy because of the relatively small number of cases in the discovery set. Although successful replication of the significance of this PDE4D polymorphism in the independent set suggests the reliability of our study and compensated for its shortcomings, further larger scale studies will be required to verify the PDE4D signals associated with COPD. Second, individuals in the control group were phenotypically normal but they had not undergone pulmonary function testing, so we cannot rule out the possibility that some had abnormal lung function.
This ‘contamination’ of controls is likely to reduce the effect sizes of SNPs and may lead to masking true associations. In other words, since we may underestimate associations by using contaminated controls, but not exaggerate them, it does not undermine the validity of our results. Third, we did not verify whether these PDE4D polymorphisms really accelerate enzyme activity. Although our subgroup analysis results suggest that PDE4D polymorphisms might be involved in blocking the anti-inflammatory effect of cAMP, further study will be required to probe this mechanism. Fourth, we cannot conclude whether the PDE4D polymorphism identified in this study is specific to Korean non-emphysematous COPD patients or not. However, our result may add more support via GWAS to Homma et al.'s finding of East-Asian specific association of PDE4D with COPD (Citation7). Further larger-scaled GWAS studies including diverse ethnic groups will be useful to determine this.
Conclusion
From GWAS, we identified that a novel SNP in the PDE4D gene was significantly associated with COPD and confirmed the association in the independent set. From subgroup analysis, we additionally observed that the risk allele (T allele) of rs16878037 in PDE4D was significantly more common in the mild mixed and obstruction-dominant groups than in the severe mixed and emphysema-dominant groups in this Korean population. To our knowledge, this is the first report identifying the association between COPD and PDE4D polymorphism through GWAS and also the first report that this polymorphism is specifically involved in the susceptibility to non-emphysematous subgroups. These results will be helpful in understanding the pathophysiology of COPD and could be useful for predicting the treatment responses to second-generation PDE4 inhibitors including roflumilast.
Acknowledgments
The authors thank the KOLD study group, who generously provided the samples of the KOLD cohort. The Consortium for Large Scale Genome Wide Association Study was provided by genotyping data of healthy Korean individuals (genome wide association analysis of community based cohort study, 2007) from Korea Association REsource (KARE), Korea National Institute of Health, Ministry for Health and Welfare, Republic of Korea. As a member of the consortium, we would like to thank KARE for providing part of genotyping data of control group for GWAS. Authors Hyoung-Kyu Yoon and Hae-Jin Hu contributed equally to this paper.
Declaration of Interest Statement
This study was supported by the grants from Korea Healthcare Technology R&D Project (A092258), (A102065), Cancer Evolution Research Canter (2012R1A5A2047939), and GRNP (KRF-2008-220-E00025) Republic of Korea. The authors report no conflicts of interest. The authors are responsible for the content and writing of this paper.
Supplementary materials are available in the online version of this article.
References
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3(11):e442.
- Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance — United States, 1971–2000. MMWR Surveill Summ 2002; 51(6):1–16.
- Ingebrigtsen T, Thomsen SF, Vestbo J, van der Sluis S, Kyvik KO, Silverman EK, Svartengren M, Backer V. Genetic influences on Chronic Obstructive Pulmonary Disease — a twin study. Respir Med 2010; 104(12):1890–1895.
- Hubert HB, Fabsitz RR, Feinleib M, Gwinn C. Genetic and environmental influences on pulmonary function in adult twins. Am Rev Respir Dis 1982; 125(4):409–415.
- Lewitter FI, Tager IB, McGue M, Tishler PV, Speizer FE. Genetic and environmental determinants of level of pulmonary function. Am J Epidemiol 1984; 120(4):518–530.
- Gillum RF. Chronic obstructive pulmonary disease in blacks and whites: pulmonary function norms and risk factors. J Natl Med Assoc 1991; 83(5):393–401.
- Homma S, Sakamoto T, Hegab AE, Saitoh W, Nomura A, Ishii Y, Morishima Y, Iizuka T, Kiwamoto T, Matsuno Y, Massoud HH, Massoud HM, Hassanein KM, Sekizawa K. Association of phosphodiesterase 4D gene polymorphisms with chronic obstructive pulmonary disease: relationship to interleukin 13 gene polymorphism. Int J Mol Med 2006; 18(5):933–939.
- Beavo JA, Brunton LL. Cyclic nucleotide research — still expanding after half a century. Nat Rev Mol Cell Biol 2002; 3(9):710–718.
- Berndt A, Leme AS, Shapiro SD. Emerging genetics of COPD. EMBO Mol Med 2012; 4(11):1144–1155.
- Himes BE, Hunninghake GM, Baurley JW, Rafaels NM, Sleiman P, Strachan DP, Wilk JB, Willis-Owen SA, Klanderman B, Lasky-Su J, Lazarus R, Murphy AJ, Soto-Quiros ME, Avila L, Beaty T, Mathias RA, Ruczinski I, Barnes KC, Celedon JC, Cookson WO, Gauderman WJ, Gilliland FD, Hakonarson H, Lange C, Moffatt MF, O’Connor GT, Raby BA, Silverman EK, Weiss ST. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet 2009; 84(5):581–593.
- Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995; 75(4):725–748.
- Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med. 1998; 157(2):351–370.
- Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Br J Pharmacol 2006; 147 Suppl 1:S252–257.
- Barber R, Baillie GS, Bergmann R, Shepherd MC, Sepper R, Houslay MD, Heeke GV. Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol Lung Cell Mol Physiol 2004; 287(2):L332–343.
- Pillai SG, Kong X, Edwards LD, Cho MH, Anderson WH, Coxson HO, Lomas DA, Silverman EK. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 182(12):1498–1505.
- Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, Ruppert A, Lodrup Carlsen KC, Roses A, Anderson W, Rennard SI, Lomas DA, Silverman EK, Goldstein DB. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet 2009; 5(3):e1000421.
- Wilk JB, Walter RE, Laramie JM, Gottlieb DJ, O’Connor GT. Framingham Heart Study genome-wide association: results for pulmonary function measures. BMC Med Genet 2007; 8 Suppl 1:S8.
- Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua AA, Sparrow D, Lange C, Won S, Murphy JR, Beaty TH, Regan EA, Make BJ, Hokanson JE, Crapo JD, Kong X, Anderson WH, Tal-Singer R, Lomas DA, Bakke P, Gulsvik A, Pillai SG, Silverman EK. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet 2010; 42(3):200–202.
- Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, Stockley RA, Sin DD, Rodriguez-Roisin R. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187(4):347–365.
- Yoo KY, Shin HR, Chang SH, Choi BY, Hong YC, Kim DH, Kang D, Cho NH, Shin C, Jin YW. Genomic epidemiology cohorts in Korea: present and the future. Asian Pac J Cancer Prev. 2005; 6(3):238–243.
- Affymetrix (II). BRLMM–P: a genotype calling method for the SNP 5.0 array. Technical Report. Affymetrix White Paper. 2006. Affymetrix, Inc., Santa Clara, CA, USA (http://media.affymetrix.com/support/technical/whitepapers/brlmmp_whitepaper.pdf).
- Hu HJ, Jin EH, Yim SH, Yang SY, Jung SH, Shin SH, Kim WU, Shim SC, Kim TG, Chung YJ. Common variants at the promoter region of the APOM confer a risk of rheumatoid arthritis. Exp Mol Med 2011; 43(11):613–621.
- Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004; 364(9435):709–721.
- Han MK, Agusti A, Calverley PM, Celli BR, Criner G, Curtis JL, Fabbri LM, Goldin JG, Jones PW, Macnee W, Make BJ, Rabe KF, Rennard SI, Sciurba FC, Silverman EK, Vestbo J, Washko GR, Wouters EF, Martinez FJ. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010; 182(5):598–604.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374(9691):685–694.
- Fan Chung K. Phosphodiesterase inhibitors in airways disease. Eur J Pharmacol 2006; 533(1–3):110–117.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, Rabe KF. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009; 374(9691):695–703.