
Abstract
Background: Cigarette smoking contributes to epithelial-mesenchymal transition (EMT) in COPD small bronchi as part of the lung remodeling process. We recently observed that roflumilast N-oxide (RNO), the active metabolite of the PDE4 inhibitor roflumilast, prevents cigarette smoke-induced EMT in differentiated human bronchial epithelial cells. Further, statins were shown to protect renal and alveolar epithelial cells from EMT. Objectives: To analyze how RNO and simvastatin (SIM) interact on CSE-induced EMT in well-differentiated human bronchial epithelial cells (WD-HBEC) from small bronchi in vitro. Methods: WD-HBEC were stimulated with CSE (2.5%). The mesenchymal markers vimentin, collagen type I and α-SMA, the epithelial markers E-cadherin and ZO-1, as well as β-catenin were quantified by real time quantitative PCR or Western blotting. Intracellular reactive oxygen species (ROS) were measured using the H2DCF-DA probe. GTP-Rac1 and pAkt were evaluated by Western blotting. Results: The combination of RNO at 2 nM and SIM at 100 nM was (over) additive to reverse CSE-induced EMT. CSE-induced EMT was partially mediated by the generation of ROS and the activation of the PI3K/Akt/β-catenin pathway. Both RNO at 2 nM and SIM at 100 nM partially abrogated this pathway, and its combination almost abolished ROS/ PI3K/Akt/β-catenin signaling and therefore EMT. Conclusions: The PDE4 inhibitor roflumilast N-oxide acts (over)additively with simvastatin to prevent CSE-induced EMT in WD-HBEC in vitro.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease characterized by airflow limitation that is progressive and not fully reversible. Currently, it is the fourth leading cause of death worldwide and its impact will continue to rise over the decades to come (Citation1). Cigarette smoking is considered as a key risk factor for COPD and contributes to chronic inflammation and small airway (< 2 mm of internal diameter) remodeling, thus increasing airflow limitation. It has now been revealed that the accelerated decline of FEV1 in COPD is attributed to narrowing and a loss in terminal bronchioles (by up to 90% in very severe COPD) that accounts for severe airway obstruction in the advanced stages of this ailment (Citation2).
Further, based on the observation that terminal bronchiolar architectural remodeling precedes emphysematous changes it was inferred that early emphysematous destruction would predict a rapid FEV1 decline (Citation2). Aside from chronic inflammation, small airway fibrosis may contribute to small airway remodeling. Such process could origin in an activation of resident bronchial fibroblasts, recruitment of fibrocytes or bronchial epithelial to mesenchymal/myofibroblast-like transition (Citation3). The latter is present in small airways in COPD, correlates with lung function and may be reproduced in vitro when well-differentiated human bronchial epithelial cells are exposed to cigarette smoke (Citation4).
The current first-line maintenance treatment for COPD encompasses long-acting bronchodilators (LABA, LAMA) and inhaled corticosteroids (ICS). These therapies confer partial benefits by improving airflow limitation or reducing acute exacerbations. Several emerging classes of anti-inflammatory drugs are in development for COPD treatment, such as inhibitors of phosphodiesterase-4 (PDE4), phosphoinositide-3-kinase-delta (PI3Kδ), p38 kinase or antagonists at the CXCR2 receptor (Citation5).
Roflumilast is the first PDE4 inhibitor approved for COPD. It is indicated as a treatment to reduce the risk of COPD exacerbations in patients with severe COPD associated with chronic bronchitis and a history of exacerbations. Anti-inflammatory effects are currently considered as the key to understand the clinical benefit of roflumilast but results from cellular and animal studies may indicate that the PDE4 inhibitor would also mitigate mucociliary malfunction, the burden of oxidative stress and lung architectural remodelling in COPD. In line, previous reports have shown that PDE4 inhibitors may reduce activation of lung fibroblasts, fibroblast to myofibroblast transition and alveolar epithelial to mesenchymal transition (EMT) induced by TGF-β1 (Citation6–8).
Statins have also been effective in cellular and animal models related to COPD mainly attributed to reducing the activity of small guanosine triphosphatases (GTPases) (Citation9). Indeed aside from lowering the generation of cholesterol the HMG-CoA reductase inhibitors attenuate prenylation of RhoA, Rac1, Cdc42 as prominent examples. Prenylation of these small GTPases would be imperative for their membrane targeting and activity. Given the upstream location of these GTPases in many pathways fostering inflammation and structural remodeling statins exert an array of “pleiotropic effects”. Indeed, statins suppress ROCK by inhibiting RhoA or attenuate NOX1/2 enzymatic activities and reduce reactive oxygen species (ROS) by diminishing activated Rac1. Such mechanisms may account for observations in vivo that statins alleviate emphysema, lung parenchymal and airway inflammation and pulmonary vascular remodeling following long- or short-term tobacco smoke exposure (Citation10–12) or prevent from cigarette smoke-induced airway epithelial injury (Citation13).
Further simvastatin reduced LPS-induced airway neutrophil recruitment in mice (Citation14) and healthy volunteers (Citation15). Observational studies in COPD may herald to some benefit from statins (Citation16). Further, an observational study indicated that the use of statins may limit the risk to develop bronchiolitis obliterans (BOS) following lung transplantation in man (Citation17). Recent studies have shown that statins may also inhibit EMT in renal or TGFβ1-stimulated lung alveolar (A549) epithelial cells in vitro (Citation18, 19).
The current study shall explore the effects of simvastatin alone but then focus on the potential interaction between the PDE4 inhibitor roflumilast N-oxide and simvastatin in terms of EMT in well-differentiated human bronchial epithelial cells (WD-HBEC) exposed to cigarette smoke extract (CSE) in vitro. In a preceding report we described that CSE (2.5% over 3 days) induced EMT in such HBEC cultures contingent on ROS and autocrine TGFβ1 (Citation4). The effects of roflumilast N-oxide on EMT under these experimental conditions were recently described in another manuscript showing inhibitory effects on the EMT process (Citation20). The current study further investigated the effects of statins and the PDE4 inhibitor on a Rac1/PI3K/Akt/β-catenin pathway involved in EMT. Roflumilast N-oxide is the active metabolite largely accounting for the clinical efficacy of roflumilast.
Material
Roflumilast N-oxide (N-(,5-Dichloro-1-oxypyridin-4-yl)-3-cyclopropylmethoxy-4-difluoromethoxybenzamide) was synthesized by Nycomed GmbH, Konstanz, Germany (now Takeda Pharmaceutical Company Ltd., Osaka, Japan). Unless indicated otherwise, all other reagents used were obtained from Sigma Aldrich Quimica SL (Madrid, Spain). Roflumilast N-oxide, simvastatin, ITX3 (2-[(2,5-Dimethyl-1-phenyl-1H-pyrrol-3-yl)methylene]-thiazolo[3,2-a]benzimidazol-3(2H)-one) or LY294002 (2-(4-Morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride) were dissolved in dimethyl sulfoxide (DMSO) at 10 mM stock concentration. Several dilutions of the stocks were performed with cell culture medium. The final concentrations of DMSO (0.1%) in the cell culture did not affect cell viability and functions.
Methods
Isolation of primary human bronchial epithelial cells (HBEC)
Primary HBECs were isolated and cultured from human lung tissue of patients undergoing surgery for lung cancer (approval of the local ethics committee and informed consent was obtained) as previously outlined (Citation4, Citation21). In brief, small pieces of human bronchi (0.5–1 mm internal diameter) were excised from microscopically normal lung areas, carefully dissected free from lung parenchyma and plated on culture dishes in bronchial epithelial growth medium (BEGM, comprising bronchial epithelial basal medium (BEBM) supplemented as previously outlined (Citation4, Citation21). After a period of about 1 week to 12 days, bronchial epithelial cells were observed around bronchi. Cell viability was assessed by vital trypan blue exclusion analysis using the Countness automated cell counter (Life Technologies, Madrid, Spain). Cell viability was > 98% in all cell cultures tested in this work.
Culture of air liquid interface bronchial epithelial cells
Primary HBECs were trypsinized and subpassaged on 12-well polyester Transwell inserts (Millipore) at 1.5 ×105 cells per insert as previously outlined (Citation4, Citation21). Cells were left for 7 days submerged in BEGM/DMEM (1:1) culture medium. From day 7 the air-liquid interface culture was initiated by removing the medium from the upper well leaving the apical side of the cells exposed to air and changing the final epidermal growth factor (EGF) concentration to 0.5 ng/ml (differentiation medium). Cells were maintained at 37°C with 5% CO2 and medium changed every other day. At this stage a pseudo-stratified bronchial epithelium comprising basal cells, ciliated cells and goblet cells was obtained and considered as “well-differentiated” (WD-HBEC) as previously outlined (Citation4, Citation21).
Preparation of cigarette smoke extract and incubations
CSE solutions were prepared as previously described (Citation22). Briefly, the smoke of a research cigarette (2R4F, from Tobacco Health Research, University of Kentucky) was bubbled into a flask containing 25 ml of pre-warmed (37°C) differentiation medium using a respiratory pump model (Harvard Apparatus Rodent Respirator 680, Harvard Apparatus) that operates through a puffing mechanism corresponding to the human smoking pattern (3 puffs min−1; 35 ml per each puff of 2 seconds' duration with a volume of 0.5 cm above the filter). The cigarette smoke solution was then drawn into a syringe through a 0.22-μm pore size filter to remove particles and the tar phase and to obtain a sterile solution (Corning, NY).
The resulting solution was defined as CSE at 100% and used in the different experiments within 30 minutes of preparation following appropriate dilution as indicated. Cytotoxicity possibly emanating from CSE was analysed by exposing differentiated bronchial epithelial cells to 10% CSE for up to 7 days (CSE and culture medium were replaced every Citation24 hours) based on the release of lactate dehydrogenase (LDH) in culture supernatants. No significant differences in LDH activities were observed in the culture supernatants between the CSE and control group (LDH cytotoxicity assay, Cayman, Spain, data not shown).
WD-HBECs were stimulated with CSE (2.5%), replacing culture medium and stimulus every 24 h. Roflumilast N-oxide (2 nM; corresponds to the free plasma concentrations (unbound to plasma protein) after repeated, oral, once-daily dosing of roflumilast at the clinical dose of 500 μg d−1) (Citation23), simvastatin (100 nM–10 μM), (Citation18) ITX3 (10 μM; an inhibitor of TrioRhoGEF→RhoG/Rac1 pathway), (Citation24) LY294002 (10 μM; a non-selective inhibitor of phosphoinositide 3-kinases (PI3K))(Citation25) was added 30 minutes before stimulus and replaced every 24 hours, together with the stimulus.
Both test compounds and CSE were added to the basolateral media (500 μl) and at the apical surface (25 μl). As manipulations at the apical surface may affect pseudo-stratified epithelium, all incubations with vehicle controls were run under identical conditions as with CSE and test compounds (for example identical volumes of medium were added to the apical surface throughout all conditions, same for the basolateral compartment). In control experiments where vehicles/medium (for test compounds/CSE) were added to the apical surface of WD-HBECs over a maximum of 3 days (including daily replacement procedures, as indicated) the number of ciliated cells and expression of cilia markers were found to be not different from cultures of differentiated human bronchial epithelial cells in the absence of the manipulations at the apical surface.
In experiments designed to evaluate EMT markers or NADPH oxidase NOX1, NOX2 or NOX4 units, cells were exposed to CSE and test compounds over 3 days. To determine active, GTP-bound Rac1, Akt phosphorylation (pAkt), cytoplasmic and nuclear β-catenin cells were stimulated with CSE (2.5%) over 15, 30 and 60 min, respectively following pre-incubation with test compounds or vehicle for 30 min.
Immunofluorescence
WD-HBECs were washed three times with PBS and fixed with 4% paraformaldehyde for 30 minutes at room temperature. After another three washes with PBS, differentiated HBECs were permeabilized (20 mM HEPES pH 7.6, 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100), blocked (10% goat serum in PBS) and incubated with the primary antibodies mouse anti-human E-cadherin (cat. No. CM1681; ECM BioScience) and rabbit anti-human collagen type I (cat. No. PA1-26204; Affinity Bioreagents) overnight at 4°C, followed by a secondary anti-rabbit/mouse-IgG FITC antibody (1:100, Molecular Probes). Cells were visualized by epifluorescence microscopy (×200; Nikon eclipse TE200 inverted microscope, Tokyo, Japan). Fluorescence intensity was quantified using the MetaFluor V.5.0 software (Molecular Devices, Sunnyvale, CA) and data shown as Relative Fluorescence Units (RFU).
Real-time RT-PCR
Total RNA was isolated from WD-HBEC in air liquid interface by using TriPure Isolation Reagent (Roche, Indianapolis, USA). The integrity of the extracted RNA was confirmed with Bioanalyzer (Agilent, Palo Alto, CA, USA). The reverse transcription was performed in 300 ng of total RNA with TaqMan reverse transcription reagents kit (Applied Biosystems, Perkin-Elmer Corporation, CA, USA). cDNA was amplified with predesigned kits of forward and reverse primers and probes (Applied Biosystems) for alpha-smooth muscle actin (α-SMA; Hs00559403_m1), α1(I)-collagen (collagen type I; cat. no. Hs00164004_m1), vimentin (cat. no. Hs 00958116_m1), E-cadherin (cat. no. Hs01023894_m1), zona occludens-1 (ZO-1; cat. no. Hs01551861_m1), NOX1 (cat. no. Hs01071088_m1), NOX2 (cat. no. Hs00166163_m1), NOX4 (cat. no. Hs00276431_m1), and GAPDH (pre-designed by Applied Biosystems, cat. no. 4352339E) as a housekeeping gene in a 7900HT Fast Real-Time PCR System (Applied Biosystem) using Universal Master Mix (Applied Biosystems). Relative quantification of these different transcripts was determined with the 2−δδCt method using GAPDH as endogenous control and normalized to the control group.
Western blot
Western blot analysis was instrumental to detect changes in active GTP-bound Rac1, pAkt, nuclear and cytoplasmic β-catenin, vimentin, collagen type I, E-cadherin, ZO-1 in lysates of WD-HBECs. Nuclear and cytoplasmic protein extraction was performed with the nuclear extraction kit from Active Motif (Rixensart, Belgium) according to the manufacturer's instructions. Dilutions of antibodies were 1:1000 unless indicated otherwise. The Bio-Rad assay (Bio-Rad Laboratories Ltd., Herts, UK) was used to quantify the level of protein in each sample to ensure equal protein loading. Sodium dodecyl sulphate polyacrylamide gel electrophoresis was used to separate the proteins according to their molecular weight.
Briefly, 10 μg proteins (denatured) along with molecular weight protein markers (Bio-Rad Kaleidoscope marker, Bio-Rad Laboratories), were loaded onto an acrylamide gel consisting of a 5% acrylamide stacking gel stacked on top of a 10% acrylamide resolving gel and run through the gel by application of 100 V for 1 h. Proteins were transferred from the gel to a polyvinylidene difluoride membrane using a wet blotting method. The membrane was blocked with 5% Marvel in PBS containing 0.1% Tween 20 (PBS-T) and then probed with a mouse anti-human vimentin (cat. no. V6389; Sigma), rabbit anti-human collagen type I (cat. no. PA1-26204; Affinity Bioreagents), mouse anti-human E-cadherin (cat. no. CM1681; ECM BioScience), rabbit anti-human ZO-1 (cat. no. ab59720; Abcam), rabbit anti-human NOX4 (cat. no. NB110-58849; Novus Biologicals, Cambridge, UK), rabbit anti-human β-catenin (cat. no: NBP1-89989; Novus Biologicals) and rabbit anti-human pAkt (Thr308; cat. no. 2965; Cell Signalling) antibody.
Active GTP-bound Rac1 was determined with the active Rac1 detection kit from Cell Signalling (cat. no. 8815) according to manufacturer's instructions. Briefly, GTP-bound Rac1 was captured by a GST-PAK1-PBD fusion protein and the complex immunoprecipated with GSH-resin. Rac1 in the immunoprecipitate was detected by Western blotting using the provided mouse antibody specifically raised against human Rac1 and a secondary HRP-coupled anti mouse IgG antibody. Protein expression was normalised to total β-actin (detected with mouse anti-human β-actin, cat. no.A1978; Sigma), Akt (detected with rabbit anti-human Akt, cat. no. 4691; Cell Signalling) and Rac1 (detected with rabbit anti human Rac1 cat. no. 2465; Cell Signalling) for cytoplasmic protein expression and to lamin A (rabbit anti human antibody cat. no. L1293; Sigma) for nuclear protein expression.
ECL plus (Amersham GE Healthcare, Buckinghamshire, UK) was used to detect labelled proteins. Quantification of protein expression was performed by densitometry relative to β-actin, Akt, Rac1, or lamin A expression using the GeneSnap software, version 6.08.
Measurements of reactive oxygen species
The compound 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCF-DA, Molecular Probes, UK) is a cell-permeable analogue of H2DCF that following intracellular ester hydrolysis is oxidized to fluorescent 2′, 7′-dichlorofluorescein (DCF) by O2− and H2O2, and can therefore be used to monitor intracellular generation of ROS (Citation26). To quantify ROS levels, WD-HBEC were washed twice with PBS and incubated for 30 minutes with 50 μM H2DCF-DA diluted in Opti-MEM in presence or absence of roflumilast N-oxide or simvastatin.
Then, cells were again washed twice with PBS to remove remaining H2DCF-DA and stimulated with CSE for 30 minutes in presence or absence of the same compounds indicated before. Five randomly selected fields per condition were measured for fluorescent intensity using an epifluorescence microscope (Nikon Eclipse TE 200, Tokyo, Japan) with filter set for FITC. Subsequent image capture and analysis was performed using Metafluor 5.0 software (Molecular Devices, Sunnyvale, CA). Results were expressed as DCF fluorescence in relative fluorescence units.
Statistical analysis
Data are presented as mean ± SEM of n experiments. Statistical analysis of data was performed by analysis of variance (ANOVA) followed by Bonferroni test (GraphPad Software Inc., San Diego, CA, USA). Significance was accepted when p < 0.05.
Results
Roflumilast N-oxide and simvastatin alleviated cigarette smoke-induced epithelial to mesenchymal transition
Simvastatin (100 nM–10 μM) concentration-dependently reversed the loss in E-cadherin and ZO-1 epithelial phenotype mRNA transcripts and reduced the increase in αSMA, type I collagen and vimentin mesenchymal phenotype mRNA transcripts following exposure of WD-HBEC to CSE at 2.5% over 3 days indicating that the statin may attenuate CSE-induced EMT in these cells (Figures and ). As previously reported, roflumilast N-oxide at 2 nM (a concentration in the range of therapeutic plasma concentrations) partly mitigates the CSE-induced EMT in WD-HBEC in vitro (Citation20).
Figure 1. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on cigarette smoke extract (CSE)-induced loss of epithelial markers in WD-HBECs, mRNA analyses. WD-HBECs were incubated with SIM (100 nM–10 μM), RNO (2 nM), or RNO (2 nM) and SIM (100 nM) together for 30 minutes before exposure to CSE (2.5%) for 72 h. Total RNA was isolated for real-time RT-PCR analysis of epithelial markers E-cadherin and ZO-1. Data are expressed as the ratio to GAPDH and normalized to the vehicle control group. Results are expressed as means ± SEM of n = 3–4 independent experiments per condition. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; # p < 0.05 related to CSE. *# p < 0.05 related to RNO (2 nM) or SIM (100 nM) alone.
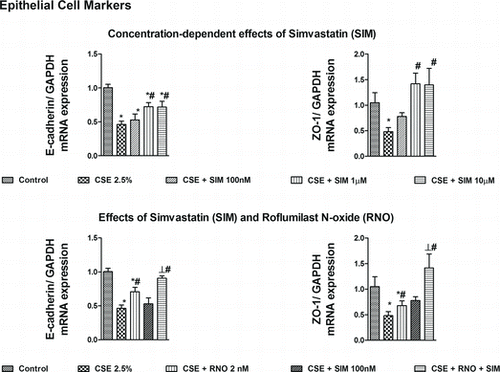
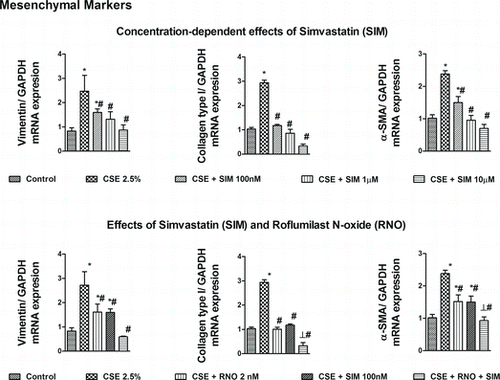
Figure 2. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on cigarette smoke extract (CSE)-induced mesenchymal markers in WD-HBECs, mRNA analyses. WD-HBECs were incubated with SIM (100 nM–10 μM), RNO (2 nM), or RNO (2 nM) and SIM (100 nM) together for 30 minutes before exposure to CSE (2.5%) for 72 hours. Total RNA was isolated for real time RT-PCR analysis of mesenchymal markers vimentin, collagen type I and alpha smooth muscle actin (α-SMA). Data are expressed as the ratio to GAPDH and normalized to the vehicle control group. Results are expressed as means ± SEM of n = 3–4 independent experiments per condition. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. *# p < 0.05 related to RNO (2 nM) or SIM (100 nM) alone.
When this concentration of roflumilast N-oxide and simvastatin at 100 nM were added together the effects to reverse EMT markers were higher than with either the PDE4 inhibitor or the statin alone (Figures and ). For example, roflumilast N-oxide (2 nM) or simvastatin (100 nM) reversed the compromised expression of E-cadherin transcripts following CSE by 45% or 12%, while the combination of both resulted in a recovery by 82%. In case of ZO-1 when the PDE4 inhibitor and the statin where added together, a 164% rescue from the CSE-induced loss in transcripts was observed (which is in excess compared to the expression at baseline), yet roflumilast N-oxide (2 nM) and simvastatin (100 nM) alone resulted in a reversal by 34% and 53%, respectively.
For the mesenchymal markers roflumilast N-oxide (2 nM) and simvastatin (100 nM) both partially reduced vimentin and αSMA, while their combination abolished the increment in these transcripts secondary to CSE. The expression of collagen type I that was abrogated by either roflumilast N-oxide (2 nM) or simvastatin (100 nM) was suppressed below baseline when these were incubated together (Figure ).
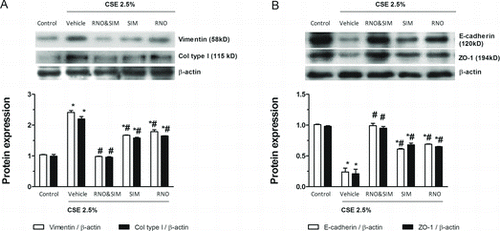
Comparable effects of roflumilast N-oxide (2 nM) and simvastatin (100 nM) were confirmed in terms of expressed proteins. As depicted in Figure , densitometric analyses of Western blots for vimentin and collagen type I or E-cadherin and ZO-1 respresenting epithelial and mesenchymal markers revealed that co-incubation with roflumilast N-oxide and simvastatin fully reversed the loss in E-cadherin or ZO-1 or the increase in vimentin and collagen type 1 secondary to CSE (2.5%) over 3 days. And the PDE4 inhibitor and the statin, on their own, were only partially effective.
Figure 3. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on cigarette smoke extract (CSE)-induced epithelial to mesenchymal transition (EMT) in WD-HBECs, protein analyses. WD-HBECs were incubated with RNO (2 nM), SIM (100 nM–10 μM), or RNO (2 nM) and SIM (100 nM) together for 30 minutes before exposure to CSE (2.5%) for 72 hours (A, B). Cells were lysed and protein was detected using antibodies against vimentin, collagen type I, E-cadherin or ZO-1 with β-actin as control by Western blotting as detailed in Methods. A representative Western blot as well as summaries from densitometric evaluations of n = 3 independent experiments are illustrated. Results are expressed as means ± SEM. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; # p < 0.05 related to CSE.
These findings were supported and complemented by immunofluorescence (IF) analyses (Figure ). First of all, WD-HBEC exposed to CSE (2.5%) over 3 days adopted a flattened and elongated morphology characteristic of a myofibroblast-like phenotype. The epithelial cell phenotype appeared protected when cells were co-incubated with roflumilast N-oxide (2 nM) and simvastatin (100 nM) in spite of the presence of CSE. Each on their own the statin or the PDE4 inhibitor partially prevented the CSE-induced morphological changes (left column of microphotographs in Figure and quantification).
Figure 4. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on cigarette smoke extract (CSE)-induced phenotype alterations in WD-HBECs. (A) WD-HBECs exposed to air exhibit typical cobblestone morphology. Following exposure to CSE (2.5 %) for 72 hours a mesenchymal morphology emerged with reduced cell-cell contact in phase contrast microscopy. These morphological changes were partially prevented by RNO (2 nM) and SIM (100 nM) but abolished by their combination. In line, immunofluorescence analyses revealed reduced E-cadherin but increased collagen type I following CSE (2.5%). RNO (2 nM) and SIM (100 nM) fully prevented these effects while on their own they were partially effective. Representative images are shown. Scale bar: 20 μm. (B) Quantification of immunofluorescence images in relative fluorescence units of n = 3 experiments per condition. Results are expressed as the mean ± SEM. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. *# p < 0.05 related to drug monotherapy.
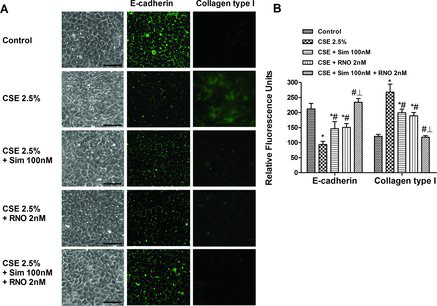
In IF, the expression of the tight junction protein E-cadherin was visualized as a continuous line of fluorescence around the margin of the cells in the controls. In contrast, cells exposed to CSE showed a reduction of E-cadherin fluorescence intensity with sporadic and diffuse expression. The mesenchymal marker collagen type I was absent in control cells, while it increased following exposure to CSE. Either roflumilast N-oxide (2 nM) or simvastatin (100 nM) on their own partially attenuated the loss of E-cadherin and the increase of collagen type I, while their co-incubation almost abolished the effect of CSE on these EMT markers (Figure ).
Roflumilast N-oxide and simvastatin reduced reactive oxygen species and NADPH oxidase isoenzymes increased by cigarette smoke
Generation of oxidative stress by cigarette smoke partially mediates EMT in airway epithelial cells (Citation4). Therefore, it would be of interest to explore the effects of roflumilast N-oxide and simvastatin on cigarette smoke-induced ROS and NADPH oxidase expression. When WD-HBEC were exposed to CSE at 2.5% over 3 days the expression of the NADPH oxidase isoenzymes NOX1, NOX2 (gp91phox) (mRNA) and NOX4 (mRNA and protein) increased (). This up-regulation was partially reduced by either roflumilast N-oxide at 2 nM or simvastatin at 100 nM but abolished when the PDE4 inhibitor and the statin were added together ().
Figure 5. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on the increase in reactive oxygen species (ROS) and NADPH oxidases NOX1, NOX2 and NOX4 induced by CSE in WD-HBEC. (A-D) WD-HBEC were pre-incubated with RNO (2 nM), SIM (100 nM) or their combination for 30 minutes and then exposed to CSE (2.5%) over 72 hours. NOX1, NOX2 and NOX4 mRNA and NOX4 protein were quantified by real-time quantitative RT-PCR and Western blot, respectively as described in Methods. (E, F) WD-HBECs were loaded with H2DCF-DA in presence of RNO (2 nM), SIM (100 nM) or their combination for 30 minutes. Excess H2DCF-DA was removed by washing with PBS. Compounds were replenished before cells were exposed to CSE at 2.5%. Following a 30-minute incubation period accumulation of fluorescent DCF was assessed with fluorescence microscopy in a total of 5 fields per condition as detailed in Methods. (E) Quantitative evaluation of DCF fluorescence. (F) Representative microphotographs (scale bar corresponds to 50 μm). Results are given as means ± SEM of n = 4–5 independent experiments per condition. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. *# p < 0.05 related to RNO or SIM alone. CTR: control.
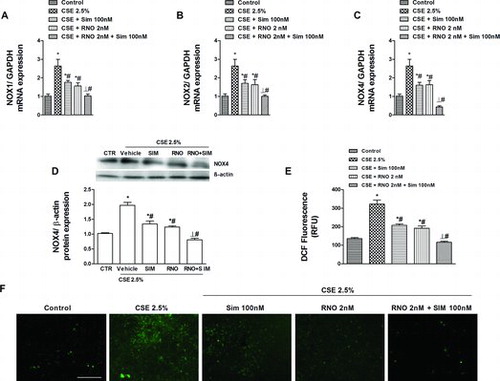
Intracellular ROS was quantified as fluorescent 2′,7′-dichlorofluorescein (DCF) following oxidation from H2DCF-DA. Exposure of WD-HBECs to CSE at 2.5% over 30 minutes resulted in an about 2.4-fold increase in DCF accumulation over control. Pre-incubation with either simvastatin (100 nM) or roflumilast N-oxide (2 nM) decreased this increment in DCF accumulation by about 60% and 70%, respectively while the combination suppressed intracellular ROS below control levels ().
Effects of roflumilast N-oxide, simvastatin and its combination on cigarette smoke-induced Rac1 and Akt activation: Implications on EMT
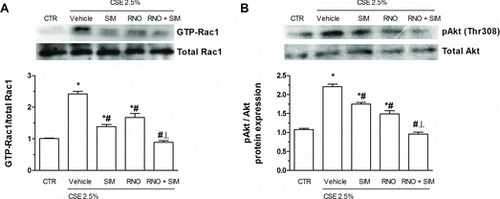
Activation of Rac1 or of the phosphoinositide 3-kinase (PI3K)/Akt pathway is considered to impart oxidative stress or EMT entailed by cigarette smoke in bronchial epithelial cells at least in part (Citation27). In this study, when exposed to CSE (2.5%) activated, GTP-bound Rac1 (measured at Citation15 minutes of CSE) and Akt phosphorylation (measured at 30 minutes of CSE) were increased (Fig A and B). Either roflumilast N-oxide (2 nM) or simvastatin (100 nM) partially reduced this increment in activated, GTP-bound Rac1 and Akt phosphorylation while the PDE4 inhibitor and the statin together reversed these increases to baseline ().
Figure 6. Effects of roflumilast N-oxide (RNO) and simvastatin (SIM) on active, GTP-bound Rac1 and Akt phosphorylation enhanced by cigarette smoke extract (CSE). WD-HBEC were pre-incubated with RNO (2 nM), SIM (100 nM) or their combination for 30 minutes and then exposed to CSE (2.5%) over (A) 15 minutes or (B) 30 minutes. Cells were lysed and proteins detected using a GTP-Rac1 detection kit and pAkt antibody as detailed in Methods. A representative Western blot as well as results from densitometric evaluations are illustrated. Means ± SEM of n = 3–4 independent experiments per condition. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. *# p < 0.05 related to RNO or SIM alone. CTR: control.
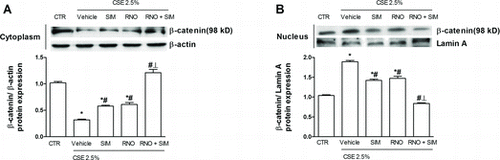
β-catenin is involved in EMT and activated in primary HBECs from small bronchi in COPD (Citation4). In the current study CSE 2.5% (1 hour) enhanced the nuclear translocation of β-catenin in WD-HBEC. This is illustrated by increased nuclear yet reduced cytoplasmic β-catenin (). Pre-incubation with either roflumilast N-oxide (2 nM) or simvastatin (100 nM) partly attenuated the nuclear translocation of β-catenin, while the combination retained β-catenin in the cytosol ().
Figure 7. Effects of roflumilast N-oxide and simvastatin on cigarette smoke extract (CSE)-induced β-catenin nuclear translocation. WD-HBEC were pre-incubated with RNO (2 nM), SIM (100 nM) or their combination for 30 minutes and then exposed to CSE (2.5%) over 60 min. Cells were lysed and (A) cytoplasmic or (B) nuclear protein were detected using a β-catenin antibody as detailed in Methods. A representative Western blot as well as results from densitometric evaluations against β-actin (cytoplasma) or lamin A (nucleus) are shown. Means ± SEM of n = 3 independent experiments per condition. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. *# p < 0.05 related to RNO and SIM alone. CTR: control.
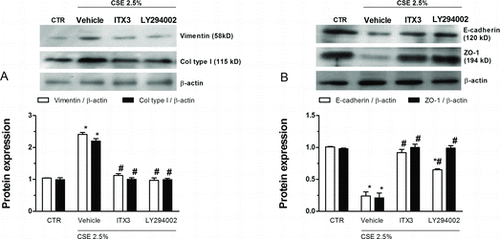
To explore whether Rac1 and PI3K/Akt pathways may be involved in CSE-induced EMT in WD-HBEC ITX3 (10 μM) and LY294002 (10 μM) were instrumental to inhibit active, GTP-bound Rac1 and PI3K. Both, ITX3 or LY294002 were able to reverse the enhanced vimentin and collagen type I mesenchymal and the reduced ZO-1 epithelial EMT marker (protein) secondary to CSE exposure over 3 day (). The CSE-induced loss in E-cadherin was partly reversed by LY294002 while it was fully restored by ITX3 ().
Figure 8. Effects of Rac1 or PI3K inhibition on cigarette smoke extract (CSE)-induced epithelial to mesenchymal transition. WD-HBEC were pre-incubated with either the TrioRhoGEF inhibitor ITX3 (10 μM), or the pan-PI3K inhibitor LY294002 for 30 minutes followed by exposure to CSE (2.5%) over 72 hours. Cells were lysed and protein was detected using antibodies against vimentin, collagen type I, E-cadherin, ZO-1 as detailed in Methods. Results from densitometric evaluations of Western blots are shown as the means from n = 3 independent experiments per condition ± SEM. One-way ANOVA followed by post hoc Bonferroni tests. *p < 0.05 related to vehicle controls; #p < 0.05 related to CSE. CTR: control.
Discussion
Expanding on previous work from others and our own group, in the current study the HMG-CoA reductase inhibitor simvastatin concentration-dependently prevented EMT following long-term exposure of bronchial epithelial cells to cigarette smoke. Simvastatin (100 nM) was mostly additive to the PDE4 inhibitor roflumilast N-oxide (2 nM) to reverse the CSE-induced loss in the epithelial markers E-cadherin and ZO-1 and increase in the mesenchymal markers collagen type I, vimentin and αSMA. Simvastatin (100 nM) and roflumilast N-oxide (2 nM) on their own partly reduced CSE-induced activated GTP-bound Rac1, ROS, NOX1, 2 and 4, pAkt and nuclear β-catenin while these increments were abolished with the statin and the PDE4 inhibitor together.
Previously this laboratory showed evidence for EMT associated with increased nuclear β-catenin in small airways of smokers and COPD (Citation4). In the current study, nuclear accumulation of β-catenin paralleled EMT in the in vitro model of CSE-exposed WD-HBEC. In fact, β-catenin activates TCF/Lef1 supporting mesenchymal transition (Citation28). CSE augmented pAkt in WD-HBEC and the panPI3K inhibitor LY294002 prevented EMT. pAkt increases nuclear β-catenin (Citation27). A critical role of oxidative stress and NOX isoenzymes to impart CSE-induced EMT was described before by us (Citation4) in WD-HBEC and others (Citation29) in HBEC.
The increase in activated, GTP-bound Rac1 following short-term exposure of WD-HBEC to CSE corroborating previous observations (Citation30) can be expected to augment ROS generation from NOX(1&2) isoenzymes. That activated Rac1 is involved in EMT secondary to CSE in WD-HBEC is supported by the finding that ITX3 prevented the increased vimentin and loss in E-cadherin expression. ITX3 ([1,3] thiazolo [3,2-a]benzimidazole-3(2H)-one) was described as an inhibitor, albeit modest (IC50 to inhibit TrioN-medicated GDP→GTP exchange on RhoG at 76 μM) of the interaction between the GTP Exchange Factor (GEF) TrioN with RhoG or Rac1 (Citation31).
Accommodating these results others showed that both a small molecule inhibitor and siRNA against Rac1 mitigate EMT secondary to CSE in A549 cells (Citation32). These authors further demonstrated that an enhanced pAkt/Akt ratio following CSE was blocked by Rac1 suppression with the pharmacological inhibitor or the siRNA in A549 cells. Finally, in leukocytes (U937 monocytic cells, neutrophils) CSE rapidly enhances PI3Kδ activity, hence Akt phosphorylation (Citation33–35). Taken together, as a plausible hypothesis EMT secondary to CSE in WD-HBEC evolves by a sequential activation of Rac1, generation of ROS, increase in PI3K activity, hence phosphorylated Akt and nuclear β-catenin. In line, it was shown that CSE enhances nuclear β-catenin in A549 cells (Citation36).
As both PDE4 inhibitors and statins are able to diminish CSE-induced EMT in WD-HBEC (Citation4, Citation19, Citation20) their effect when added together was addressed. In clinical practice patients with severe COPD on roflumilast may occasionally receive statins given that cardiovascular and metabolic ailments are common comorbidities (Citation37). Further, statins may display favorable effects in COPD (Citation9, Citation16). In the COPDGene study, the use of statins was associated with less small airway narrowing as determined by HRCT scanning supporting the notion that statins may improve small airways disease (Citation38). However, STATCOPE, a prospective, randomized, placebo-controlled, parallel group did not show a therapeutic benefit of simvastatin (Citation40 mg/d PO) in patients with moderate-severe COPD in terms of the rate of acute exacerbations, the time to first exacerbations, lung function, quality of life or mortality over an at least 1 year's treatment period (Citation39).
To explore effects of roflumilast N-oxide and simvastatin together the PDE4 inhibitor was used at 2 nM and the statin at 100 nM. Roflumilast N-oxide at 2 nM corresponds to plasma levels (unbound to protein) following the clinical dose of 500 μg/d. Simvastatin at 100 nM is at the lower end of common concentrations in in vitro studies (0.1–10 μM) but higher than peak plasma concentrations in man at standard doses of 20 or 40 mg/d and the IC50 to inhibit HMG-CoA reductase. On the other hand, in vivo simvastatin was taming airspace enlargement, lung parenchymal inflammation, pulmonary hypertension and pulmonary vascular remodelling following 4 months of tobacco smoke exposure in rats at doses resulting in plasma concentrations close to exposure at standard doses used for lipid lowering in man (Citation10). Why higher concentrations of simvastatin are required to observe effects in in vitro studies (except hepatocytes) is not clear.
Overall, the effects from roflumilast N-oxide (2 nM) and simvastatin (100 nM) together to reverse the cigarette smoke-induced loss in epithelial markers E-cadherin and ZO-1 and the increase in mesenchymal markers vimentin, collagen type I, α-SMA transcripts or (where measured) protein in WD-HBEC where higher than achieved with the PDE4 inhibitor or the statin alone, in most cases additive. Such additive effects extended to the morphologic phenotype as the CSE-induced flattening and elongation of the cell pattern, loss in plasma membrane E-cadherin and emergence of cytosolic collagen type I (measured by IF) was fully prevented by the presence of both simvastatin and the PDE4 inhibitor.
That the PDE4 inhibitor roflumilast N-oxide (2 nM or 1 μM) reduces the rapid (likely GTP-Rac1 → NOX1/2 mediated) ROS generation as well as the long-term upregulation of NOX1, 2 and 4 secondary to CSE has been disclosed before by this laboratory (Citation20, 21). Attenuation of activated, GTP-bound Rac1 by cAMP is described in U46619-stimulated human vascular smooth muscle cells (Citation40). In line, roflumilast N-oxide reduces GTP-Rac1 and ensuing NOX1-dependent ROS generation in human pulmonary artery smooth muscle cells stimulated with U46619 (Citation41). Why a PDE4 inhibitor/cAMP curbs GTP-bound Rac1 is not explored in detail. A discussed mechanism is a PKA-dependent inhibitory phosphorylation of a Rac1-activated GEF, such as P-rex1 (Citation42). Effects of cAMP on Rac1 are cell-type specific, as in endothelial cells, an increase in cAMP enhances GTP-bound Rac1 (Citation43). Reduced GTP-bound Rac1 with simvastatin is explained by the prenylation inhibition of small G proteins imparted by statins (Citation9).
Roflumilast N-oxide and simvastatin may prevent the CSE-induced increase of GTP-bound Rac1 by complementary mechanisms resulting in an additive interaction. While the statin limits geranylgeranylation hence, Rac1 membrane docking the PDE4 inhibitor would curb the GDP→GTP exchange. Given the critical role of GTP-Rac1 for NOX1/NOX2 activity the additive effects of roflumilast N-oxide and simvastatin to suppress CSE-induced ROS may reflect their inhibition of GTP-Rac1. As previously shown removal of ROS protects from CSE-induced EMT in WD-HBEC (Citation4). Therefore, that roflumilast N-oxide and simvastatin were additive to prevent CSE-induced EMT could be attributed to their suppression of CSE-induced ROS in WD-HBEC.
That simvastatin and roflumilast N-oxide prevented CSE-induced activation of the PI3K/Akt pathway and the ensuing increase in nuclear β-catenin in WD-HBEC is a novel finding. One plausible explanation however, may be their ability to attenuate CSE-induced ROS in WD-HBEC.
In conclusion the current work shows additive effects of the PDE4 inhibitor roflumilast N-oxide and the HMG-CoA reductase inhibitor simvastatin to mitigate EMT secondary to tobacco smoke in well-differentiated human bronchial epithelial cells. The PDE4 inhibitor and the statin may act on different pathways involved in CSE-induced EMT reflected by inhibition of GTP-Rac1, ROS, PI3K/Akt and nuclear β-catenin.
Declaration of Interest Statement
JC received a research grant from Nycomed. HT and JA are full-time employees of Takeda. The other authors declare no conflict of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
Both authors Milara and Peiró contributed equally to this work.
Additional information
Funding
References
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364:613–620.
- Hogg JC, McDonough JE, Suzuki M. Small airway obstruction in COPD: New insights based on micro-ct imaging and mri imaging. Chest 2013; 143:1436–1443.
- Burgel PR, Bourdin A, Chanez P, Chabot F, Chaouat A, Chinet T, de Blic J, Devillier P, Deschildre A, Didier A, Garcia G, Jebrak G, Laurent F, Morel H, Perez T, Pilette C, Roche N, Tillie-Leblond I, Verbanck S, Dusser D. Update on the roles of distal airways in COPD. Eur Respir Rev 2011; 20:7–22.
- Milara J, Peiro T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013; 68:410–420.
- Matera MG, Calzetta L, Segreti A, Cazzola M. Emerging drugs for chronic obstructive pulmonary disease. Expert Opin Emerg Drugs 2012; 17:61–82.
- Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A. Inhibition of tgf-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol 2007; 572:12–22.
- Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, Seeger W, Banat GA, Schermuly RT, Pullamsetti SS. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: The role of phosphodiesterase 4. Mol Biol Cell 2009; 20:4751–4765.
- Selige J, Hatzelmann A, Dunkern T. The differential impact of pde4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. J Cell Physiol 2011; 226:1970–1980.
- Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: Potential utility in COPD. Eur Respir Rev 2009; 18:222–232.
- Lee JH, Lee DS, Kim EK, Choe KH, Oh YM, Shim TS, Kim SE, Lee YS, Lee SD. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med 2005; 172:987–993.
- Wan WY, Morris A, Kinnear G, Pearce W, Mok J, Wyss D, Stevenson CS. Pharmacological characterisation of anti-inflammatory compounds in acute and chronic mouse models of cigarette smoke-induced inflammation. Respir Res 2010; 11:126.
- Wright JL, Zhou S, Preobrazhenska O, Marshall C, Sin DD, Laher I, Golbidi S, Churg AM. Statin reverses smoke-induced pulmonary hypertension and prevents emphysema but not airway remodeling. Am J Respir Crit Care Med 2011; 183:50–58.
- Davis BB, Zeki AA, Bratt JM, Wang L, Filosto S, Walby WF, Kenyon NJ, Goldkorn T, Schelegle ES, Pinkerton KE. Simvastatin inhibits smoke-induced airway epithelial injury: Implications for COPD therapy. Eur Respir J 2013; 42(2):350–361.
- Grommes J, Vijayan S, Drechsler M, Hartwig H, Morgelin M, Dembinski R, Jacobs M, Koeppel TA, Binnebosel M, Weber C, Soehnlein O. Simvastatin reduces endotoxin-induced acute lung injury by decreasing neutrophil recruitment and radical formation. PLoS One 2012; 7:e38917.
- Shyamsundar M, McKeown ST, O'Kane CM, Craig TR, Brown V, Thickett DR, Matthay MA, Taggart CC, Backman JT, Elborn JS, McAuley DF. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am J Respir Crit Care Med 2009; 179:1107–1114.
- Janda S, Park K, FitzGerald JM, Etminan M, Swiston J. Statins in COPD: A systematic review. Chest 2009; 136:734–743.
- Johnson BA, Iacono AT, Zeevi A, McCurry KR, Duncan SR. Statin use is associated with improved function and survival of lung allografts. Am J Respir Crit Care Med 2003; 167:1271–1278.
- Patel S, Mason RM, Suzuki J, Imaizumi A, Kamimura T, Zhang Z. Inhibitory effect of statins on renal epithelial-to-mesenchymal transition. Am J Nephrol 2006; 26:381–387.
- Yang T, Chen M, Sun T. Simvastatin attenuates tgf-beta1-induced epithelial-mesenchymal transition in human alveolar epithelial cells. Cell Physiol Biochem 2013; 31:863–874.
- Milara J, Peiro T, Serrano A, Guijarro R, Zaragoza C, Tenor H, Cortijo J. Roflumilast n-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther 2014, in press.
- Milara J, Armengot M, Banuls P, Tenor H, Beume R, Artigues E, Cortijo J. Roflumilast n-oxide, a pde4 inhibitor, improves cilia motility and ciliated human bronchial epithelial cells compromised by cigarette smoke in vitro. Br J Pharmacol 2012; 166:2243–2262.
- Ortiz JL, Milara J, Juan G, Montesinos JL, Mata M, Ramon M, Morcillo E, Cortijo J. Direct effect of cigarette smoke on human pulmonary artery tension. Pulm Pharmacol Ther 2010; 23:222–228.
- Bethke TD, Bohmer GM, Hermann R, Hauns B, Fux R, Morike K, David M, Knoerzer D, Wurst W, Gleiter CH. Dose-proportional intraindividual single- and repeated-dose pharmacokinetics of roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor. J Clin Pharmacol 2007; 47:26–36.
- Yano T, Yamazaki Y, Adachi M, Okawa K, Fort P, Uji M, Tsukita S. Tara up-regulates e-cadherin transcription by binding to the trio rhogef and inhibiting rac signaling. J Cell Biol 2011; 193:319–332.
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 2000; 275:36803–36810.
- Trayner ID, Rayner AP, Freeman GE, Farzaneh F. Quantitative multiwell myeloid differentiation assay using dichlorodihydrofluorescein diacetate (h2dcf-da) or dihydrorhodamine 123 (h2r123). J Immunol Meth 1995; 186:275–284.
- Dasari V, Gallup M, Lemjabbar H, Maltseva I, McNamara N. Epithelial-mesenchymal transition in lung cancer: Is tobacco the “smoking gun”? Am J Respir Cell Mol Biol 2006; 35:3–9.
- Wang Q, Wang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res 2013; 14:67.
- Gorowiec MR, Borthwick LA, Parker SM, Kirby JA, Saretzki GC, Fisher AJ. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a tgf-beta1-dependent mechanism. Free Radic Biol Med 2012; 52:1024–1032.
- Zhang L, Gallup M, Zlock L, Finkbeiner WE, McNamara NA. Rac1 and cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and independent pathways. Am J Pathol 2013; 182(6):1982–1995.
- Bouquier N, Vignal E, Charrasse S, Weill M, Schmidt S, Leonetti JP, Blangy A, Fort P. A cell active chemical gef inhibitor selectively targets the trio/rhog/rac1 signaling pathway. Chem Biol 2009; 16:657–666.
- Shen HJ, Sun YH, Zhang SJ, Jiang JX, Dong XW, Jia YL, Shen J, Guan Y, Zhang LH, Li FF, Lin XX, Wu XM, Xie QM, Yan XF. Cigarette smoke-induced alveolar epithelial-mesenchymal transition is mediated by rac1 activation. Biochim Biophys Acta 2014; 1840:1838–1849.
- Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, Ito K, Adcock IM, Kirkham PA, Papi A. Inhibition of pi3kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med 2009; 179:542–548.
- Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast n-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2014; 134(2):314–322.
- To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, Elliott WM, Hogg JC, Adcock IM, Barnes PJ. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 182:897–904.
- Tian D, Zhu M, Li J, Ma Y, Wu R. Cigarette smoke extract induces activation of beta-catenin/tcf signaling through inhibiting gsk3beta in human alveolar epithelial cell line. Toxicol Lett 2009; 187:58–62.
- Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J 2009; 33:1165–1185.
- Young RP, Hopkins RJ. Statins and small airways disease in COPD. Am J Respir Cell Mol Biol 2013; 49:501.
- Criner GJ, Connett JE, Aaron SD, Albert RK, Bailey WC, Casaburi R, Cooper JA Jr., Curtis JL, Dransfield MT, Han MK, Make B, Marchetti N, Martinez FJ, Niewoehner DE, Scanlon PD, Sciurba FC, Scharf SM, Sin DD, Voelker H, Washko GR, Woodruff PG, Lazarus SC. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med 2014; 370:2201–2210.
- Muzaffar S, Shukla N, Bond M, Sala-Newby G, Angelini GD, Newby AC, Jeremy JY. Acute inhibition of superoxide formation and rac1 activation by nitric oxide and iloprost in human vascular smooth muscle cells in response to the thromboxane a2 analogue, u46619. Prostaglandins Leukot Essent Fatty Acids 2008; 78:247–255.
- Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Roflumilast N-oxide inhibits NADPH oxidase expression and activity in human pulmonary artery smooth muscle cells. Proc Br Pharmacol Soc 2008; 6:027P.
- Mayeenuddin LH, Garrison JC. Phosphorylation of p-rex1 by the cyclic amp-dependent protein kinase inhibits the phosphatidylinositiol (3,4,5)-trisphosphate and gbetagamma-mediated regulation of its activity. J Biol Chem 2006; 281:1921–1928.
- Birukova AA, Burdette D, Moldobaeva N, Xing J, Fu P, Birukov KG. Rac gtpase is a hub for protein kinase a and epac signaling in endothelial barrier protection by camp. Microvasc Res 2010; 79:128–138.