
Abstract
Background: Acute exacerbation of chronic obstructive pulmonary disease (AECOPD) remains a major cause of mortality. Clinical criteria of AECOPD are subjective. Biomarkers for AECOPD may aid in the initiation of early treatment. Increased production of asymmetric and symmetric dimethylarginine (ADMA, SDMA) is related to hypoxia. In COPD, a rise in ADMA results in a shift of L-arginine breakdown, contributing to airway obstruction. We aimed to compare serum levels of ADMA, SDMA and L-arginine in patients with and without AECOPD.
Methods: L-arginine metabolites quantified by high-performance liquid chromatography in venous blood samples and partial capillary oxygen pressure were prospectively investigated in 32 patients with COPD, 12 with AECOPD and 30 healthy subjects.
Results: Both ADMA and SDMA were significantly higher in AECOPD compared to stable COPD (p = 0.004 and p < 0.001, respectively). Oxygen content in capillaries correlated with serum ADMA concentration. However, the concentration of L-arginine was not different between AECOPD and stable COPD. Both ADMA and SDMA separated AECOPD with high sensitivity and specificity (AUC: 0.81, p = 0.001; AUC: 0.91, p < 0.001, respectively). A cut-off value ≥0.57 for SDMA was an independent variable to confirm AECOPD in a regression model (OR: 1.632, p = 0.001). All markers were significantly higher in the sera of both patient groups compared to the controls (p < 0.05, respectively).
Conclusions: COPD is associated with elevated L-arginine, ADMA and SDMA serum levels. In patients with AECOPD, production of ADMA and SDMA are more pronounced presumably due to more severe hypoxic insult. Methylated arginine derivatives in the sera may help early recognition of AECOPD.
Introduction
COPD is an obstructive airway disease associated with chronic airway inflammation (Citation1). Recent studies indicated an association of COPD with an abnormal inflammatory response of the lung leading to hypoxia, in addition to a systemic inflammation which coexist with vascular remodeling (Citation2). Acute exacerbations aggravate these pathological processes (Citation3). This highlights the importance of potential biomarkers for early recognition of patients with acute exacerbation.
The relationship between hypoxia and the L-arginine-NO pathway is dual: hypoxia not only induces nitric oxide (NO) overproduction (Citation4), but also facilitates protein arginine methylation (Citation4). NO is produced by the conversion of L-arginine into L-citrulline by nitric-oxide synthase (NOS) (Citation5). Three NOS isoforms have been identified: endothelial NOS (eNOS), inducible NOS (iNOS) and neuronal NOS (nNOS). Whereas eNOS and nNOS are mostly calcium/calmodulin-dependent, iNOS can be induced in response to hypoxia (Citation6) or inflammation (Citation7). NOS activity can be reduced by the accumulation of endogenous inhibitors such as asymmetric dimethylarginine (ADMA), which has recently been recognized as being associated with lung disease (Citation8–Citation10). Due to the effect of hypoxia on protein arginine methylation, ADMA is generated.
Although ADMA causes endothelial dysfunction by inhibiting eNOS, it also increases hypoxic tolerance (Citation11). Arginine methylation of cellular proteins is catalyzed by protein arginine methyltransferases (PRMT). Recently, increased lung ADMA levels and ADMA/L-arginine ratios were found due to PRMT2 over-expression under chronic hypoxic conditions. This suggests that structural and functional changes caused by hypoxia may be linked to ADMA metabolism (Citation12). Recent data suggest that the metabolism of methylarginine by the pulmonary system significantly contributes to circulating ADMA and SDMA levels (Citation13). Circulating ADMA levels have been assessed in a variety of systemic cardiovascular diseases, and are increased in conditions associated with hypoxia, ischemic stroke, renal failure, pulmonary hypertension, heart failure, and hypercholesterolemia (Citation14). Despite these lines of evidence pointing towards ADMA and SDMA as potential systemic biomarkers of different clinical stage of COPD, this has not been tested.
C-reactive protein (CRP) is increased in most forms of tissue damage, inflammation, and/or infection (Citation15). CRP predicts death and cardiovascular complications in several diseases including inflammatory diseases (Citation16), sepsis (Citation17), pulmonary diseases (Citation18), coronary heart diseases (Citation19) and ischemic stroke (Citation20). The interplay between ADMA and inflammation is a relevant issue because both factors, ADMA/SDMA and CRP, have been implicated in endothelial dysfunction in humans (Citation21–Citation23). Patients with COPD have elevated CRP levels compared to controls (Citation24), and CRP was extensively evaluated as potential diagnostic and predictive biomarker in patients with AECOPD. However the wide range of variation in elevated CRP level was probably due either to different causes of AECOPD (bacterial vs. non-bacterial) or other influential factors such as inhaled corticosteroids (Citation25).
Identifying novel biomarkers for recognition of patients with AECOPD seems to be highly valuable. To our knowledge, there is no data on comparing serum L-arginine, ADMA and SDMA levels between patients with stable COPD and AECOPD. We therefore aimed to compare the serum level of the precursor molecule L-arginine and its metabolites (ADMA and SDMA) as markers of vascular changes due to hypoxia and potential markers of acute inflammation/infection, and the acute-phase reactant hsCRP as a classic infection marker in patients with/without AECOPD.
Material and Methods
The study protocol was approved by the Regional Local Ethics Committee at University of Pecs, Faculty of Medicine (approval number: 3950, issued on 28th January, 2011) and informed consent was obtained from each patient. Anonymous blood donors served as age-matched healthy controls (n = 30).
Subjects
A total of 44 patients with COPD were recruited into this prospective study. Patients with COPD were enrolled from Department of Pulmonology at University of Pecs, Hungary. COPD was defined according to the Global initiative for chronic Obstructive Lung Disease (GOLD) guidelines (Citation26). Exclusion criteria were chronic renal failure (eGFR < 50 and/or creatinine >120 μmol/l) and decline to participate in the study.
Severity of COPD was classified as follows: I = mild, FEV1 ≥ 80% predicted (n = 3); II = moderate, 50% ≤ FEV1 < 80% predicted (n = 23); III = severe, 30% ≤ FEV1 < 50% predicted (n = 9); IV = very severe, FEV1 < 30% predicted or FEV1 < 50% predicted plus chronic respiratory failure (CRF) (n = 9). CRF (n = 16) was defined when PaO2 ≤ 55mmHg, or oxygen saturation ≤ 88% was measured twice over a three week period.
Exacerbations of COPD (AECOPD, n = 12) were defined as “an event in the natural course of the disease characterized by a change in the patient's baseline dyspnea, cough and/or sputum that is beyond normal day-to-day variations, acute onset, which may warrant a change in regular medication” (Citation1). In this subgroup, three patients were classified into class II and III respectively, and six patients into class IV according to the above-mentioned criteria.
The medical history concerning previous coronary artery disease, myocardial infarction, diabetes mellitus, hypertension, dyslipidaemia, stroke, current cigarette smoking and orally taken medication was obtained from all patients. A history of hypertension was defined as the use of antihypertensive drugs or blood pressure more than 140/90 mmHg on at least two separate occasions. Diabetes mellitus was defined as the use of anti-diabetic drugs or a fasting plasma glucose value ≥7.0 mmol/L.
Blood sampling and determination of arginine derivatives and hsCRP
Partial oxygen pressure (capillary pO2) was analyzed from capillary blood taken from the earlobe. Fasting venous blood samples were drawn and serum samples were frozen within 60 minutes and stored at -70°C until analysis. Serum high-sensitivity C-reactive protein (hsCRP) was measured by automated fluorescence immunoassay (BRAHMS Kryptor, Hennigsdorf, Germany). The amino-acid content of the blood serum samples was retrieved by solid-phase extraction (SPE) method and was quantified by high-performance liquid chromatography (HPLC) after derivatization. SPE of the analytes was performed as previously described (Citation27, Citation28). Arginine and homoarginine were detected at λex = 337 nm, λem = 520 nm, and λem = 454 nm was used for asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA).
Statistical analysis
Data were evaluated using SPSS software package (Version 19.0, SPSS Inc, Chicago, USA). Categorical data were summarized by means of absolute and relative frequencies (counts and percentages). Quantitative data were presented as mean and 95% confidence interval, as well as mean ± SD. The Kolmogorov–Smirnov test was applied to check for normal distribution. Due to normally distributed data, parametric methods (chi-square test for categorical data, Student-t test for continuous data) were used for demographic and clinical data. Non-parametric method (Mann–Whitney test) was used for hsCRP, L-arginine, ADMA and SDMA. Multiple logistic regression analysis was applied to confirm independent predictors. Cut-off value was calculated by a ROC analysis. Correlation analysis was performed calculating Spearman's correlation coefficient (r). A p-value < 0.05 was considered statistically significant.
Results
Demographic and clinical data are summarized in
Table 1. Demographic and clinical data
COPD vs. healthy subjects
Levels of L-arginine were significantly higher in recruited patients (n = 44) compared to healthy subjects (p < 0.05). Concentration of ADMA and SDMA were also significantly higher in patients compared to healthy controls (p < 0.001, respectively). Serum level of hsCRP was also significantly higher in the COPD group (p < 0.05) (see Table ).
Table 2. Serum concentration of the L-arginine pathway metabolites and hsCRP in patients with COPD
AECOPD vs. stable COPD
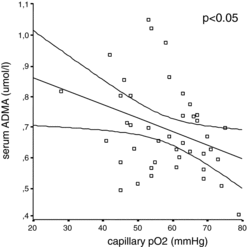
Both ADMA and SDMA were significantly higher (p < 0.001, respectively) in AECOPD compared to stable COPD patients (Figures and ); however, neither the levels of precursor molecule L-arginine (Figure ) nor hsCRP showed significant difference (see Table ). Based on a ROC analysis, both ADMA (cut-off ≥ 0.69 μmol/l, AUC: 0.81, p = 0.001) with a sensitivity of 75% and specificity of 73%, and particularly SDMA (cut-off ≥0.57 μmol/l, AUC: 0.91, p < 0.001) with a sensitivity of 92% and specificity of 86%, differentiated patients with AECOPD from stable COPD. SDMA was found as an independent variable in a multiple regression model including age, gender, co-morbidities, serum creatinine and L-arginine to verify AECOPD (OR: 1.632, 95% CI:0.75–2.5, p = 0.001). Using a ROC analysis, a cut-off value of ≥0.57 μmol/l for SDMA discriminated AECOPD with a sensitivity of 92% and specificity of 86% (AUC: 0.91, p < 0.001). Capillary pO2 was significantly lower in patients with AECOPD compared to stable COPD (p = 0.01) (Table ). We also found a significant negative correlation between partial capillary O2 pressure reflecting severity of hypoxia and serum ADMA level (p < 0.05) (Figure ).
Figure 1. Serum ADMA levels in COPD patients with and without acute exacerbation compared to healthy subjects. Serum concentration of ADMA (μmol/l) is shown in COPD patients with acute exacerbation (AECOPD), stable COPD and in healthy subjects (HS). Data are presented as mean and 95% confidence interval.
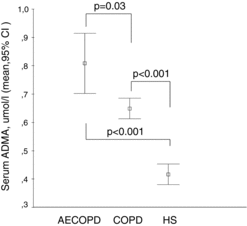
Figure 2. Serum SDMA levels in COPD patients with and without acute exacerbation compared to healthy subjects. Serum concentration of SDMA (μmol/l) is shown in COPD patients with acute exacerbation (AECOPD), stable COPD and in healthy subjects (HS). Data are presented as mean and 95% confidence interval.
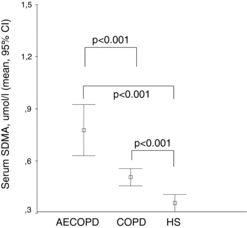
Figure 3. Serum L-arginine levels in COPD patients with and without acute exacerbation compared to healthy subjects. Serum concentration of L-arginine (μmol/l) is shown in COPD patients with acute exacerbation (AECOPD), stable COPD and in healthy subjects (HS). Data are presented as mean and 95% confidence interval.
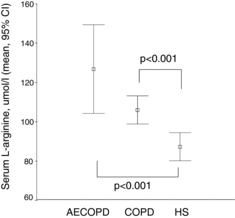
Figure 4. Correlation between partial capillary oxygen pressure and ADMA in patients with COPD. Correlation between partial capillary oxygen pressure (mmHg) and serum concentration of ADMA (μmol/l) is shown in COPD patients. (Pearson correlation, r = -0.34, p < 0.05, scatter plot diagram).
COPD patients with chronic respiratory failure (CRF)
Levels of L-arginine or its derivatives (ADMA, SDMA) in patients with CRF (n = 16) were not different from the levels in patients without CRF. Nevertheless, concentration of hsCRP was significantly higher in COPD patients with CRF (mean: 25.2, 95% CI: 5.9–44.4 vs. 6.5, 2.9–10.0, p = 0.008). CRF was not associated with AECOPD.
Discussion
To our knowledge this is the first study to demonstrate that methylarginine derivates (ADMA and SDMA) and their precursor L-arginine, are increased in the blood of patients with COPD compared to healthy subjects. We also demonstrated that the levels of SDMA and ADMA are further elevated in patients with AECOPD, and provide surrogate information in the diagnosis of COPD exacerbation with high sensitivity and specificity.
Our data obtained by analyzing systemic circulation extend observations of other studies demonstrating elevated ADMA levels in airway samples (i.e., exhaled breath condensate and induced sputum supernatant) (Citation29,Citation30). Although blood ADMA levels were also compared among patients with COPD, obstructive sleep apnea and overlap syndrome, healthy subjects were not included in the previous analyses (Citation31).
Association between L-arginine and hypoxia
We found that serum level of the precursor molecule L-arginine was elevated in the peripheral blood in patients with COPD. In experimental model of airway hyper reactivity, the exogenous administration of L-arginine resulted in a protective effect, though it was different depending on airway level and pre-treatment duration (Citation32). In addition, the total length of vessels within the gas exchange region of the hypoxic lungs was significantly increased after L-arginine supplementation (Citation32). Taking that into consideration, an elevated serum L-arginine level might be an adaptive response to chronic hypoxia associated with local and systemic vascular remodeling. As COPD is accompanied by altered gas-exchange which causes sustained systemic hypoxemia because of an increased diffusion distance, poor ventilation, or loss of alveolar structures, the beneficial effect of L-arginine can also be important in prevention of systemic hypoxic insults (Citation33,Citation34).
In accordance with this, a recent study reported that endothelial cell dysfunction induced by hypoxia due to smoking was attenuated by the addition of L-arginine or by overexpression of DDAH-2 (Citation35). However, the effect of L-arginine on the NO-pathway under chronic hypoxia remains controversial. Interestingly, the long-term oral L-arginine administration has failed to improve the hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy (Citation36), presumably because the intracellular arginine is not increased during L-arginine supplementation (Citation5). Several studies have shown that L-arginine stimulates NO-overproduction in oxidative stress, inflammation and ischemia (Citation37). On the contrary, a recent in vitro study reported a dose-dependent inhibition of DDAH activity by L-arginine, which competes with ADMA for binding to DDAH resulting in decreased NO production (Citation38).
Link between L-arginine and ADMA in pulmonary diseases
In addition to L-arginine, ADMA was also significantly higher in the serum of patients with COPD in our cohort. L-arginine can be metabolized by NOS and arginase, and arginase is the first step in polyamine production necessary for cellular proliferation. ADMA inhibits NOS, but it does not inhibit arginase activity, which result in a greater L-arginine bioavailability to arginase and therefore increases viable pulmonary endothelial cell number (Citation39). Although endothelial dysfunction and increased risk of various cardio- and cerebrovascular incidents are associated with high ADMA-concentrations, several studies point towards a potential beneficial effect of ADMA, mainly in the context of angioproliferative diseases such as cancer and pulmonary fibrosis (Citation32).
It has also been reported that ADMA infusion results in significantly enhanced lung resistance and decreased dynamic compliance in response to methacholine in an animal model (Citation40). These aphysiologic changes were associated with significantly increased lung collagen content. Furthermore, a significant decrease of nitrite in lung fluid was accompanied by elevated lung fluid urea and arginase activity in lung homogenates (Citation40). These changes were reversed after completion of ADMA administration. In addition to this, arginase activity and collagen formation was stimulated by treatment with ADMA in mouse lung fibroblasts (Citation40). These data support the hypothesis that ADMA may play a role in airway diseases through the inhibition of NOS and enhancement of arginase activity (Citation40).
Superimposed hypoxic events in AECOPD and ADMA/SDMA production
We observed that levels of ADMA, and particularly SDMA, were higher in patients with AECOPD compared to stable COPD. This indirectly suggested that acute hypoxia may contribute to elevation of these molecules in the serum. Therefore, we correlated serum levels of ADMA and SDMA with oxygen content in capillaries. This indicated a direct correlation between the severity of an acute hypoxic episode in patients with COPD and their serum ADMA-concentration. We may hypothesize that such an elevation of ADMA in acute hypoxia may be beneficial. Animal data suggests that acute hypoxic conditioning increases hypoxic tolerance (Citation12). Furthermore, inhibition of eNOS, but not iNOS, by endogenous ADMA also mediates enhancement of hypoxic tolerance (Citation12). Several data support that ADMA exerts its inhibitory effect in a concentration-dependent manner on different isoforms of NOS (Citation39,Citation40). This protective effect of ADMA may be exerted via a dose-dependent inhibition of inflammation induced iNOS activity, preventing the deleterious consequences that such hypoxic insults provoke in the lung (Citation41).
Our observation that ADMA and SDMA are elevated in the acute exacerbation of COPD may also reflect that this pathway is not only linked to hypoxia, but also to inflammation: proinflammatory pathways are activated in COPD and also in acute exacerbation (Citation42). although ADMA may play a beneficial role by improving hypoxic tolerance, it also promotes inflammation by inducing oxidative and nitrosative stress in lung epithelial cells, which can be deleterious in COPD (Citation12,Citation43). ADMA increases the generation of the downstream proinflammatory mediators TNF-α and IL-8, and it activates the NF-κβ pathway and the binding of monocytes to endothelial cells (Citation34). During the acute phase of infection, a substantial increase of SDMA was observed parallel to inflammatory/infection markers, while ADMA concentration was similar to healthy subjects (Citation23).
Interestingly, the resolution of acute inflammation secondary to infectious process was associated with a clear-cut rise in ADMA levels coincident with a marked decline in biomarkers of inflammation and nitrosative stress, while SDMA levels remained unchanged throughout (Citation23). Cytokines like IL-1 (Citation44), IL-6 (Citation45) and procalcitonin (Citation46) all amplify the expression of iNOS, as well as NO-production. Pro-inflammatory cytokines in vitro have a dual effect on the ADMA-iNOS system (Citation47). IL-1 stimulates iNOS and activates parallel DDAH resulting in decreased ADMA levels, with a net effect of NO overproduction (Citation47). The effect of pro-inflammatory cytokines on SDMA levels has still not been clarified. In a recent study, SDMA serum levels were significantly elevated in patients with sepsis and closely correlated with biomarkers of inflammation (Citation48).
Limitation of C-reactive protein in diagnostics of AECOPD
In our cohort, hsCRP did not distinguish patients with AECOPD from the stable COPD group. CRP is increased in response to a number of infectious and inflammatory conditions, but is not specific to either COPD or acute exacerbation of COPD (Citation49). CRP was not able to discriminate infectious from noninfectious causes of AECOPD (Citation49), but serves as a useful monitoring marker indicating resolution of the primary insult and inflammation. We have recently shown the time-dependent differential role of hsCRP in risk assessment related to ischemic stroke: the early increase in hsCRP level reflects atherosclerosis and therefore a risk for ischemic stroke, while its late elevation in the subacute phase of stroke was a risk factor for post-stroke infection, presumably reflecting the initial subclinical phase of infection (Citation20). A similar situation may characterize COPD, where CRP is elevated due to its association with infection, and depends on the time of investigation. There is also a great need for longitudinal studies that could prove the role of hsCRP and L-arginine metabolites (ADMA, SDMA) as predictors of AECOPD, similar to stroke (Citation50).
Importantly, COPD is a special clinical entity, where chronic hypoxia, chronic lung inflammation and systemic inflammation coexist, leading to vascular inflammation and acute exacerbations even aggravate these pathological processes (Citation2,Citation3). Therefore, our findings suggest that ADMA and particularly SDMA are not only determinants of endothelial dysfunction, but also biomarkers of AECOPD. Longitudinal studies are required to determine the role of methylated arginine derivatives as prospective biomarkers that may aid in prediction of acute exacerbation and its outcome in COPD.
| Abbreviations | ||
| AECOPD | = | COPD with acute exacerbation |
| ADMA | = | asymmetric dimethylarginine |
| AUC | = | area under the curve |
| COPD | = | chronic obstructive pulmonary disease |
| CRP | = | C-reactive protein |
| DDAH | = | dimethylarginine dimethylaminohydrolase |
| HPLC | = | high-performance liquid chromatography |
| IL | = | interleukin |
| PCT | = | procalcitonin |
| ROC | = | receiver operating characteristic |
| NO | = | nitric oxide |
| NOS | = | nitric oxide synthase |
| PRMT | = | protein arginine methyltransferase |
| SDMA | = | symmetric dimethylarginine |
Declaration of Interest Statement
Author contributions: TM had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. LN and SK contributed substantially to study design, measurement of the biomarkers and interpretation of the results. IR, VS, BI contributed substantially to recruitment of patients, blood sampling and study design. TM, LB, IR, ZI and IH contributed substantially to data analysis and interpretation, and the writing of the manuscript. TM takes responsibility for the content of the manuscript, including the data and analysis.
Acknowledgments
The authors thank Elise Steen for copyediting the revised manuscript.
Funding
This study was supported by a grant from University of Pecs AOK-KA- 34039/10-17 to T. Molnar.
References
- Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004; 23:932–946.
- Van Eeden S, Leipsic J, Paul Man SF, Sin DD. The relationship between lung inflammation and cardiovascular disease. Am J Respir Crit Care Med 2012; 186:11–16.
- Wedzicha JA, Donaldson GC. Exacerbations of chronic obstructive pulmonary disease. Respir Care 2003; 48:1204–1213.
- Chen K, Inoue M, Wasa M, et al. Expression of endothelial constitutive nitric oxide synthase mRNA in gastrointestinal mucosa and its downregulation by endotoxin. Life Sci 1997; 61:1323–1329.
- Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care 2010; 13:97–104.
- Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev 2004; 84:731–765.
- Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ Res. 1996; 79:363–380.
- Luiking YC, Poeze M, Ramsay G, Deutz NE. Reduced citrulline production in sepsis is related to diminished de novo arginine and nitric oxide production. Am J Clin Nutr 2009; 89:142–152.
- Jeyabalan G, Klune JR, Nakao A, et al. Arginase blockade protects against hepatic damage in warm ischemia-reperfusion. Nitric Oxide 2008; 19:29–35.
- Luiking YC, Hallemeesch MM, van de Poll MC,Dejong CH,de Jonge WJ, Lamers WH, Deutz NE. Reduced citrulline availability by OTC-deficiency in mice is related to reduced nitric oxide production. Am J Physiol Endocrinol Metab 2008; 295:E1315–1322.
- Song MY, Zwemer CF, Whitesall SE, D'Alecy LG. Acute and conditioned hypoxic tolerance augmented by endothelial nitric oxide synthase inhibition in mice. J Appl Physiol 2007;102:610–5.
- Yildirim AO, Bulau P, Zakrzewicz D, et al. Increased protein arginine methylation in chronic hypoxia: role of protein arginine methyltransferases. Am J Respir Cell Mol Biol 2006; 35:436–443.
- Bulau P, Zakrzewicz D, Kitowska K, et al. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am J Physiol Lung Cell Mol Physiol 2007; 292:L18–24.
- Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine: dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol 2004; 24:1023–1030.
- Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest 2003; 111:1805–1812.
- Goodson NJ, Symmons DP, Scott DG, Bunn D, Lunt M, Silman AJ. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: a ten-year followup study of a primary carebased inception cohort. Arthritis Rheum 2005; 52:2293–2299.
- Lobo SM, Lobo FR, Bota DP et al. C-reactive protein levels correlate with mortality and organ failure in critically ill patients. Chest 2003; 123:2043–2049.
- Man P, Connett JE, Anthonisen NR, Wise RA, Tashkin DP, Sin DD. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax 2006; 61:849–53.
- Blake GJ, Ridker PM. Inflammatory bio-markers and cardiovascular risk prediction. J Intern Med. 2002; 252:283–294.
- Molnar T, Papp V, Banati M, et al. Relationship between C-reactive protein and early activation of leukocytes indicated by leukocyte antisedimentation rate (LAR) in patients with acute cerebrovascular events. Clin Hemorheol Microcirc 2010; 44:183–192.
- Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol 2000; 20:2032–2037.
- Memon L, Spasojevic-Kalimanovska V, Bogavac-Stanojevic N, et al. Assessment of endothelial dysfunction: the role of symmetrical dimethylarginine and proinflammatory markers in chronic kidney disease and renal transplant recipients. Dis Markers 2013; 35:173–180.
- Zoccali C, Maas R, Cutrupi S, et al. Asymmetric dimethyl-arginine (ADMA) response to inflammation in acute infections. Nephrol Dial Transpl 2007; 22:801–806.
- Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and metaanalysis. Thorax 2004;59(7):574–580.
- Sin D, Lacy P, York E, Man SF. Effects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 170:760–765.
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHLBI/WHO workshop report. Bethesda, National Heart, Lung and Blood Institute, April 2001; NIH Publication No. 2701:1-100. Last update 2008.
- Nonaka S, Tsunoda M, Imai K, Funatsu T. High-performance liquid chromatographic assay of NG-monomethyl-L-arginine, NG,NG-dimethyl-L-arginine, NG,NG’-dimethyl-L-arginine using 4-fluoro-7-nitro-2,1,3-benzoxadiazole as a fluorescent reagent. J Chromatogr A 2005; 1066:41–45.
- Mengerink Y, Kutlán D, Tóth F, Csámpai A, Molnár-Perl I. Advances in the evaluation of the stability and characteristics of the amino acid and amine derivatives obtained with the o-phthaldialdehyde/3-mercaptopropionic acid and o-phthaldialdehyde/N-acetyl-L-cysteine reagents. High-performance liquid chromatography-mass spectrometry study. J Chromatogr A 2002; 949:99–124.
- Di Gangi IM, Pirillo P, Carraro S, et al. Online trapping and enrichment ultra performance liquid chromatography-tandem mass spectrometry method for sensitive measurement of “arginine-asymmetric dimethylarginine cycle” biomarkers in human exhaled breath condensate. Anal Chim Acta 2012; 754:67–74.
- Scott JA, Duongh M, Young AW, Subbarao P, Gauvreau GM, Grasemann H. Asymmetric dimethylarginine in chronic obstructive pulmonary disease (ADMA in COPD). Int J Mol Sci 2014; 15:6062–6071.
- Nural S, Günay E, Halici B, Celik S, Ünlü M. Inflammatory processes and effects of continuous positive airway pressure (CPAP) in overlap syndrome. Inflammation 2013; 36:66–74.
- Howell K, Costello CM, Sands M, Dooley I, McLoughlin P. L-Arginine promotes angiogenesis in the chronically hypoxic lung: a novel mechanism ameliorating pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2009; 296:L1042–1050.
- Lüneburg N, Harbaum L, Hennigs JK. The endothelial ADMA/NO pathway in hypoxia-related chronic respiratory diseases. Biomed Res Int 2014; 2014:501612.
- Sommer N, Dietrich A, Schermuly RT et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 2008; 32:1639–1651.
- Jiang DJ, Jia SJ, Yan J, Zhou Z, Yuan Q, Li YJ. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem Biophys Res Commun 2006; 349:683–693.
- Laursen BE, Dam MY, Mulvany MJ, Simonsen U. Hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy is unaltered by long-term oral L-arginine administration. Vascul Pharmacol 2008; 49:71–76.
- Chen MF, Xie XM, Yang TL, et al. Role of asymmetric dimethylarginine in inflammatory reactions by angiotensin II. J Vasc Res 2007; 44:391–402.
- Wang J, Sim AS, Wang XL, Wilcken DE. L-arginine regulates asymmetric dimethylarginine metabolism by inhibiting dimethylarginine dimethylaminohydrolase activity in hepatic (HepG2) cells. Cell Mol Life Sci 2006; 63:2838–2846.
- Chen B, Strauch K, Jin Y, Cui H, Nelin LD, Chicoine LG. Asymmetric dimethyl arginine does not inhibit arginase activity and is pro-proliferative in pulmonary endothelial cells. Clin Exp Pharmacol Physiol 2014; 41:469–474.
- Wells SM, Buford MC, Migliaccio CT, Holian A. Elevated asymmetric dimethylarginine alters lung function and induces collagen deposition in mice. Am J Respir Cell Mol Biol 2009; 40:179–188.
- Rus A, Peinado MA, Castro L, Del Moral ML. Lung eNOS and iNOS are reoxygenation time-dependent upregulated after acute hypoxia. Anat Rec (Hoboken) 2010; 293:1089–1098.
- Brusselle GG, Provoost S, Bracke KR, et al. Inflammasomes in respiratory disease: from bench to bedside. Chest 2014; 145:1121–1133.
- Wells SM, Holian A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol 2007; 36:520–528.
- Ratnam S1, Mookerjea S. The regulation of superoxide generation and nitric oxide synthesis by C-reactive protein. Immunology 1998; 94:560–568.
- Klein E, Weigel J, Buford MC, Holian A, Wells SM. Asymmetric dimethylarginine potentiates lung inflammation in a mouse model of allergic asthma. Am J Physiol Lung Cell Mol Physiol 2010; 299:L816–825.
- Boota A., Zar H., Kim YM. et al. IL-1 beta stimulates superoxide and delayed peroxynitrite production by pulmonary vascular smooth muscle cells. Am J Physiol 1996; 271:L932–L938.
- Kinugawa K, Shimizu T, Yao A, et al. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ Res 1997; 81:911–921.
- Hoffmann G, Czechowski M, Schloesser M, Schobersberger W. Procalcitonin amplifies inducible nitric oxide synthase gene expression and nitric oxide production in vascular smooth muscle cells. Crit Care Med 2002; 30:2091–2095.
- Antonescu-Turcu AL, Tomic R. C-reactive protein and copeptin: prognostic predictors in chronic obstructive pulmonary disease exacerbations. Curr Opin Pulm Med 2009: 15:120–125.
- Molnar T, Pusch G, Papp V, et al. The L-arginine pathway in acute ischemic stroke and severe carotid stenosis: temporal profiles and association with biomarkers and outcome. J Stroke Cerebrovasc Dis 2014; 23:2206–2214.