
Abstract
COPD has become a more popular research area in the last 3 decades, yet the first clear descriptions of acute and chronic bronchitis were in 1808. This brief history, comprehensively referenced, leads us through the early developments in respiratory physiology and their applications. It emphasises the early history of chronic bronchitis and emphysema in the 19th and early 20th centuries, long before the dominant effects of cigarette smoking emerged. This remains relevant to developing countries today.
Introduction
The term “chronic obstructive pulmonary disease” (COPD) came into use gradually in the 1960s and 1970s replacing the previous term “chronic bronchitis and emphysema” (CB&E). Since that time the new term—COPD—has been almost universally adopted by health professionals, if not by all the patients suffering from the condition. This brief history of the earlier years of COPD was motivated by the possibility that the change in terminology might make its earlier history less familiar. In particular it emphasises the early history of CB&E in the 19th and first half of the 20th centuries, long before the effects of cigarette smoking became dominant. This experience could still be relevant for the prevalence of COPD in many parts of the world in the 21st century.
We end this history about 1980 when terms indicating the newly recognised central feature of chronic airflow obstruction were first introduced into the International Classification of Disease (ICD).
Before 1930
Dividing this review into 3 separate periods—before 1930, 1930–1960 and after 1960—is arbitrary, but it facilitates comments on key developments in diverse areas across a similar time.
Early pioneers in clinical medicine and lung function
Classical descriptions of chronic bronchitis and emphysema were made in the early 19th century in Western Europe. The first clear description of acute and chronic bronchitis, distinguishing these conditions from other pulmonary disorders, was published in 1808 by Charles Badham (Citation1), at that time a 28-year-old “observant practical physician” working in London, who later became Professor of Practice of Physic in the University of Glasgow. The remarkable French physician and clinical scientist, René Laennec (1781–1826) invented the (monaural) stethoscope in 1815 and his experience in 1819 with auscultation led to the first descriptions of the pathological changes in pulmonary oedema and comprehensive studies of emphysema using inflated lung sections (Citation2); among the features of emphysema he described were the presence of peripheral airway obstruction, collateral ventilation, loss of lung recoil, right ventricular hypertrophy in advanced disease and chronic bronchitis (bronchial “catarrh”). Laennec's knowledge of functional pathology remained far in advance of clinical application, and many aspects were not rediscovered until well into the 20th century.
In 1837 William Stokes, an Irish physician, published his classic textbook on the “Diagnosis and Treatment of Diseases of the Chest” (Citation3). Stokes used the stethoscope and was familiar with Laennec's description of emphysema, but states emphatically that:
“To the practical physician, however, the great point of consideration is that this disease of the lung is the result of bronchitis; and that for its prevention, alleviation or cure, if that were possible, the treatment must be conducted upon this principle.”
In his section on “Bronchitis,” Stokes describes the full evolution in a patient from an insidious onset, the liability to frequent exacerbations, dyspnoea in winter, on to the severely breathless patient with a stooped posture, enlargement of the heart and liver and peripheral oedema. This clinical description was to dominate the British experience for the next century or more.
James Jackson Jnr (1810–1834) was perhaps the most astute and determined follower of Laennec's diagnostic skill with the stethoscope (Citation4). Shortly after qualifying in medicine at Harvard he travelled to Paris to study for 3 years under Professor Pierre Louis (Citation4). During this time he described a hereditary form of emphysema in 26 young adults. Jackson found 18 of 26 such patients had one or both parents affected by a similar condition.
Edward Greenhow (1814–1888) a pioneering public health physician at the Middlesex Hospital in London, who was aware of Jackson's work, provided support for the existence of a hereditary form of emphysema in his book, but he agreed with Stokes that emphysema more often followed years of suffering from bronchitis. Osler, in the first edition of his textbook in 1892 (Citation5), draws attention to the earlier findings of James Jackson Jnr, without commenting on emphysema also following bronchitis. However in 1899, emphysema—almost always accompanied by bronchial catarrh— was present at post mortem examination in 11% of deaths from the medical wards of St. Bartholomew's Hospital in London (Citation6). At this time treatment was typically confined to a cough mixture and drainage of dependent oedema.
Although some earlier scientists had measured the total volume of air that could be expelled from the lungs after a full inspiration, this measurement was put on a scientific basis in the 1840s by John Hutchinson, a singlehandedly medically qualified investigator, who presciently named this volume “vital capacity” (VC) and examined its value systematically in more than 2000 men (Citation7). To measure the volume of gas expired he developed (and named) a spirometer, which he demonstrated in the Crystal Palace at the Great Exhibition of 1851 in Hyde Park. In his comprehensive article on VC (Citation7), he states his intention to establish “a precise and easy method of detecting disease by the spirometer.” Unfortunately his spirometer was never used to evaluate CB&E (chronic bronchitis and emphysema). Hutchinson also promoted VC as a simple index of lung function, physique and bodily development; he noted consistent differences in VC at a given height between “poor” and “prosperous” that might be useful in assessing longevity of subjects for life insurance, a prediction confirmed by the Framingham Study more than a century later (Citation8).
An extraordinary subsequent study was the measurement of VC, using a U.S.-manufactured spirometer, in over 20,000 soldiers of the Union Army at the end of the American Civil War in the 1860s (Citation9). This study was inspired by Hutchinson's belief in VC as a general measure of lung health and development; VC was found to be larger in White than Black soldiers, provoking disputed explanations which continue into the 21st century (Citation10). Unfortunately, after these large groundbreaking studies, there was only sporadic use of the spirometer by individual enthusiasts, and its use in assessing lung disease was not established until well into the 20th century.
Mortality
Registration of cause of death in Britain commenced in 1837; in the mid-19th century repeated bronchial infections leading to oedematous heart failure were recorded as the cause of >5% of all deaths in middle age and old age (Citation11). These deaths were most often recorded as due to “bronchitis,” but emphysema clearly was also present in some patients.
In the 19th century and the first half of the 20th century, CB&E were widely regarded as much more common in both men and women in Britain than in other European and North American countries. In particular the emphasis on disability due to bronchitis was often regarded as a peculiarly “British disease,” contrasted with greater emphasis on emphysema in the medical textbooks of other European (particularly Germany) and North American countries. Some of the reported high prevalence of CB&E in Britain may have resulted from the early development of public health and mortality records, but urbanisation, industrialisation and accompanying pollution and poverty were particularly prominent in British cities in the 19th century.
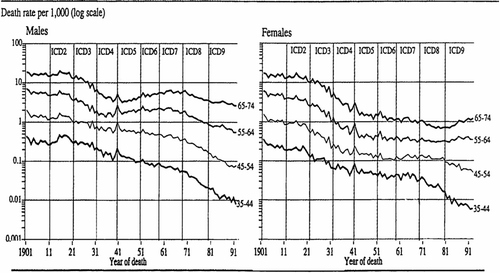
Reliable death rates attributed to “chronic bronchitis” in the United Kingdom are available from the early years of the 20th century () (Citation12). There was a 4- to 5-fold difference in mortality between unskilled labourers and the upper classes reported for 1910–1912 (Citation13), much higher than was ever reached later in the century. Some of this increase in mortality may have been due to poorer lung development in adults born in poverty, as first suggested by Hutchinson (Citation7) in the 1840s.
Figure 1. Deaths per 1,000 shown for 10-year age bands on a log scale (Citation12). The 9 headings indicate the updates to the international classifications of disease (ICD) during this period. In ICD1, “bronchitis” was used without qualification. Important conversion factors have been applied to the change from ICD 2 to 3 (which first used a sub-category of “acute bronchitis”) in the early 1920s and from ICD 4 to 5 in 1940 when rules of precedence in causes of death were changed. Nevertheless the label “chronic bronchitis,” whether or not emphysema was mentioned, was applied to the great majority of deaths from “chronic bronchitis and emphysema” in Britain throughout the century until the term “chronic airways obstruction” was introduced in ICD 9 in 1978. Note: The large falls in mortality in both men and women of all ages in the 1920s which continued in the 1930s for all age groups except middle-aged men.
Developments in spirometry and respiratory physiology/gas exchange
Early in the 20th century, interest in spirometry increased in Denmark and Germany; in particular attaching a spirometer to a recording drum allowed short periods of tidal breathing to be measured. Volhard (Citation14), a German physician, observed that expiration was slowed in emphysema contrasting with the better maintenance of inspiratory ability; his pupil Raither (Citation15) subsequently confirmed this important observation using a recording spirometer. Few chest physicians used VC to assess CB&E. In 1917 Peabody, a Boston cardiologist, published a series of papers on ventilation and basal metabolic rate in cardiac failure and popularised the use of changes in VC to assess cardiac dyspnoea (Citation16). The commercial spirometer developed in the USA at this time was remarkably similar to that developed by Hutchinson in being designed solely for measuring VC and not for dynamic spirometry [the FEV1 (forced expiratory volume in 1 second) was not developed until the end of the 1940s as explained later]. The development of chest radiographs from the end of the 19th century were useful for CB&E only for the detection of advanced disease—gross areas of emphysema or bullae, and increased total lung capacity.
Between 1890 and 1920 there were remarkable developments in knowledge of respiratory physiology; for example, in measuring the oxygen and carbon dioxide contents of arterial blood and alveolar air, defining the shape of the O2 dissociation curve with haemoglobin, and understanding the role of carbon dioxide in driving ventilation. Much of this progress developed from the competitive research of Christian Bohr and August Krogh in Copenhagen and Haldane in Oxford in disputing rival theories of whether O2 uptake by the lungs was passive.
Despite effective methods of oxygen treatment being introduced in a few academic hospitals, Collis, Professor of Preventive Medicine in Cardiff, in 1923 gave a sobering view of the more typical medical management of bronchitis in Britain (Citation13). He concluded that the two most important preventive measures would be an improvement in the socioeconomic status of the lower grades of society and smoke abatement in industrial districts and great towns.
Trends in tobacco smoking
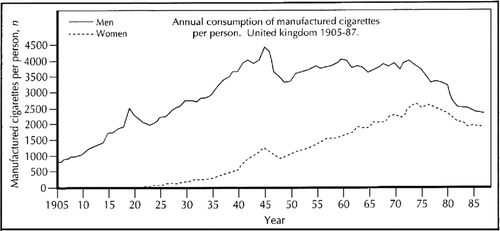
As discussed later, tobacco smoking was not considered as a possible cause of CB&E until after World War II (WW2). In the 19th century, consumption of tobacco was strongly established in British men but was smoked mainly in pipes; men started smoking cigarettes around 1890, shortly after machines were developed for their manufacture in the United States, and consumption steadily rose for many subsequent decades (). Until World War I (WW1), annual male tobacco consumption remained steady because increase in smoking cigarettes was offset by decline in pipe smoking. During WW1 and up to WW2, both cigarette and total tobacco consumption by men steadily increased with ∼50% increase in the total consumption over the level before WW1 (Citation17). Women had considerable mortality from bronchitis in the early 20th century () but active smoking of cigarettes in women only began between the two world wars.
Figure 2. Cigarette smoking in the United Kingdom in the 20th century. Figures not available beyond 1985 when tobacco smuggling into the United Kingdom became significant (P.N. Lee, personal communication, May 2013).
1930–1960
The major advance in the 1930s and the early years after WW2 was the development of many tests of lung function, which quantified the extent of lung damage and the central role of airflow obstruction in causing disability. A few attempts at relieving breathlessness were made by thoracic surgeons, but the biggest advance in treatment was the introduction of antibiotics after WW2. Advances in pathology, public health and epidemiology and causation did not appear until the 1950s.
Developments in applied pulmonary physiology
The immediate stimulus to develop tests of ventilatory capacity came from thoracic surgeons who needed tests that could predict whether their patients would tolerate lung resection for tuberculous areas or, increasingly, lung cancer. The first test of ventilatory capacity, the measurement of maximum voluntary ventilation (MVV) usually over a 15–30-second period (Citation18,Citation19), was developed in Germany using Knipping's recording spirometer. MVV was complemented by a sophisticated test of regional lung function, bronchospirometry, which provided an estimate of how much ventilated lung would be removed by surgery (Citation20).
Examination of breathing records during the MVV test led to their recognition of the obstructive pattern of tidal expiration (first described before WW1 by Volhard and Raither, see above), and the first attempts at describing obstructive types of respiratory insufficiency (Citation21). Other physiological techniques were used to diagnose CB&E in the 1930s. Measurement of total lung capacity (TLC) and its subdivisions had been attempted before WW1 by an early Boyle's law method and by multibreath gas equilibration using hydrogen. In the early1930s Christie (Citation22) standardised the technique for the nitrogen washout method and also made measurements of lung elasticity using intrapleural catheters (Citation23). Barach experimented with helium-oxygen mixtures to reduce resistance to breathing and assessed the efficacy of inhaled bronchodilators by timed vital capacity measurements (Citation24). Abdominal belts were used (Citation25) to simulate the effects of pneumo-peritoneum (Citation26) in improving diaphragm function by increasing its resting length.
By the early 1940s Eleanor Baldwin, Cournand and Richards were applying a remarkably comprehensive set of measurements of pulmonary function to a variety of chronic pulmonary diseases at Bellevue Hospital (Citation27–Citation29), measurement of TLC and subdivisions using a development of Christie's nitrogen washout method (Citation30), MVV before and after bronchodilators, an index of intrapulmonary mixing, ventilation, expired gas analysis and samples of arterial blood taken at rest (for oxygen and carbon dioxide content and sometimes acidity, pH) and at the end of a one minute step test. Of course the most famous contribution of Cournand, Richards and colleagues in this period was the clinical introduction of right heart catheterisation, for which they won the Nobel Prize for Medicine and Physiology in 1956 (Citation31).
The introduction of a catheter into the right heart (and subsequently into the pulmonary artery) allowed the measurements of mixed venous blood oxygen and carbon dioxide in the right heart rather than to assist cardiac diagnosis. Their new measurements led directly to the 3-compartment model of gas exchange as ideal alveolar air, dead space and venous admixture and the first recognition that abnormal ventilation-perfusion relationships were the major cause of arterial hypoxaemia in CB&E (chronic bronchitis and emphysema) (Citation32). Studies of the right heart were also important for understanding cor pulmonale, which was most commonly caused by CB&E but with large differences in prevalence between, and even within, countries. Cor pulmonale was common in Britain in the 19th century; in the 1950s it accounted for a remarkable 25–35% (Citation33–Citation35) of all patients admitted to hospital with congestive heart failure in Sheffield, a city dominated by heavy industry. In contrast, Richards commented it was uncommon at Bellevue in the 1930s (Citation36).
In an extraordinarily productive period in the United States in the 20 years after WW2 many additional tests were introduced into clinical practice: body plethysmography to measure airways resistance and lung volumes, the single breath N2 test, tests of CO diffusion, all at the University of Pennsylvania led by Comroe, Fowler, DuBois and Forster (Citation37); new methods of measuring resistance and the elastic properties of the lung using an oesophageal balloon-catheter as a surrogate for pleural pressure led by Mead and Whittenberger (Citation38) at the Harvard School of Public
Health; analysis of the maximum expiratory flow-volume curve by Fry and Hyatt at the National Institutes of Health (Citation39–Citation41). Measurement of arterial oxygen, carbon dioxide and pH, which had earlier been extremely laborious, was simplified by the development of electrodes.
Although the characteristic pattern of expiratory obstruction during MVV tests had been clearly described in the 1930s, these tests were tiring for patients and difficult to standardize for investigators. Analysis of the forced expiratory spirogram of a single breath, introduced by Tiffeneau (a clinical pharmacologist) in 1947 in France (Citation42) and subsequently popularised by Gaensler (a thoracic surgeon) in the United States (Citation43), probably had a greater practical effect than all the other tests, because it could be adopted by many less specialized hospital laboratories. Within a few years the MVV test was discarded and the (FEV1) and FEV1/VC (or FVC) ratio established as the standard, simple tests for diagnosis and for following the progress of pulmonary obstructive diseases.
Treatment
Knowledge of all these “physiological” abnormalities had very little effect on treatment. From 1950 antibiotics had a large effect on bacterial pneumonia and probably shortened the course of exacerbations of chronic bronchitis due to bacterial infections. Pharmacological treatment, with the exception of various respiratory stimulants which had short periods of popularity, largely followed the same succession of bronchodilators as used in asthma, moving from ephedrine, adrenaline (epinephrine), isoprenaline, atropine and derivatives by inhalation and theophylline tablets on to more selective inhaled short-acting β-agonists and anticholinergic drugs.
The response to bronchodilators differed in CB&E from that in asthma in being much smaller but with hints that response was larger to anticholinergic drugs than to β-agonists. Attempts to reduce breathlessness were occasionally made from the earliest years of thoracic surgery in the 1930s (Citation44,Citation45), most successfully by bullectomy. Several more experimental surgical procedures attempted to aid breathing by altering the relation between the lungs and thoracic cage, either by reducing the volume of the lungs or by enlarging the volume of the chest cage. In particular Brantigan et al. (Citation46), in the 1950s, explored the role of selective resection of the most overinflated areas of the lungs.
Pulmonary pathology and morphometry
After Laennec's brilliant description, there was little interest and few developments in pathological anatomy until Gough (Citation47) in the early years after WW2 developed a simple method of preparing thin slices of inflated lungs. This method, developed originally to study pneumoconiosis, allowed the severity and distribution of emphysema (and importantly of surviving normal lung) to be visualized and discussed with fellow pathologists. From such 2D slices Gough and his colleagues distinguished between a centrilobular (or centriacinar) type of emphysema, associated with inflamed and obstructed proximal respiratory bronchioles and most apparent in the upper lobes, and a panacinar type with perhaps a different pathogenesis (Citation48). In the centrilobular type Gough proposed—from meticulous reconstruction of adjacent 2D slices—that the local emphysema was formed from destruction of respiratory bronchioles distal to the obstructed proximal respiratory bronchioles. Gough also showed that anatomical changes of bullous emphysema did not occur even in the most chronic and long standing asthma, a view still widely held. His thin slices for examining inflated lungs facilitated the development of macroscopic grading systems by Thurlbeck (Citation49).
A major advance at the end of this period was the start of morphometry of the normal lung, airways and air spaces made by Gomez and Weibel, working at Bellevue Hospital. A particularly important finding was that the number and total cross-sectional area of the peripheral airways in normal lungs had been grossly underestimated in 1915 by Rohrer, who had examined uninflated lungs. Weibel's study (Citation50) predicted that peripheral airways were only responsible for a low proportion of the total airway resistance in normal lungs, a striking contrast to the predictions of Rohrer.
Contributions of emphysema and of airway disease to functional impairment
British physicians had from early in the 19th century emphasised the importance of “bronchitis”; in other countries when patients became increasingly breathless, this was usually attributed to the development of emphysema. Some functional changes initially thought to be diagnostic of CB&E, such as obstructive spirometry, hyperinflation and ventilation/blood flow mismatching were found also in exacerbations of asthma, indirectly supporting the idea that the airway component of CB&E could be important. Detection of the structural changes of emphysema during life remained difficult but by the early 1960s reduction in lung recoil pressure, a modest increase in total lung capacity and a severe fall in single breath carbon monoxide diffusion were established as its characteristic functional effects (Citation51,Citation52).
CB&E was recognised to include a range of clinical phenotypes, most strikingly the contrast between the asthenic, intensely breathless but not grossly hypoxaemic patient (“pink puffer” or “fighter”) with the severely hypoxæmic, often oedematous but less breathless patient (“blue bloater,” “non-fighter” (44(p334), 51–54). An obvious possibility was that severe emphysema (probably of the panacinar type) and predominant primary airway disease accounted for the two phenotypes, although an alternative hypothesis was that these contrasts reflected differences in the individual's ventilatory response to hypoxaemia (Citation54). These two phenotypes mirrored differences between U.K. and U.S. stereotypes; a comparison of 2 groups of 50 men aged between 45 and 65 with obstructive spirometry and FEV1 < 1.5 L, recruited from outpatient clinics in London and 50 patients in Chicago showed that both phenotypes were present and similar in each centre, the only difference being a trend to more troublesome bronchial infection and less radiological evidence of emphysema in London (Citation51,Citation52).
Until 1968 there was no information on the specific site(s) of airflow obstruction. Laennec had observed obstructive change in the peripheral airways of lungs affected by emphysema. Airflow resistance of the peripheral airways was first measured when the ‘retrograde catheter’ technique, developed by Macklem and Mead in animals at Harvard School of Public Health (Citation55), was applied in post-mortem human lungs from 5 subjects with normal lungs and 9 patients with severe CB&E in Montreal (Citation56). These studies showed that relatively modest increases in total airflow resistance were due to gross increases in the resistance of the small airways distal to the retrograde catheter (which was placed in an airway of 2–3-mm diameter).
Because the lungs were studied at a standard lung recoil pressure, some of the effects of emphysema were minimized, but other effects on destroying attachments to the airway perimeter and simple loss of functioning alveoli would contribute to the increase in peripheral airway resistance. A single in vivo study has examined the serial partitioning of flow resistance in patients with CB&E and FEV1 ∼50%pred (Citation84); this replicated the large increase in resistance of airways less than 3 mm in diameter, but also found a moderate increase in resistance of the conducting airways. Emphysema becomes increasingly common and severe as airflow obstruction worsens in CB&E (Citation11), but no detailed sequential physiological studies have been made.
The Montreal study (Citation56), also confirmed Weibel's prediction (Citation50) that the resistance of peripheral airways in normal lungs was low, their large number far outweighing the effect of the decreased diameter of each individual airway. As a result considerable pathological damage could accumulate in the “silent zone” of the peripheral airways without significant loss of total lung function or breathlessness developing. This discovery stimulated a large amount of research in the 1970s into tests which could detect functional abnormality of the peripheral airways before significant spirometric impairment developed. Subtle abnormalities could be detected early in the smoking history of almost all smokers, most simply by showing abnormality of the single breath N2 test, but no clear identification of the smoker destined to be disabled in later life was achieved. Fortunately reference values of FEV1 over the life span were well developed, allowing spirometry to be used to define progression of airflow obstruction over long periods.
Public health and epidemiology
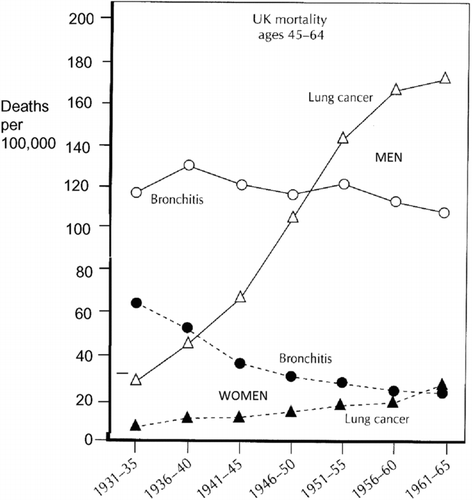
From about 1930 much more systematic data about public health were gathered in Britain and many other European and North American countries. U.K. mortality from chronic bronchitis and emphysema () (Citation57) continued to fall at all ages in women from 1931–1961 (and indeed until about 1980) but in men between 55 and 74 mortality rose between 1941 and 1961 reaching a peak increase in the late 1960s, a result compatible with the estimate that population lifetime exposure to cigarettes was greatest in men born between 1900 and 1910. As a result the previous rather small differences in mortality between men and women greatly expanded between 1940 and 1970. Although U.K. middle-aged men showed little change in CB&E mortality between 1930 and 1950 deaths from lung cancer rose rapidly ()(58).
Figure 3. Mortality for CB&E in 55–74-year-old men and women in the United States and United Kingdom. Data analysed by J. Boreham and R. Peto from the WHO Mortality Database for deaths COPD in the United States and United Kingdom for 55–74-year-old men and women (Citation57).
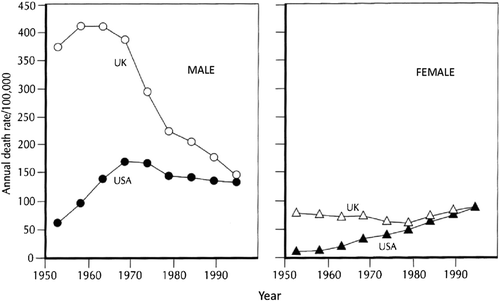
Figure 4. Death rates from lung cancer and ‘bronchitis’ per 100,000 in men and women aged 45–64 years in England and Wales. Note the considerable decline in mortality from “bronchitis” in middle-aged women between 1931 and 1965 and the dissociation between trends in death from lung cancer and bronchitis in both middle-aged men and women (reprinted from ref. (Citation58). In this period the diagnosis of “bronchitis” in Britain referred to “chronic bronchitis.”
The foundation of the World Health Organisation (WHO) at the end of WW2 encouraged international projects. An early WHO report comparing CB&E mortality in European and North American countries in 1950 (Citation34) estimated that the death rates from ‘chronic bronchitis’ in the United Kingdom were at least 15 times those reported for Canada and the United States, despite the time course of the cigarette smoking epidemic being similar in all three countries. Some of these differences may have been due to differing diagnostic habits, in particular sudden death in CB&E being attributed to cardiac disease in USA (Citation59).
Awareness of the medical and economic burden of “chronic bronchitis and emphysema” had always been greater among public health doctors than their clinical colleagues. A review of mortality, morbidity and treatment of chronic bronchitis in Britain covering the period up to 1952 (Citation60) echoed the conclusions of Collis 30 years earlier. Although both clinicians and the lay public had been aware of the chronic “smokers’ cough,” often associated with expectoration, this was widely regarded as a tiresome inconvenience. Studies in the early 1950s however documented the importance of smoking in increasing sickness absence (Citation60–Citation62).
Two major developments in these years however transformed this neglect. The first was a succession of pollution disasters in cities in Europe and the United States, culminating most dramatically in London in December 1952. Deaths occurred mainly in elderly people already suffering from chronic respiratory or cardiovascular disease. These disasters reinforced opinions that urban pollution was of great importance and stimulated rapid action by governments to promote cleaner air and financial support for epidemiological studies. The second major event was the first definitive evidence that smoking considerably increased mortality from chronic bronchitis and emphysema (Citation63), which came 6 years after the first reports of the strong relation between cigarette smoking and lung cancer (Citation64). Whereas the effect of urban pollution on established chronic respiratory disease was immediately accepted, the importance of smoking tobacco on lung cancer and chronic bronchitis was less accepted on scientific grounds and worried governments because of the major contribution tobacco made to their tax revenue.
As recounted by Doll in 2000 (Citation65), when their initial study to examine the increase in lung cancer was set up in 1948 neither urban air pollution nor smoking tobacco appealed as a possible cause. As a result patients admitted to hospital with chronic bronchitis and coronary artery disease were included in the control group to compare with patients with carcinoma of the lung. Doll adds:
…“that chronic bronchitis was not then (1948) thought to be related to smoking is now almost incomprehensible; but the cough that smokers so often had was called a (benign) ‘smokers cough’.”
To expand the evidence connecting disease with smokers Doll and Hill set up their 40 year study of the mortality of 40,000, mainly male, U.K. doctors, which confirmed that smokers had increased mortality from lung cancer, but also from chronic bronchitis and coronary artery disease (Citation63). Before 1950 male doctors smoked as much as the rest of the male population but they rapidly reduced their exposure after the lung cancer report in 1950; however reductions in population exposure to cigarettes did not begin until the late 1960s ().
Thus by the end of the 1950s, there had been considerable developments in the functional and structural assessment of the changes in chronic bronchitis and emphysema and a new emphasis on cigarette smoking and urban pollution in its pathogenesis. The recognition that expiratory airflow obstruction was the dominant functional abnormality in breathless patients led to attempts, starting productively with the Ciba Symposium set up by British investigators in 1958 (Citation66), to incorporate this concept within the older terminology.
The cumbersome phrase “chronic non-specific lung disease” (CNSLD) was suggested to include patients without airflow obstruction but with chronic bronchitis, emphysema and asthma in remission, as well as patients with “generalised obstructive lung disease”; the latter term never came into use but was intended to include asthma as well as chronic bronchitis and emphysema and was subdivided into intermittent and persistent categories. Some experts thought that emphysema should be defined functionally as an increase in residual volume/total lung capacity ratio; this was rejected in favour of an anatomical diagnosis, as first suggested by Laennec.
Emphysema—extensively illustrated by thin Gough sections—was initially defined at the Ciba symposium as ‘an increase in the size of the airspaces distal to the terminal bronchiole due to either dilatation or destruction of their walls’, but restricted to destruction after a WHO report in 1961. The clinical definition of chronic bronchitis proposed at the Ciba Symposium (Chronic bronchitis refers to the condition of subjects with chronic or recurrent excessive mucous secretion in the bronchial tree) survived untouched to the present day; a later attempt to introduce the term ‘chronic obstructive bronchitis’ to separate it from simple “chronic bronchitis” without obstruction failed, when it was recognised subsequently that while the site of increased bronchial secretions was in central airways the major site of airflow obstruction was in peripheral airways (Citation67). A definition of asthma as reversible obstructive lung disease was suggested and the possibility of an irreversible component was considered. A variety of tests of lung function were recommended, including forced expiratory spirometry before and after a bronchodilator spray, which was among the tests labelled as “essential,” but no specific diagnostic boundaries were specified.
After 1960
By the early 1960s smoking had acquired a central role in the causation of CB&E but the precise mechanisms by which it damaged the lung were uncertain.
Pathogenesis
Possible pathogenetic factors in emphysema were reviewed in 1958 at the first Aspen “Symposium of emphysema and the ‘chronic bronchitis’ syndrome” (Citation68); these included mechanical damage, perhaps by cough, of hyperinflated and obstructed lung, premature ageing, heredity, primary loss of pulmonary capillaries and enlargement of the thoracic cage. Probably the most popular hypothesis was that smoking and other irritants weakened airway defences, promoted chronic inflammation and recurrent infection, which eventually caused widespread airflow obstruction and emphysema; this became known as the “British Hypothesis.” In North America there was more emphasis on emphysema and less on infection (Citation69).
An unexpected and dramatic development in 1963 was the discovery of an association between severe deficiency of a serum protein, α1-antitrypsin, and early onset emphysema by Laurell and Eriksson in Malmo (Citation70,Citation71). Although severe α1-antitrypsin deficiency is only responsible for a small proportion of patients with emphysema, the novel idea of an inherited “systemic” disorder, which upset protease/antiprotease balance defines the start of the modern era of applying the techniques of genetics, cell biology and molecular medicine to pulmonary disease.
The Swedish observations were rapidly followed by experiments producing emphysema in animals by intratracheal instillation of proteolytic enzymes (Citation72) and the extension of the protease/antiprotease imbalance hypothesis to the causation of common types of emphysema, in which it was postulated the protease burden was increased and the antiprotease defence impaired by inflammatory cells commonly stimulated by direct or indirect effects of smoking. Although the early emphasis was on damage to elastin, subsequently the concepts have expanded to include mechanisms destroying other components of the extracellular matrix and the effects of collagen deposition during repair.
A wide range of animal models, some of which produced emphysema without accompanying airway disease, were developed to dissect out specific mechanisms underlying CB&E. Most of the early models did not involve exposing animals to tobacco smoke and it has proved difficult to induce disease in the peripheral airways of small animals. Probably the pathogenesis of centrilobular emphysema, where there is always associated bronchiolitis, differs from that of panacinar emphysema. A recent important study from Hogg and colleagues (Citation75,Citation76) confirms the pathogenesis proposed by Leopold and Gough in 1957 (Citation48) that the primary process in centrilobular emphysema is obstruction of proximal respiratory bronchioles which is followed later by local emphysema associated with destruction of more distal respiratory bronchioles.
Developments in oxygen treatment
The management of acute and chronic hypoxaemia of CB&E began in the first decade after WW1. In the 1930s it was found that hypercapnia could occur if high inspired oxygen concentrations were given (Citation73), leading Barach (Citation74) to recommend that oxygen treatment should begin cautiously. Nevertheless after WW2 high concentrations of inspired oxygen continued to be used and frequently led to unconsciousness and severe respiratory academia, necessitating intubation and intermittent positive pressure ventilation. Some experts suggested that this problem could be avoided by giving oxygen treatment intermittently (for instance only for 20mins in each half-hour) despite evidence that withdrawal of oxygen was often followed by more severe hypoxaemia than found before treatment (Citation77,Citation78).
Moran Campbell (Citation79) in the 1960s vigorously rejected this approach—famously rejected by Haldane during WW1. Instead Campbell recommended that continuous treatment should start with a small increase in inspired oxygen concentration which in most patients relieved dangerous levels of hypoxaemia, while avoiding deterioration in consciousness. This new approach required the development of face masks using the Venturi principle to provide 24 and 28% O2 (Citation79). Flenley (Citation80), who adopted this approach, emphasised that the aim of oxygen treatment should be to obtain an adequate intracellular mitochondrial Po2- which he estimated as ∼10 mmHg- not to restore normal arterial oxygen saturation.
The evidence that hypoxaemia in CB&E could be effectively treated by small increases in inspired oxygen concentration was very important for long term development of home oxygen treatment of chronic hypoxaemia. In the USA home reservoirs of liquid oxygen were developed which supplied 3–4 days of gaseous oxygen at 2–4 L/min; the patient filled a 3-kg canister from this reservoir, which supplied 2–4 hours of treatment away from home (Citation81). A more widely used source of home oxygen was provided by the development of oxygen concentrators in the 1970s.
These were used in the landmark multi-centre North American (Citation82) and British (Citation83) studies of home long-term oxygen treatment (LTOT). Both studies gave LTOT by nasal prongs, usually at 2 L/min. In the North American study 102 patients were treated for 12 hours daily and compared with 101 patients who were treated with continuous oxygen for a mean of 19.3 months. Annual mortality for those treated with continuous oxygen was 11.9% compared with 20.6% for those only receiving nocturnal oxygen. The British study of 87 patients was confined to patients who had had at least one episode of heart failure and oedema attributed to cor pulmonale; although the patients in both trials had similar obstructive spirometry and arterial Po2 while breathing air, mean values of chronic arterial Pco2 in the British trial were considerably higher (59 mmHg) than in the North American study (43 mmHg).
British patients were randomised to receive either at least 15 hours (including the sleeping hours) of oxygen or to the control group without home oxygen for a 3 year period. In the first 18 months of the trial there was no difference in survival but subsequently annual mortality was 12% in those treated with home oxygen and 29% in the control group. These ambitious studies in severely disabled patients, published in the early 1980s, were the first controlled trials to show any improvement in survival in CB&E.
Natural history of airflow obstruction; Dutch and British hypotheses
Historically British and North American investigators both regarded CB&E and asthma as separate conditions with a different clinical course and pathogenesis. In contrast Orie (Citation84) at the first Bronchitis symposium in Groningen in 1961 proposed that all chronic airway diseases, including asthma, emphysema and chronic bronchitis were different manifestations of a single disease with common genetic origins. Orie suggested that the minority of smokers who developed chronic and largely irreversible airflow obstruction shared with asthmatic subjects a common allergic constitution and over-reactive airways, bronchial hyper-responsiveness (BHR), which predated the development of important disease. No clear mechanism was suggested whereby hyper-responsiveness led to irreversible obstructive changes or the development of emphysema. This theory was termed the “Dutch hypothesis” by Fletcher in 1969 (Citation85).
The first prospective studies to test these hypotheses were begun in the United Kingdom and the Netherlands in 1961. Fletcher set up a 7-year follow up of middle-aged working men with mild airflow obstruction to study the infective (or ‘British’) hypothesis in London. The decline in spirometry, unexpectedly, was not accelerated in men who suffered from frequent infections and was slowly progressive without step changes; the presence of chronic bronchitis and progression of airflow obstruction could be dissociated, and decline in FEV1 in susceptible smokers reverted to a normal rate on quitting smoking (Citation85,Citation86). The much-quoted diagram showing the change of FEV1 over the whole adult life span (Citation86) with evidence that subtle changes in the lungs of many smokers occurred early in their smoking history provided convincing evidence in support of early intervention.
In the Netherlands, the Dutch hypothesis was investigated by the Vlagtwedde-Vlaardingen Study (VVS). Many smokers had BHR but evidence of allergy/atopy was less striking (Citation87). The Dutch and English hypotheses have been explored by subsequent observational studies. An epidemiological project, set up by Burrows and sustained over 25 years, of all types of obstructive airway disease in the population of Tucson, Arizona (Citation80), studied the effects of smoking and quitting smoking on lung function, on the distribution of spirometric abnormality in relation to pack-years, on the role of childhood respiratory disease and subsequent impaired lung development during growth, and emphasized the overlap between asthma and CB&E. In most countries CB&E and asthma are considered separate diseases, so almost all trials of pharmacological treatment have been made either on selected patients with classical features of asthma—childhood onset, atopy, never-smokers and near-complete reversibility, or of CB&E with a smoking history and onset in middle age, with little reversibility, thus limiting their applicability for many patients with chronic airway disease.
Subsequently the North American Lung Health Study (LHS) confirmed the sustained spirometric benefit of quitting smoking (Citation88) and the importance of BHR as a risk factor for progression of CB&E (Citation89). How often the underlying mechanism of BHR in CB&E is the same as in atopy and asthma remained uncertain. Some of the BHR is probably a consequence rather than a cause of airway narrowing. The LHS (Citation90) and other North American (Citation91) and European studies (Citation92) have supported the original ‘British hypothesis’ that repeated bronchial infections accelerate decline in FEV1.
New terminology
In 1979 the 9th revision of ICD added the term “chronic airflow obstruction” (see , legend) to the previous terms of chronic bronchitis and emphysema and this has become the most widely used coding for CB&E and its successor term, COPD. The proposed new terms implied a condition diagnosed solely by demonstrating chronic airflow obstruction. “Chronic obstructive pulmonary disease” (COPD) emerged in the 1970s, without “official” endorsement, as the preferred English language and international term used by health professionals.
There are several curiosities about the new term COPD; First, that the diagnosis would be made by spirometry but no boundary was defined until the first GOLD criteria in 1999 (Citation93). Second, no formal definition of inclusions and exclusions was developed; it was ‘presumed’ that cystic fibrosis, and some causes of obliterative bronchiolitis were excluded and that “chronic” implied persistent rather than intermittent obstruction. Also the term COPD was used even when there was clearly overlap with asthma and other airway diseases. Third, although COPD was historically intended as a substitute for CB&E it was unclear whether this label included subjects without abnormal spirometry. In 1989 Snider (Citation94) suggested COPD
“was a process characterized by the presence of chronic bronchitis or emphysema that may lead to development of airways obstruction; airways obstruction need not be present at all stages of the process; the airways obstruction may be partially reversible.”
A look forward
Most developed countries have now passed through their peak of smoking induced disease. But even at that peak the smoking-attributed risk for COPD only reached 30% in men compared to over 90% in lung cancer (Citation94).
In the last two decades, numerous studies of COPD prevalence and mortality have been made in less ‘developed’ parts of the world, most notably in China, where, as in Britain in the 19th and early 20th century, there are many deaths attributed to COPD in women who had no exposure to active tobacco smoking (Citation95,Citation96). Passive exposure to tobacco smoke from the increasing number of male smokers is only a small effect (Citation97). A more likely cause may be intense exposure to fumes from domestic cooking and heating. More generally, many developing countries have large segments of the population brought up in poor socioeconomic conditions, which are now being exposed to rapid urbanisation and industrial pollution. In addition cigarette smoking in men is increasing rapidly. This raises the fear that the situation in many “developing” countries in the early 21st century may be a magnified replay on a compressed time scale of Britain's unfortunate experience of CB&E more than a century earlier.
Declaration of Interest Statement
The authors declare they have no conflict of interest, financial or otherwise in relation to this manuscript. The authors alone are responsible for the content and writing of the paper.
References
- Badham C. Observations on the Inflammatory Affections of the Mucous Membranes of the Bronchi. Callow: London, 1808.
- Laennec RTH. A Treatise on the Diseases of the Chest and on Mediate Auscultation (translated by John Forbes), 2nd ed. Underwood: London, 1827.
- Stokes W. A Treatise on the Diagnosis and Treatment of Diseases of the Chest. Hodges and Smith: Dublin, Ireland, 1837.
- Knudson RJ, Jackson J. Jr. The young pulmonologist who described familial emphysema. An historical footnote. Chest 1985; 87:673–676.
- Osler W.. The Principles and Practice of Medicine: Designed for the Use of Practioners and Students of Medicine, 5th ed. D. Appleton: New York, London, 1903.
- Gee S. Lumleian lectures on bronchitis, pulmonary emphysema and asthma; delivered before the Royal College of Physicians of London, Lecture II. Br Med J 1899; 1:715–719.
- Hutchinson J. On the capacity of the lungs, and on the respiratory functions, with a view on establishing a precise and easy method of detecting disease by the spirometer. Med Chir Trans 1846; 29:137–252.
- Ashley F, Kannel WB, Sorlie PD, Masson R. Pulmonary function: relation to ageing cigarette habit and mortality. The Framingham Study. Ann Intern Med 1975; 82:739–745.
- Braun L. Spirometry, measurement, and race in the nineteenth century. J Hist Med Allied Sci 2005; 60:135–169.
- Harik-Khan RI, Fleg JL, Muller DC, Wise RA. The effect of anthropometric and socioeconomic factors on the racial difference in lung function. Am J Respir Crit Care Med 2001; 164:1647–1654.
- Tanner TH. The Practice of Medicine. 6th edition, vol 1, part 5, section 10. Bronchitis p554–561. Renshaw: London, 1869.
- Marks GB, Burney PGJ. Diseases of the respiratory system. Ch 20. In: Charlton J, Murphy M, eds. Health of adult Britain: 1841 to 1994 Part II. HMSO London, 1997; 93–113.
- Collis EL. The general and occupational prevalence of bronchitis and its relation to other respiratory diseases. J Ind Hyg Toxicol 1923; 5:264–276.
- Volhard F. Discussion following paper by M. Bonniger. Verhandlungen Dtsch Gesellschaft fur Innere Medezin 1908; 25:530.
- Raither E, Studienüber Emphysem. Beitr Klin Tuberk 1912; 22:137–164.
- Peabody FW, Wentworth JA. Clinical studies on respiration: IV. The vital capacity of the lungs and its relation to dyspnoea. Arch Int Med 1917; 20:443–467.
- Royal College of Physicians. Smoking and Health: Smoking and Chronic Bronchitis. Pitman: London, 1962: 27–31.
- Jansen K, Knipping HW, Stromberger K. Untersuchungen über atmung und blutgase. Beitr Klin Tuberk 1932; 80:304–373.
- Hermannsen J. Untersuchungen über die maximale Ventilationsgrösse (Atemgrenzwert). Z ges Exp Med 1933; 90:130–137.
- Jacobæus HC, Frenckner P, Björkman S. Some attempts at determining the volume and function of each lung separately. Acta Med Scand 1932; 79:174–215.
- Cournand A, Richards DW. Pulmonary insufficiency. 1. Discussion of a physiological classification and presentation of clinical tests. Am Rev Tuberc 1941; 44:26–41.
- Christie RV The lung volume and its subdivisions. I. Methods of measurement. J Clin Invest 1932; 11:1099–1322.
- Christie RV. The elastic properties of the emphysematous lung and their significance. J Clin Invest 1934; 13:295–321.
- Barach AL. Physiological methods in the diagnosis and treatment of asthma and emphysema. Ann Intern Med 1938; 12:454–481.
- Alexander HL, Kountz WB. Symptomatic relief of emphysema by an abdominal belt. Am J Med Sci 1934; 187:687–692.
- Reich L. Der Einfluss des Pneumoperitoneum auf das Lungenemphysem. Wien Arch Int Med 1924; 8:245–252.
- Baldwin E de F, Cournand A, Richards DW, Jr. Pulmonary insufficiency. I. Physiological classification, clinical methods of analysis, standard values in normal subjects. Medicine 1948; 27:243–278.
- Baldwin E de F, Cournand A, Richards DW, Jr. Pulmonary insufficiency, III. A study of 122 cases of chronic pulmonary emphysema. Medicine 1949; 28:201–237.
- Baldwin E de F, Harden KA, Greene DG, Cournand A, Richards, DW Jr. Pulmonary insufficiency, IV. A study of 16 cases of large pulmonary air cysts or bullae. Medicine 1950; 29:169–194.
- Darling RC, Cournand A, Richards DW. Studies on the intrapulmonary mixture of gases, III. An open circuit method for measuring residual air. J Clin Invest 1940; 19:609–618.
- Richards DW. Right heart catheterisation. Its contribution to physiology and medicine. Science 1957; 125:1181–1185.
- Riley RL, Cournand A. Ideal alveolar air and the analysis of ventilation-perfusion relationships in the lungs. J Appl Physiol 1949; 1:825–847.
- Flint FG. Cor pulmonale. Incidence and aetiology in an industrial city. Lancet 1954; ii:510–518.
- Stuart-Harris CH, Hanley T, Clifton M. Chronic Bronchitis, Emphysema and Cor Pulmonale. John Wright and Sons: Bristol, UK, 1957: 199–236.
- Stuart-Harris CH, and Physicians of the Sheffield Hospital Region. A hospital study of congestive heart failure, with special reference to cor pulmonale. Brit Med J 1959; 2:201–208.
- Richards DW. Pulmonary emphysema: etiologic factors and clinical forms. Ann Intern Med 1960; 53:1105–1120.
- Comroe JH Jnr, Forster RE Jnr, DuBois AB, Briscoe WA, Carlsen E. The Lung. Clinical Physiology and Pulmonary Function Tests, 1st ed. Year Book Medical Publishers: Chicago, IL, 1955.
- Mead J, Whittenberger JL. Physical properties of human lungs measured during spontaneous respiration. J Appl Physiol 1953; 5:779–796.
- Fry DL, Hyatt RE. Pulmonary mechanics. Am J Med 1960; 29:672–689.
- Hyatt RE, Schilder DP, Fry DL. Relationship between maximum expiratory flow and degree of lung inflation. J Appl Physiol 1958; 13:331–336.
- Pride NB, Permutt S, Riley RL, Bromberger-Barnea B. Determinants of maximal expiratory flow from the lungs. J Appl Physiol 1967; 23:646–662.
- Tiffeneau R, Pinelli A. Air circulant et air captif dans l'exploration de la fonction ventilatrice pulmonaire. Paris Med 1947; 133:624–628.
- Gaensler EA. Analysis of the ventilatory defect by timed vital capacity measurements. Am Rev Tuberc 1951; 64:256–278.
- Knudson RJ, Gaensler EA. Surgery for emphysema. Ann Thorac Surg 1965; 1:332–362.
- Kountz WB, Alexander HL. Emphysema. Medicine (Balto). 1934; 13:251–316.
- Brantigan OC, Muller E, Kress MB. A surgical approach to pulmonary emphysema. Am Rev Respir Dis 1959; 50;194–201.
- Gough J, Wentworth JE. The use of thin sections of entire organs in morbid anatomical studies. J Royal Micr Soc 1949; 69:231–235.
- Leopold JG, Gough J. The centrilobular form of hypertrophic emphysema and its relation to chronic bronchitis. Thorax 1957; 12:219–235.
- Thurlbeck WM. Chronic Airflow Obstruction in Lung Disease. W.B. Saunders: Philadelphia, PA, 1976.
- Weibel ER. Morphometry of the Human Lung. Springer-Verlag: Berlin/ Academic: New York, 1963.
- Burrows B, Niden AH, Fletcher CM, Jones NL. Clinical types of chronic obstructive lung disease in London and in Chicago. Am Rev Respir Dis 1964; 90:14–27.
- Fletcher CM, Jones NL, Burrows B, Niden AH. American emphysema and British bronchitis. A standardised comparative study. Amer Rev Resp Dis 1964; 90:1–27.
- Burrows B, Bloom JW, Traver GA, Cline MG. The course and progression of different forms of chronic airway obstruction in a sample from a general population. N Engl J Med 1987; 317:1309–1314.
- Robin ED, O'Neill RP. The fighter versus the non-fighter. Control of ventilation in chronic obstructive pulmonary disease. Arch Environ Health 1963; 7:125–129.
- Macklem PT, Mead J. Resistance of central and peripheral airways measured by a retrograde catheter. J Appl Physiol 1967; 22:395–401.
- Hogg JC. Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 1968; 278:1355–1360.
- Peto R, Lopez AD, Boreham J, Thun M, Heath C. Mortality from smoking in developed countries 1950–2000. 1994, Oxford University Press: Oxford, UK, updated to 2010. Available from: http://www.ctsu.ox.ac.uk/research/mega-studies/mortality-from- smoking-in-developed-countries-1950-2010/). (Accessed May 2013).
- Royal College of Physicians. Smoking and Health Now. Chapter 5. Smoking, Chronic Bronchitis and Emphysema. Pitman: London, 1971.
- Reid DD, Fletcher CM. International studies in chronic respiratory disease. Br Med Bull 1971; 27:59–64.
- Goodman N, Lane RE, Rampling SB. Chronic bronchitis: an introductory examination of existing data. Br Med J 1953; 2:237–243.
- Oswald NC, Harold JT, Martin WJ. Clinical pattern of chronic bronchitis. Lancet 1953; ii:639–643.
- Oswald NC, Medvei VC. Chronic bronchitis. The effect of cigarette smoking. Lancet 1955; 2:843–844.
- Doll R, Hill AB. Lung cancer and other causes of death in relation to smoking;a second report on the mortality of British doctors. Br Med J 1956; ii:1071–1081.
- Doll R, Hill AB. Smoking and carcinoma of the lung: a preliminary report. Br Med J 1950; ii:739–748.
- Doll R. Smoking and lung cancer. How it really happened. Am J Respir Crit Care Med 2000; 162:4–6.
- Ciba Guest Symposium. Terminology, definitions and classification of chronic pulmonary emphysema and related conditions. Thorax 1959; 14:286–299.
- Fletcher CM, Pride NB. Definitions of emphysema, chronic bronchitis, asthma and airflow obstruction: 25 years on from the Ciba symposium. Thorax 1984; 39:81–85.
- Mitchell RS. Theories of the pathogenesis of emphysema. Am Rev Respir Dis 1959; 50(7, part 2); 2–4.
- Gaensler EA, Lindgren I. Chronic bronchitis as an etiologic factor in obstructive emphysema. Preliminary report. Am Rev Resp Dis 1959; 50:185–193.
- Eriksson S. Pulmonary emphysema and α1-antitrypsin deficiency. Acta Med Scand 1964; 175:197–205.
- Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum of α1-antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Gross PM, Babyak A, Tolker E, Kaschak M. Enzymatically produced pulmonary emphysema: a preliminary report. J Occup Med 1964; 6:481–484.
- Richards DW Jnr, Barach AL. Prolonged residence in high oxygen atmospheres. Effects on normal individuals and on patients with chronic cardiac and pulmonary insufficiency. Quart J Med 1934; 3:437–466.
- Barach AL. The treatment of anoxia in clinical medicine. Bull New York Acad Med 1950; 26:370–383.
- McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott, WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Paré PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Eng J Med 2011; 365:1567–1575.
- Hogg JC, McDonough JE, Suzuki M. Small airway obstruction in COPD. New insights based on micro-CT imaging and MRI imaging. Chest 2013; 143:1436–1443.
- Campbell EJM. Respiratory failure. The relation between oxygen concentration of inspired air and arterial blood. Lancet 1960; ii:10–12.
- Massaro DJ, Katz S, Luchsinger PC. Effect of various modes of oxygen administration on the arterial gas values in patients with respiratory acidosis. Br Med J 1962; 2:627–629.
- Campbell EJM. The J. Burns Amberson Lecture. The management of acute respiratory failure in chronic bronchitis and emphysema. Am Rev Respir Dis 1967; 96:626–639.
- Flenley DC. The rationale of oxygen treatment. Lancet 1967; i:270–273.
- Petty TL, Finigan MM. Clinical evaluation of prolonged ambulatory therapy in chronic airway obstruction. Amer J Med 1968; 45:242–252.
- Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive pulmonary disease. Ann Intern Med 1980; 93:391–398.
- Medical Research Council Working Party. Long term domiciliary oxygen therapy in chronic cor pulmonale complicating chronic bronchitis and emphysema. Lancet 1981; 1:681–686.
- Orie NGM, Sluiter HJ, de Vries K, Tammeling GJ, Witkop J. The host factor in bronchitis. In: Bronchitis. An international symposium, 27–29 April 1960. University of Groningen. Assen: Royal Van Gorcum, 1961; 43–59.
- Fletcher CM, Peto R, Tinker C, Speizer FE. The natural history of chronic bronchitis and emphysema: an eight-year study of early chronic obstructive lung disease in working men in London. Oxford University Press: Oxford, UK, 1976.
- Fletcher CM, Peto R. The natural history of chronic airflow obstruction. Br Med J 1977; 1:1645–1648.
- Sluiter HJ, Koeter GH, de Monchy JGR, Postma DS, de Vries K, Orie NG. The Dutch hypothesis (chronic non-specific lung disease) revisited. Eur Resp J 1991; 4:479–489.
- Anthonisen NR, Connett JE, Murray RP, for Lung Health Study Research Group. Smoking and lung function of Lung Health Study participants after 11 years. Am J Respir Crit Care Med 2002; 166:675–679.
- Tashkin DP, Altose MD, Connett JP, Kanner RE, Lee WW, Wise RA. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive lung disease. Am J Respir Crit Care Med 1996, 153:1802–1811.
- Kanner RE, Anthonisen NR, Connett JE for Lung Health Study Research Group. Lower respiratory illnesses promote FEV1 decline in current but not ex-smokers with mild chronic obstructive lung disease. Am J Respir Crit Care Med 2001; 164:358–364.
- Sherman CB, Xu X, Speizer FE, Ferris BG Jr, Weiss ST, Dockery DW. Longitudinal lung function decline in subjects with respiratory symptoms. Am Rev Respir Dis 1992; 146:855–859.
- Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagan City Heart Study Group. Am J Respir Crit Care Med 1996; 153:1530–1535.
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for diagnosis, management and prevention of COPD (internet). Available from: http://www.goldcopd.org. (Accessed May 2013).
- Snider GL. Chronic obstructive pulmonary disease: a definition and implications of structural determinants of airflow obstruction for epidemiology. Am Rev Respir Dis 1989; 140:S3–S8.
- Chen JC, Mannino DM. Worldwide epidemiology of COPD. Current Opin Pulm Med 1999; 5:93–99.
- Liu B-Q, Peto R, Chen Z-M, Boreham J, Wu, Y-P, Li J-Y, Colin Campbell T, Chen J-S. Emerging tobacco hazards in China: Retrospective proportional mortality study of one million deaths. Brit Med J 1998; 317:1411–1422.
- Yin P, Jiang CQ, Cheng KK, Lam TH, Lam KH, Miller MR, Zhang WS, Thomas GN, Adab P. Passive smoking exposure and risk of COPD among adults in China: the Guangzhou Biobank Cohort Study. Lancet 2007; 370:751–757.