
Abstract
A Belgian alpha-1-antitrypsin (AAT) deficiency registry has been established in 2003. Currently 55 patients are included. At the same time, a working group has been set up for publishing national guidelines. In 2014, several Belgian patients founded Alpha-1 Global. We hope that the integrated activities of all the stakeholders involved in AAT deficiency will permit a high quality care for all patients suffering from this disabling disease.
Prevalence of the disease in Belgium
Alpha-1 antitrypsin (AAT) deficiency is a hereditary recessive disorder. Up to now, more than 100 genetic variants have been recognized. Pi*M is the normal allele.
In Belgium, as in most countries, patients suffering from AAT severe deficiency are essentially Pi*ZZ homozygotes (in clinical practice, 96% of the related diseases are linked to this phenotype) and, to a lesser extent, some Pi*SZ heterozygotes.
International publications concerning the distribution of the gene frequencies in Europe have mentioned only one specific study in Belgium.Citation1 This only source concerned a neonatal screening in Limburg.Citation2 The AAT phenotype was determined in cord blood of 1345 Belgian newborns. The prevalence of the Pi*S and Pi*Z deficiency alleles were estimated to 54.3 and 16.7 cases per 1000 inhabitants, respectively. Another Belgian study screened systematically for AAT deficiency in 39.289 consecutive births in Liège. Data suggested a prevalence of 3.6% and 1.4% of Pi*S and Pi*Z alleles (36 and 14 cases for 1000 inhabitants, respectively).Citation3
In 2014, the population of Belgium is estimated to 11.203.992 inhabitants.Citation4 Therefore, taking the two Belgian studies into account, the number of Pi*ZZ patients in Belgium would be between 2196 and 2896. This figure can seem high, compared with the number of estimated Pi*ZZ patients in The Netherlands (frequency of PI*Z allele = 10 cases per 1000 inhabitants). Would the estimation of 1120 Pi*ZZ patients in Belgium not have been more realistic?
Belgian participation to the adult Alpha1-International Registry (AIR)
During the WHO meeting on AAT deficiency in 1996,Citation5 the guidelines recommended the establishment of national and international registries of AAT deficiency, favoring collaborative research and development of adequately powered clinical trials. The Alpha-1 International Registry (AIR) was created in Malmö, Sweden (1997).Citation This registry is web-based and includes, on November 2014, 18 countries and 5147 patients.
In 1999, Belgium joined AIR, which resulted in 2001, in a first Belgian working group settled by the Belgian Thoracic Society. The objectives of the group were to disseminate information concerning AATD, to promote the recommended screening for AATD, to promote preventive measures and to promote registration of cases.
Each Belgian chest physician can receive a web access to the Belgian AAT registry after inscription by the Belgian Thoracic Society. After introduction of new patients, the registry administrator will validate the case if the data have been correctly entered. The first Belgian patients were included in the AIR registry in 2003.
In December 2005, R. Stockley, chairman of AIR, was invited for participating to the Annual Meeting of the Belgian Thoracic Society. His presentation of the topic “COPD interventions: lessons from AATD” gave an important impulse to the activities of the Belgian Working Party. In September 2005, 18 patients were included in the Belgian registry and, in March 2006, 27 Belgian patients were included (50% new cases in 6 months). Nevertheless, only 0.93% of the 2,627 expected severe deficient patients were registered.
Currently, 55 patients are included in the Belgian registry, 36 males and 19 females. Of them 46 phenotyped patients are also included in AIR. There were only 9 non-smokers but 36 had stopped smoking. Nearly all patients tested for AAT were suffering from lung disease. Six patients were diagnosed in a context of family screening. In no patient first analysis was done for a hepatic disease but 2 had a history of neonatal hepatitis. Eighteen patients were lung transplanted, 1 liver transplanted. Sixteen patients had, on registration, benefited from augmentation therapy, and 5 from long-term oxygenotherapy. Between 1.6 and 2.1% of the estimated cases are currently registered.
Belgian participation to the pediatric Alpha1-International Registry (AIR)
Thirty-one severe Pi*ZZ AAT deficient patients are followed in Brussels in “Cliniques Universitaires St Luc” by the Gastroenterology and Hepatology Pediatric Service.Citation7 Since 2014, most of these patients were included in the Alpha one International Registry (AIR).
Position paper of the Belgian Thoracic Society
In December 2009, during the annual meeting of the Belgian Thoracic Society, Belgian Guidelines for Diagnosis and Management of Patients with alpha1-Antitrypsin Deficiency were presented and published on the official site of the Society.Citation8
The recommendations were summarized as follows:
1. Screening for alpha1-Antitrypsin (AAT) deficiency should be systematically performed on patients with emphysema (regardless of smoking history), COPD, bronchiectasis without evident aetiology, asthma with persistent obstruction despite treatment, liver disease without evident aetiology, offspring or parents of an individual with an AAT deficiency (first degree collaterals), absence of an AAT peak on electrophoresis, and unexplained panniculitis or anti-proteinase3 vasculitis.
2. The result of a normal or abnormal AAT serum level should be saved in the appropriate section of the patient's record to avoid needless retesting.
3. Looking at the AAT serum level, we can decide that the classical threshold above which a patient will not be sick or even carrier of a Z allele is above 150 mg/dL. Looking at the patients from the Spanish and Italian registries, Costa et alCitation9 and Gorrini et alCitation10 tried to clarify the level of normality above which they did not have to request complementary blood analyses for detecting deficient individuals or carriers. They proposed similar threshold values, 114 and 113 mg/dL, respectively. They only recommended phenotyping or genotyping patients whose serum levels were under these thresholds. We can adopt this strategy which permits, at a lower cost, to detect efficiently patients suffering of severe AAT deficiency and carriers.
4. Patients with an AAT serum level <114 mg/dL, should undergo a pheno- and/or genotyping by sending appropriate samples to one of the reference labs in Belgium or by sampling three large blood droplets (0,5 mL of blood in total) on a special card (dried blot spots).
5. Cards can be obtained at no cost from the secretariat of the Belgian Society of Pneumology.
6. Cards to be sent by post to the Belgian Thoracic Society secretariat, after filling in clinical and administrative data. They will be forwarded to one European reference laboratory (Marburg, Germany) for pheno- and genotyping.
7. The Marburg Laboratory will communicate the result to the physician.
8. In return, physicians are requested to register their patients with severe AAT deficiency (serum level < 50 mg/dL, Pi*Null-Null, Pi*Z-Null, Pi* ZZ, Pi*SZ, Pi*S-Null), once the phenotype is known, via the web site (http://www.air-registry.org/be/).
9. Patients with severe AAT deficiency should be informed about the natural history of the disease and the importance of stopping smoking. Management of severe AAT deficiency should focus on COPD treatment according to guidelines, aggressive treatment of pulmonary infections, vaccinations against influenza, hepatitis A and hepatitis B, annual follow-up of pulmonary function and imaging, liver tests, α-fetoprotein, liver echography. Augmentation therapy should be considered in non-or ex-smokers with AAT serum level under 50 mg/dL (11 μmol/L) and with moderate to severe obstruction (FEV1 between 30–65% predicted) or with a rapid decline in FEV1.
10. Genetic testing should be performed in all first degree relatives of a homozygous or heterozygous AAT deficient patient (Z or Null allele carrier). Genetic counselling could be provided.
AAT Augmentation Therapy in Belgium
The substitution therapy with purified AAT obtained from human serum has been approved by the US Food and Drug Administration (FDA) in 1987. It remains the only applicable specific therapy for the disease but controversy remains concerning efficacy.
Augmentation infusions increase circulating trough levels of AAT to levels approximating those seen in heterozygous. A protective level of active AAT is restored in the epithelial lining fluid.
Augmentation therapy was available and almost completely reimbursed in Belgium until mid-2010 and 26 patients were treated. Thereafter the National Institute for Disease and Invalidity Insurance decided to continue to reimburse the treatment only for those patients who were already receiving it. No additional patient could be reimbursed for AAT substitution.Citation11 Therefore, since 2010, augmentation therapy is no longer reimbursed in Belgium for new patients.
The Belgian Thoracic Society's workgroup has been recently requested to express an opinion on the selection of patients who could benefit from the augmentation therapy. A discussion is ongoing for defining eventual new conditions of reimbursement by the social security.
To effectively limit the costs of treatment, recent studies aimed at targeting better the patients particularly at risk and the most susceptible to benefit from AAT substitution. Stockley et al.Citation12 suggested the importance of a personalized approach considering the severity of the lung disease, the rate of decline in lung function, age and frequency of exacerbations.
At the moment, we propose to consider substitution therapy in non-smokers or ex-smokers suffering from a severe AAT deficiency with a plasma level <50 mg/mL, with a FEV1 in the range of 30–60% predicted and a FEV1 decline of at least 0.5% predicted per year.
For estimating the decline of functional parameters, a longitudinal analysis for at least 2–3 years is required. The measures of lung physiology must be done at least 6 weeks after any exacerbation and when smoking has ceased.
Patients with an FEV1 in range of 60–80% predicted would be followed up unless they demonstrate a particularly rapid decline of FEV1 in excess of 1.0% predicted per year.
No recommendation has been published concerning the interpretation of lack of efficacy and treatment withdrawal criteria. It is evident that a longitudinal follow-up of at least 3 years would be necessary while on therapy to determine the subsequent FEV1 decline. Continued decline at the pretreatment level or an increase in the rate of decline would require reassessment of augmentation strategy.
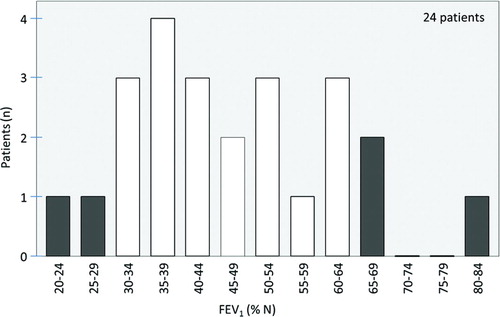
We analyzed in 2012 the patients actually benefiting from augmentation therapy. We have collected the functional data and the follow-up values by questioning the treating chest physicians individually. We included 26 substituted patients and we were able to obtain the initial parameters of 24 of them.
Looking at the FEV1 measured in 24 patients before initiating augmentation therapy (see Figure ), 19 were already in the limits suggested by the studies concerning efficacy of substitution therapy and the ATS/ERS recommendations.
Figure 1. FEV1 measured before augmentation therapy.
Lung and liver transplantation in Belgium
In 2010, Van Raemdonck and VerledenCitation13 discussed the indications and the results of lung transplantation for respiratory failure. Belgium is amongst the world leading countries in terms of lung donors and lung transplants per inhabitants. To date, the Belgian Lung Transplantation Registry accounts 821 transplants. Thirty-two patients are alphas (3.9%).
The “Cliniques Universitaires St Luc/Service de Gastroentérologie et Hépatologie pédiatrique” is a tertiary referral pediatric liver transplantation (LT) center under the supervision of Pr Etienne Sokal. In their center, they follow a cohort of 37 PiZZ AAT deficiency patients.Citation7 In the cohort (from 1974 to 2014), male predominance was observed (24 males and 13 females). At the time of AATD diagnosis, 23 of 37 (62%) PiZZ patients had neonatal cholestasis and/or cirrhosis. Of course, the AATD phenotype distribution is biased as they are a referral LT center. Neonatal cholestasis was the initial presentation in 18 of 23 (11 males and 7 females) while cirrhosis was the initial presentation in 5 males. Thirteen (56%) of these 23 patients developed end-stage liver disease requiring LT, 11 of these 13 (85%) were males, accounting for 46% of all male patients and 2 were PiZZ females (15% of all female patients). Of 24 who did not require LT, 10 (42%) had presented with neonatal cholestasis (5 males and 5 females) and 14 had no history of severe liver disease and diagnosis was an incidental finding. These results suggest a more severe evolution of liver disease in male children. All these patients did not have pulmonary symptoms at last follow-up.
Belgian patients group
The number of alpha patients in Belgium remained for a long time too small to permit the creation of a structure allowing an easy communication between Alphas. Having three national languages (French, Flemish and German) also does not help for simplifying a kind of exchange platform.
In 2012, when it was known that augmentation therapy would not be further reimbursed by the Public Health System for newly diagnosed Alphas, several Belgian patients met for trying to find solutions. As in other countries where alpha patients’ organizations are established, they decided to found a group and named it Alpha-1 Belgium. The association remained with no legal base or structure or subsides, but in contact with patients and medical doctors. The most important goals were to create awareness about the disease and to contact the Public Health Structures.
In 2013, the group was represented at the 4th international Alpha-1 congress for patients hold in Barcelona and met other patient organizations but also attended the scientific program and conferences.
Another event in summer 2013 was the meeting with two members of the European Parliament to create awareness about the Belgian lack of reimbursement. The Belgian Minister of Public Health was contacted a month later.
In autumn 2013 the necessity for a stand-alone, legal and registered structure led the patient group to create a new not-for-profit association called Alpha-1 Plus asbl/vzw. The missions and goals were to:
Provide support and information for Alpha patients;
Promote education and awareness of the disease;
Advocate for Alpha patients and community;
Communicate with other health-related structures in different countries.
Basic orientation about the disease is available in French, Flemish, English and German.
Unlike other countries there are no specific clinical reference centers for Alpha-1 in Belgium yet. Most of the members of the Alpha-1 Plus association were diagnosed before June 2010, so they are therefore still being augmented. Several meetings of a round table discussion, moderated by a professional therapist, allow the members to communicate with other patients about treatment and personal experiences.
Alpha-1 Plus asbl/vzw is also a member of:
The European Alpha-1 Federation;
Radiorg (rare diseases in Belgium);
Eurodis (European rare disease organization);
EFA (European Federation of Allergy and Airway diseases Patient Associations).
There are contacts with patient organizations in other countries (Germany, France, Denmark).
Finally, in June 2014, the Belgian association assisted to create another new structure called Alpha-1 Global, a group of 7 volunteers who are patient representatives of Alpha patient groups. The mission of Alpha-1 Global is to create a collaborative network of Alpha-1 organizations and patients to increase awareness, detection and access to care for Alphas around the world. The first tool is to create an Internet platform for patients, medical and care professionals and researchers to communicate about the disease.
Conclusions
The Belgian activities concerning AAT deficiency are growing. The screening and registration remain insufficient as it is also the case in nearly all other countries.
Nevertheless we can say that since 1999 and our participation in AIR we have put Belgium on the map. We will continue to boost the integrated activities of all the stakeholders involved in AAT deficiency, with the ultimate goal to appropriately screen advice and eventually treat all AAT patients suffering from this disabling disease.
Also, feedback from patients and communication with patients’ organizations stimulate awareness and knowledge of this rare disease.
Declaration of Interest Statement
The authors report no conflicts of interest in this work. The authors alone are responsible for the content and writing of the paper.
References
- de Serres FJ, Blanco I. Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A comprehensive review. Ther Adv Respir Dis 2012; 6(5): 277–295.
- Kimpen J, Legius E, Bosmans E, et al. Alpha-1-proteinase inhibitor gene frequencies in Belgium. Gene Geogr 1990; 4:159–163.
- Schoos R, Dodinval-Versie J, Verloes A, et al. enzyme immunoassay screening of α1-antitrypsin in dried blood spots from 39.289 newborns. Clin Chem 1991; 37(6):821–825.
- European Commission. http://epp.eurostat.ec.europa.eu/ tgm/table.do?tab=table&init=1&language= fr&pcode=tps00001&plugin=1. Accessed December 2014.
- World Health Organization. Alpha-1 antitrypsin deficiency: memorandum from a WHO meeting, available on their web site. Bull World Health Organ 1997; 75(5):397–415.
- Stockley RA, Luisetti M, Miravitlles M, et al. on behalf of the Alpha One International Registry (AIR) group. Eur Respir J 2007; 29:582–586.
- Stephenne X, Eta SL, Scheers I, et al. Liver disease evolution in Pi*ZZ Alpha1-Antitrypsin Deficiency to end-age liver disease predominates in male children. JPGN 2014, under review.
- Belgian Th oracic Society Belgian Thoracic Society. http://bvpsbp.mygreencloud.be/images/ stories/bvp/guidelines/guidelinesFR/GuidelinesAlfaantitrypsinedeficientiedec2009.pdf. Accessed December 2014.
- Costa X, Jardi R, Rodriguez F, et al. Simple method for α1-antitrypsin deficiency screening by use of dried blood spot specimens. Eur Respir J 2000; 15:1111–1115.
- Gorrini M, Ferraroti I, Lupi A, et al. Validation of a rapid, simple method to measure alpha1-antitrypsin in human dried blood spots. Clin Chem 2006; 52(5):899–901.
- European Parliament. Alpha-1 in the European Union. Expert recommendations. October 2011. http://www.alpha1awareness.org.uk/wp-content/uploads/2013/08/Alpha1-in-the-European-Union-Expert-Recommendations_electonic-copy.pdf. Accessed December 2014.
- Stockley RA, Miravitlles M, on behalf of Alpha One International Registry (AIR). Augmentation therapy for alpha1-antitrypsin deficiency: towards a personalized approach. Orphanet J Rare Dis 2013; 8:149. http://www.ojrd.com/content/8/1/149.
- Van Raemdonck D, Verleden G.M. Lung transplantation for respiratory failure: Belgium amongst the world leaders. Verh K Acad Geneesk Belg 2011; 73:41–63.