
Abstract
Airway inflammation in chronic obstructive pulmonary disease (COPD) is refractory to corticosteroids and hence COPD treatment is hindered and insufficient. This study assessed the effects of oral treatment with Montelukast (10 and 30 mg/kg) or dexamethasone (20 mg/kg) for 20 days on COPD model induced by chronic exposure to lipopolysaccharide (LPS). Six groups of male guinea pigs were studied. Group 1: naïve group, group 2: exposed to saline nebulization. Groups 3, 4, 5, and 6: exposed to 9 nebulizations of LPS (30 μg/ml) for 1 hour, 48 hours apart with or without treatment with Montelukast or dexamethasone. Airway hyperreactivity (AHR) to methacholine (MCh), histopathological study and bronchoalveolar lavage fluid (BALF) as well as lung tissue analyses were performed 48 hours after the final exposure to LPS (day 20). LPS-induced pulmonary dysfunction was associated with increased neutrophil count, leukotriene (LT) B4, and tumor necrosis factor (TNF)-α in BALF. Moreover, there was an increase in malondialdehyde (MDA) level and a decrease in histone deacetylases(HDAC) activity in the lung tissue. Both Montelukast (10 or 30 mg /kg) and dexamethasone significantly reduced neutrophil count in BALF and inflammatory cells in lung parenchyma as well as TNF-α, and MDA levels. However, dexamethasone was more effective (p < 0.05). Montelukast, at a dose of 30 mg /kg, significantly reduced specific airway resistance after the 9th LPS exposure, attenuated AHR to MCh, decreased LTB4 and increased HDAC activity in comparison to dexamethasone. These results suggest that treatment with Montelukast can be useful in chronic airway inflammatory diseases including COPD poorly responsive to glucocorticoids.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory airway disease with progressive and irreversible airflow limitation. Because airway inflammation in COPD is refractory to corticosteroids, COPD treatment is hindered and insufficient (Citation1). New investigations into the mechanisms of glucocorticoid action have broadened and deepened our understanding of glucocorticoid resistance. Cellular and molecular factors, receptors, and complex signaling pathways have all been implicated in glucocorticoid resistance. Indeed, based on molecular biological studies, excessive activation of intracellular transcription factors, impaired histone deacetylase activity, and epigenetic factors (such as miR-18 and miR-124a) may result in glucocorticoid resistance (Citation2).
The major anti-inflammatory effects of glucocorticoids appear to be largely due to interaction between the activated glucocorticoid receptor and transcription factors, notably nuclear factor-kappa B (NF-kappa B) that mediates the expression of inflammatory genes. NF-kappa B switches on inflammatory genes via a process involving recruitment of transcriptional coactivator proteins and changes in chromatin modifications such as histone acetylation. The interactions between NF-kappa B and the activated glucocorticoid receptor result in differing effects on histone acetylation and deacetylation processes (Citation3).
Acetylation of histones by histone acetyltransferases activates inflammatory genes, whereas histone deacetylation results in inflammatory gene repression. Glucocorticoids exert their anti-inflammatory effects partly by inducing acetylation of anti-inflammatory genes, but mainly by recruiting histone deacetylase-2 (HDAC2) to activated inflammatory genes. HDAC2 deacetylates acetylated glucocorticoid receptors so that they can suppress activated inflammatory genes in asthma. In COPD, there is resistance to the anti-inflammatory actions of glucocorticoids, which is explained by reduced activity and expression of HDAC2 (Citation4).
Leukotrienes (LTs), including cysteinyl LTs (CysLTs) and LTB (Citation4), are potent lipid mediators that have a role in the pathophysiology of asthma (Citation5). Leukotrienes have been measured in several biological fluids and their concentrations were found to be elevated in patients with asthma which supports their pathophysiological role (Citation6).
Neutrophils provide a defense against infections that cause exacerbations of COPD, but persistent neutrophilia in the absence of infection in COPD reflects the chronic inflammatory state of the airways in this condition (Citation7). Leukotriene (LT) B4 is a potent chemoattractant and activator of neutrophils and eosinophils (Citation8). Neutrophils rapidly release large amounts of LTB4 in response to activating stimuli and have a high density of cell-surface LTB4 receptors (Citation9). This suggests a potential involvement of LTB4 in the induction of neutrophil survival by lipopolysaccharide (LPS) activation (Citation10). Apart from neutrophils, BLT1 and BLT2 receptors are expressed in numerous airway inflammatory cells including eosinophils, mast cells and macrophages (Citation11).
Montelukast antagonizes effectively the proasthmatic/proinflammatory/priming activities of cysteinyl leukotrienes (CysLTs) and forms part of numerous international guidelines for asthma therapy (Citation12). Interestingly, recent evidence suggests that Montelukast possesses a range of secondary anti-inflammatory activities, apparently unrelated to antagonism of cysLTs receptors (Citation13). These CysLT receptor-independent, anti-inflammatory mechanisms of action of montelukast may be particularly effective in controlling the glucocorticoid-insensitive inflammation and are the major focus of the current study.
The aim of the present study was to determine the influence of therapeutic (10 mg/kg) (Citation14) and high (30 mg/kg) (Citation15) doses of montelukast in comparison to dexamethasone (20 mg/kg) on the functional and histopathological effects of chronic pulmonary inflammation induced by repeated exposure of guinea pigs to LPS as a model for the progressive inflammatory processes of COPD (neutrophilic inflammation) (Citation16). The possible molecular mechanisms of results was determined by measuring different mediators that may be involved and implicated in thease results. The outcomes measured were airway hyperreactivity (AHR) to methacholine (MCh), histopathological study and bronchoalveolar lavage fluid (BALF) as well as lung tissues analyses were performed. LTB4 and tumor necrosis factor (TNF)-α released in bronchoaveolar lavage fluid (BALF) were measured. The effects of Montelukast on malondialdehyde (MDA) level, and HDAC activity in lung tissue in comparison to dexamethasone were also assessed.
Material and Methods
Animals
Male guinea pigs, weighing 300 to 400 g, were used in this study. Animals were kept under standard laboratory conditions (12/12 h light/dark cycle, 22 ± 2°C room temperature, 50–60% humidity) for at least 1 week before starting the experiments. All animal procedures were approved by the Institutional Animal Ethics Committee for Ain Shams University, Faculty of Medicine.
Drugs and chemicals
Lipopolysaccharides from Escherichia coli serotype 055-85, methacholine chloride, carboxymethylcellulose sodium and dexamethasone sodium phosphate were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Montelukast sodium was generously provided by Merck, Sharp and Dohme, USA. LPS and methacholine were dissolved in phosphate-buffered saline (PBS) with pH 7.4. Dexamethasone and Montelukast were suspended in a 0.5% solution of carboxymethylcellulose sodium as a vehicle (Citation17).
Guinea pigs were randomly allocated into six groups (each consisted of 6 animals) as follows:
| N: | naive group | ||||
| C: | control; saline exposed and administered orally 1 ml solution of carboxy methylcellulose sodium (0.5%) daily. | ||||
| LPS: | LPS exposed and administered orally 1 ml solution of carboxy methyl cellulose (0.5%) daily. | ||||
| LPS/Mont (10): | LPS-exposed and orally treated with Montelukast (10 mg/kg/day). | ||||
| LPS/Mont (30): | LPS-exposed and orally treated with Montelukast (30 mg/kg/day). | ||||
| LPS/Dex: | LPS- exposed and pretreated with dexamethasone (20 mg/kg/day). | ||||
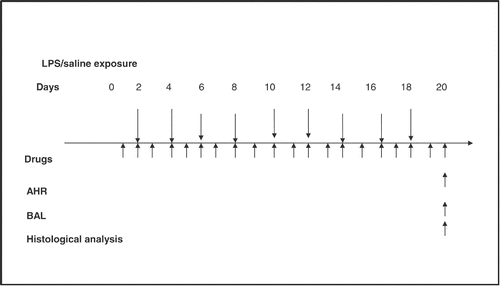
All groups (except the naïve group) were exposed for 1 h to an aerosolized solution of LPS (30 μg/ml) or saline. Animals were exposed to the aerosolized solution 9 times, 48 hours apart (Citation16). The aerosol was generated by PARI Jet nebulizer (HSE, Germany), at a rate of 0.5 ml/min. The average of two specific airway resistance (sRaw) measurements was obtained prior to exposure (baseline) and 48 hours after the first and final exposure. Treatment with the test drugs and vehicle were started 1 day before the first LPS exposure and continued for 2 days after the last LPS exposure i.e., for 20 days as illustrated in . Methacholine (MCh)-induced airway hyperreactivity (AHR) was measured 48 h after the last exposure to LPS, and then BALF was collected and the lungs were isolated. BALF was used for estimation of LTB4 and TNF α and the isolated lungs were used for histological studies and for estimation of MDA level, as well as HDAC activity.
Figure 1. Schedule of the study: Chronic (9 exposures, 48 hours apart) exposure (60 min) to nebulized lipopolysaccharides (LPS) (30 μg/ml) or vehicle (pathogen-free saline) of conscious guinea pigs, with and without Montelukast or dexamethasone treatment. Airway hyperreactivity (AHR) was measured at 48 hours after the final exposure to LPS, bronchoalveolar lavage fluid (BALF) was conducted for cellular analysis and tumor necrosis factor (TNF) α level measurement. Lungs for malondialdehyde, histone deacetylase activity and histological analysis) were collected at 48 hours. Test drugs were administered every day, usually 30 minutes before exposure on days of LPS challenge.
Measurement of respiratory function
Respiratory function was measured according to Pennock et al. (Citation18). Conscious guinea pigs were placed in cylindrical double box body plethysmograph. Respiratory flow in the body chamber of plethysmograph was measured indirectly by changes in the thoracic gas volume (body box) during respiration. These primary flow signals (nasal and thoracic) were transmitted via a differential pressure transducer (Validyne D P 45–14) and amplifier in the form of PLUGSYS with CFBA module to a data acquisition/analysis system (Pulmodyn software) from HSE (Hugo Sachs Electronic). Specific airway resistance (sRaw) is determined from the phase displacement between the nasal and thoracic flow. A minimum of 5 breaths were analyzed for each animal at each time point. Before each experiment, the animals were handled and familiarized with the equipment to reduce stress. sRaw was expressed as mmHg/s and increases in sRaw were expressed as percentage of baseline values before drug treatment and were compared with control values. Furthermore, changes in sRaw were expressed as percentage of value of drug-treated group against that of LPS control.
Measurement of airway hyper-reactivity
MCh-induced AHR were determined by modification of the method of Tulić et al. (Citation19) and Kasahara et al. (Citation20). Increasing concentrations of MCh (0.125, 0.25, 0.5, 1, 2, and 4 mg/ml) were delivered by a PARI Jet nebulizer (HSE; Germany), at a rate of 0.5 ml/ min. Each concentration of MCh was delivered for 1 min at 5 min interval. Peak values of sRaw after each concentration of MCh were recorded. The AHR of each group was expressed as PC100 MCh (concentration of MCh that causes an increase of specific airway resistance by 100% over the baseline value). The PC100 was calculated by linear interpolation of graphed data.
Bronchoalveolar lavage fluid analysis
Bronchoalveolar lavage was performed as follows: Guinea pigs were sacrificed with an overdose of pentobarbital sodium (400 mg/kg; i.p.). The lungs were lavaged via tracheal cannula with 50 ml of PBS (5 × 10 ml), which were aspirated after a gentle chest massage. The pooled BALF was centrifuged at 500g, at 4°C for 10 min, and the pellet was resuspended in 0.25% NaCl to lyse residual erythrocytes; after centrifugation, the pellet was resuspended again in 1 ml of 0.9% NaCl. A total cell count (cells/ml) of the pooled BALF was determined using a hemocytometer (Neubauer). A Cytospin smear (Shandon Centrifuge: 1000 rpm 7 min) of the BALF samples was differentially stained (Leishman’s: 1.5% in methanol, 6 min), and a minimum of 200 cells were counted. The results were expressed as a percentage of total cells and as the actual number of each cell type (Citation21).
Biochemical parameters
Bronchoalveolar lavage fluid
| A. | Measurement of Tumor necrosis factor-α The levels of TNF-α in BALF supernatants were assessed by commercially available ELISA using Rat Biosource International (California USA) micro titer strips diagnostic kit. The assay was performed according to the instructions of the manufacturers. The detection limit was 4.5 pg/ml. | ||||
| B. | Measurement of Leukotriene B4 LTB4 was measured by enzyme immunoassay (Cayman Chemicals, Ann Arbor, MI, USA). The assay was performed according to the instructions of the manufacturers. The detection limit was 7 pg/ml. | ||||
Lung tissue homogenates
After measurement of AHR, the lungs were removed. Care was taken to remove adhering tissues such as bronchia and vessels. The right lung lobe and the trachea of each lung were cut for histological studies. The other parts of the lung were then homogenized in 10 ml PBS. An aliquot of 0.5 mL of homogenate was hydrolyzed by 0.5 mL of 12N HCl at 120°C for 24 h and used for the measurement of malodialdehyde, and histone deacetylase.
| A. | Measurement of lung malondialdehyde Lipid peroxides (LP) levels were determined as thiobarbituric acid reactive substances (TBARS) according to the method of Buege and Aust (Citation22). Results were expressed as nanomoles of malondialdehyde (MDA) / mg wet tissue weight. | ||||
| B. | Measurement of lung histone deacetylase Histone deacetylase (HDAC) activity was measured using a fluorometric assay kit (Sigma- Aldrich) based on a two-step enzymatic reaction. The measured fluorescence is directly proportional to the deacetylation activity of the sample (Citation23). HDAC activity was monitored with excitation at 490 nm and emission at 525 nm. | ||||
Lung histopathology
Histological examination of the lungs was undertaken to determine whether the inflammatory cell profile obtained from BALF was representative of tissue inflammation. After lavage, the lungs were removed from the thoracic cavity, the right lung lobe and the trachea of each lung were cut as mentioned above and immersed in neutral-buffered formalin for at least 72 hours. After fixation, samples were cut, dehydrated in 70 to 100% ethanol/xylene, and embedded in paraffin wax. Sections were cut (6 μm), deparaffinized, and stained with hematoxylin and eosin. Inflammatory cells were identified by standard morphometry. Numbers of cells in tissue and air spaces were counted in 10 random, non-overlapping parenchymal fields at × 100 magnifications under bright field illumination (Citation19).
Statistical analysis
Changes in sRaw from the baseline values (taken before the procedure) are presented as a percentage of the mean baseline value preceding the first LPS challenge. Changes in airway function between groups were compared using analysis of variance, followed by Newman–Keuls Multiple Comparison Test. Differences were considered statistically significant when p < 0.05. The airway hyper-reactivity of each group is expressed as PC100 methacholine. The PC100 was calculated by linear interpolation. BAL fluid cell counts, MDA, TNF-α, LTB4, and HDAC activity were compared using analysis of variance, followed by Newman–Keuls Multiple Comparison Test. Differences were considered statistically significant when p < 0.05.
Results
Airway function studies
Effects of Montelukast and dexamethasone on respiratory function
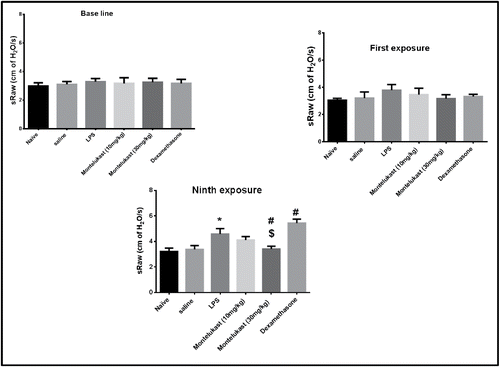
First exposure to LPS caused an immediate bronchoconstriction lasting 30 min; however, this effect was not significantly different (p > 0.05) from the response to saline. All subsequent LPS exposures caused more increase in the response. The 9th exposure to LPS caused a significant elevation in sRaw from saline group value; this outcome persisted for more than 2 hours. Montelukast (10 and 30 mg/kg) and dexamethasone (20 mg/kg) treatment did not significantly affect (p > 0.05) the initial LPS-induced bronchoconstriction after the first exposure. Moreover, Montelukast (10 mg/kg) did not significantly affect (p > 0.05) the LPS-induced bronchoconstriction after the ninth exposure. On the other hand, dexamethasone tended to significantly exaggerate the bronchoconstrictor response to LPS after the 9th ninth LPS exposure. In contrast, guinea pigs treated with Montelukast (30 mg/kg) developed significant (p < 0.05) bronchodilation after the 9th exposure to LPS ().
Figure 2. Effect of oral treatment with Montelukast (10 and 30 mg/kg) and dexamethasone on airway function of conscious guinea pigs during base line, after first exposure and after chronic exposure (9 times) to LPS (30 mg/ml) or vehicle (saline) inhalation, for 1 hour, 48 hours apart. Drug treatment started 1 day before LPS exposure and continued daily for 20 days. Airway function was expressed as specific airway resistance (sRaw). Each point represents the mean ± S.E.M. (n = 6). Significance of differences from saline exposure (*, p < 0.05), significance of differences from LPS exposures (#, p < 0.05) and significance of differences from dexamethasone treatment ($, p < 0.05) were determined by analysis of variance (single factor), followed by Newman–Keuls Multiple Comparison Test.
Effect of Montelukast and dexamethasone on airway hyperreactivity
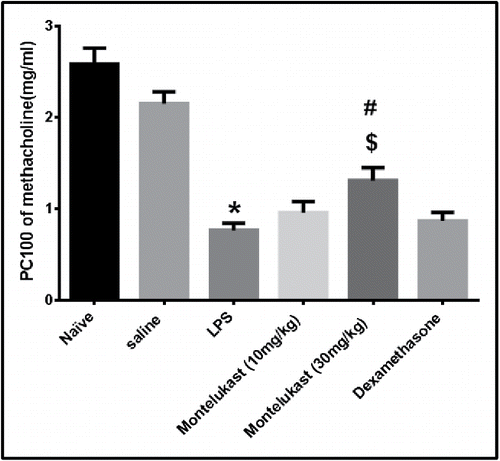
Airway hyperreactivity to MCh was determined 48 h after the 9th exposure to LPS. Inhalation of various concentration of MCh (0.125-4.0 mg/ml) caused an increase in the sRaw over the baseline in all groups (). The PC100 value of the LPS group was significantly reduced, compared with that of the saline group (P < 0.05).Treatment with Montelukast (10 mg/kg) and dexamethasone caused no significant elevation of the mean PC100 of inhaled MCh compared to that of LPS group. Moreover, there was no significant between these two groups (P > 0.05). The PC100 value of Montelukast (30 mg/kg) treated group was raised significantly compared to that of LPS and dexamethasone-treated groups ().
Table 1. Effect of pretreatment with montelukast and dexamethasone on airway hyperresponsiveness (AHR) to methacholine (Mch) in chronic LPS exposed guinea pigs
Figure 3. Effect of oral treatment with Montelukast (10 and 30mg/kg) and dexamethasone on airway hyperreactivity (AHR). AHR was examined 48 hours after the 9th exposure to LPS with increasing concentration of methacholine. Airway hyperreactivity is expressed as PC100 (the methacholine dose, in mg/kg, at which 100% bronchoconstriction was induced. Values given are means ± S.E.M. (n = 6). Significance of differences from saline exposure (*, p < 0.05), significance of differences from LPS exposures (#, p < 0.05) and significance of differences from dexamethasone treatment ($, p < 0.05) were determined by analysis of variance (single factor), followed by Newman–Keuls Multiple Comparison Test.
Bronchoalveolar lavage fluid
The total leukocyte count in BALF in the control LPS group was significantly (p < 0.05) amplified after the 9th exposure to LPS, compared with cells in the saline exposed group. Differential cell counts revealed that the number of macrophages, eosinophils, and neutrophils (p < 0.05) were significantly more in the LPS group compared with those in the saline group. Treatment with Montelukast (10 and 30 mg/kg) and dexamethasone significantly reduced the number of macrophages (41.03%, 49.16%, and 58.1%, respectively), eosinophils (65%, 78.93%, and 87.5%, respectively), and neutrophils (25.85%, 33.58% and 45.69%, respectively) in the BALF, at 48 h after the 9th LPS exposure. The resultant airway neutrophilia was significantly (P < 0.05) reduced in a dose-related fashion by administration of Montelukast ().
Table 2. Effect of oral treatment with montelukast or dexamethasone on airway influx of inflammatory cells (leukocytes count) after chronic exposure to LPS or saline
Biochemical parameters
Tumor necrosis factor- α
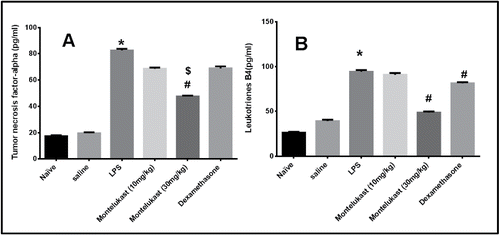
Levels of TNF-α were significantly (p < 0.05) higher in LPS-exposed guinea pigs as compared to saline-exposed guinea pigs (A). Pretreatment with Montelukast (10 and 30 mg/kg) and dexamethasone significantly (p < 0.05) reduced the TNF-α levels in the BALF as compared to LPS-exposed guinea pigs with no significant difference between Montelukast (30 mg/kg) and dexamethasone-treated groups.
Figure 4. Effect of oral treatment with Montelukast (Mont) (10 and 30 mg/kg) and dexamethasone (Dex) on release of A: Tumor necrosis factor-α. B: Leukotriene B4 in BALF 48 hours after the 9th exposure to LPS in guinea pigs. Each column shows means ± S.E.M. (n = 6). Significance of differences from saline exposure (*, p < 0.05), significance of differences from LPS exposures (#, p < 0.05) and significance of differences from dexamethasone treatment ($, p < 0.05) were determined by analysis of variance (single factor), followed by Newman–Keuls Multiple Comparison Test.
Leukotriene B4
Levels of LTB4 were significantly (p < 0.05) higher in LPS- exposed guinea pigs as compared to saline exposed guinea pigs (B). Pretreatment with Montelukast (10 mg/kg) did not significantly alter LTB4 levels in the BALF. In contrast, Montelukast (30 mg/kg) and dexamethasone (20 mg/kg) significantly (p < 0.05) reduced the LTB4 levels compared to LPS exposed guinea pigs. Montelukast (30 mg/kg) was however more effective as compared with dexamethasone (p < 0.05).
Malondialdehyde
Lung MDA contents in the LPS group were significantly increased (p < 0.05), compared with those in the saline group. Lung MDA contents in the Montelukast (10 and 30 mg/kg) and dexamethasone-treated (20 mg/kg) groups were found to be significantly reduced compared with those in the LPS exposed group. There was a significant difference between Montelukast (10 mg/kg) and dexamethasone-treated (20 mg/kg) groups but the difference between Montelukast (30 mg/kg) and dexamethasone were not significant (p > 0.05) (A).
Figure 5. Effect of oral treatment with Montelukast (10 and 30 mg/kg) and dexamethasone on A: MDA (malondialdehyde) level and B: HDAC (Histone deacetylase) activity in lung tissue homogenate 48 hours after the 9th exposure to LPS in guinea pigs. Each column shows means ± S.E.M. (n = 6). Significance of differences from saline exposure (*, p < 0.05), significance of differences from LPS exposures (#,p < 0.05) and significance of differences from dexamethasone treatment ($, p < 0.05) were determined by analysis of variance (single factor), followed by the Newman–Keuls Multiple Comparison Test.
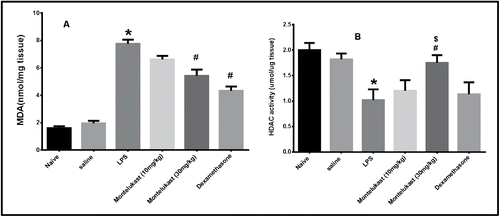
Histone deacetylase
Lung HDAC activity was significantly decreased in LPS group. Pretreatment with Montelukast (30 mg/kg) significantly (p < 0.05) increased HDAC activity. On the other hand, Montelukast (10 mg/kg) and dexamethasone (20 mg/kg) had insignificant (p > 0.05) effect on the decrease in HDAC activity induced by LPS (B).
Lung histopathology
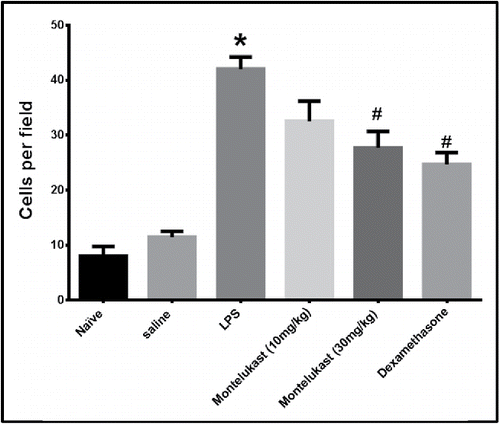
The average number of inflammatory cells in 10 randomly chosen fields (500 μm squares) counted with light microscopy is shown in . The number of cells counted in the LPS group was significantly increased (p < 0.05), compared with the cells in the saline group. The increase in the number of cells induced by LPS was significantly inhibited by Montelukast and dexamethasone (p < 0.05).
Figure 6. Effect of oral treatment with Montelukast (10 and 30 mg/kg) and dexamethasone on average number of inflammatory cells in 10 randomly chosen fields (500-μm squares) measured from 10 randomly selected lines passing through the lung in H & E stained sections taken 48 hours after the 9th exposure to LPS. Each column represents means ± S.E.M. (n = 6). Significance of differences from saline exposure (*, p < 0.05), and significance of differences from LPS exposures (#, p < 0.05) were determined by analysis of variance (single factor), followed by the Newman–Keuls Multiple Comparison Test.
Discussion
In the present study bacterial inflammation by exposure to aerosolized LPS, exhibited COPD-like pathophysiological pulmonary features similar to those reported for airway obstruction (the increase in specific airway resistance), airway hyperresponsiveness (AHR) to inhaled MCh and increased total cell count in the BALF, predominantly as a result of neutrophil influx. Many studies have documented the prominence of neutrophils after administration of LPS, either via systemic or inhaled routes (Citation24). Inflammatory response is evident in the lung parenchyma after exposure to LPS in the present study and this was demonstrated previously by Pauwels and coworkers (Citation25). In addition, LPS exposure in the present study upregulated the production of TNF-α and LTB4 in the BALF, as well as elevated MDA level and lowered HDAC activity in the lung tissue.
Dexamethasone (20 mg/kg), amplified the bronchoconstrictor response to LPS after the 9th LPS exposure. In fact, Toward and Broadler (Citation16) demonstrated that dexamethasone (20 mg/ kg, i.p.) exacerbated bronchoconstrictions in guinea pigs chronically exposed to LPS. In contrast, Montelukast (30 mg/kg)-treated guinea pigs developed significant bronchodilation after the 9th exposure to LPS.
In the present study, AHR was examined as a functional endpoint and it was found that the LPS group showed the highest response to MCh among all studied groups. Low dose of Montelukast did not significantly inhibit the airway response to gradual increasing doses of MCh. An important finding is that AHR was resistant to dexamethasone therapy. Pretreatment with high dose Montelukast (30 mg/kg), however, inhibited the development of LPS-induced AHR, indicating that Montelukast played a central role in the inhibition of steroid-resistant AHR.
Results of the present study revealed that, pretreatment with Montelukast (10 and 30 mg/kg) and dexamethasone (20 mg/kg) significantly reduced the number of macrophages, eosinophils, and neutrophils in the BALF, 48 h after the 9th LPS exposure. The resultant airway neutrophilia was significantly reduced in a dose-related fashion by administration of Montelukast. In addition, the accumulation of inflammatory cells in the lung parenchyma after the 9th exposure to LPS was inhibited by treatment with either Montelukast or dexamethasone.
Our results support the findings of Toward and Broadler (Citation16) who reported that dexamethasone reduced neutrophil influx at a dose of 20 mg/kg p.o. in a guinea pig model of pulmonary edema induced by chronic LPS exposure. Besides, Whelan (Citation26) reported inhibition of neutrophilia in guinea pigs after single exposure to LPS with treatment of 5 mg/kg dexamethasone. However, Kaneko et al. (Citation17) found that dexamethasone treatment (1 mg/kg i.p.) did not affect any increase in the total number of cells, neutrophils, and macrophages in BALF induced by chronic LPS exposure.
Belvisi (Citation27) reported that neutrophils are less sensitive to glucocorticoids than are eosinophils and T cells, and that macrophages from patients with COPD are less sensitive to steroid treatment under certain circumstances. Whelan et al. (Citation28) demonstrated that higher doses of dexamethasone were required to inhibit LPS-induced neutrophilia (ED50 10.8 mg/kg i.p.) in guinea-pig lungs. These differences in the responsiveness of activated inflammatory cells may help to explain why glucocorticoid treatment has been more beneficial for patients with asthma than for patients with COPD (Citation27,Citation29). Patients with asthma-COPD overlap syndrome (ACOS) could be more responsive to glucocorticoids (Citation30).
Zhu et al. (Citation31) reported that although neutrophils do not produce CysLTs, they do possess receptors for LTC4 and LTD4, activation of which triggers relatively modest pro-inflammatory responses in these cells. Interference with neutrophil activation by CysLTs released from other cell types, such as monocytes/macrophages, mast cells or eosinophils, may therefore underlie the neutrophil-directed therapeutic efficacy of Montelukast.
In the present study, Montelukast significantly reduced the LTB4 levels compared to LPS exposed guinea pigs. This is in agreement with Anderson et al. (Citation32), who demonstrated that Montelukast markedly attenuated LTB4 production by platelets activating factor-activated neutrophils with maximal inhibition (89%) observed at concentrations of 2 μM.
TNF-α is one of the important inflammatory mediators in COPD and its level is overstressed in the sputum of patients with COPD (Citation33). In this study, guinea pigs exposed to LPS, showed significantly higher levels of TNF-α in BALF than saline-exposed guinea pigs. Pretreatment with Montelukast (10 and 30 mg/kg) and dexamethasone (20 mg/kg), significantly reduced TNF-α levels in the BALF. Both Montelukast (30 mg/kg) and dexamethasone were more effective than Montelukast (10 mg/kg); but there was no significant difference between Montelukast (30 mg/kg) and dexamethasone treated groups.
Basyigit et al. (Citation34) evaluated the effects of Montelukast (0.1 mg/kg i.p) in smoke-induced COPD in Wistar albino rats. They found that Montelukast significantly decreased serum TNF-α levels and total histopathological damage score of the lung. There was no statistically significant difference between the Montelukast group and healthy controls. The authors concluded that Montelukast might have a protective effect on smoke-induced lung injury in rats both from a histopathological and inflammatory point of view.
Moreover, Maeba et al. (Citation35) examined the inhibitory effect of Montelukast on LPS-induced TNF-α production in peripheral blood mononuclear cells. Montelukast (Citation10-Citation5) M) significantly inhibited LPS-induced TNF-α production in the peripheral blood mononuclear cells of controls and patients with asthma. In addition the study conducted by Can et al. (Citation36) showed that Montelukast improves clinical parameters and shows anti-inflammatory response by decreasing serum TNF-α level. Dexamethasone was less effective in reducing TNF-α release in guinea pigs exposed to LPS than Montelukast (30 mg/kg). This confirms previous data showing that neither inhaled nor oral glucocorticoids had any suppressive effect on TNFα regulation in COPD patients (Citation37).
The present results showed that lung MDA contents in the Montelukast (10 and 30 mg/kg) and dexamethasone-pretreated (20 mg/kg) groups were significantly inhibited compared with those in the LPS- exposed group. There was a significant difference between Montelukast (10 mg/kg) and dexamethasone pretreated groups, but the differences between Montelukast (30 mg /kg) and dexamethasone were not significant. Similarly, the studies conducted by Cuciureanu et al. (Citation38) and Sener et al. (Citation39) indicate that Montelukast can protect against experimental organ damage through its marked antioxidant effect by reducing MDA besides its anti-inflammatory effects by reducing TNF-α. Furthermore, the study conducted by Du et al. (Citation40) showed that MDA in the serum of asthmatic patients and in the serum and lung tissue of asthma models of guinea pigs were significantly decreased by dexamethasone inhalation.
In the present study HDAC activity was significantly less in lung homogenate from guinea pigs exposed to LPS as compared with the saline-exposed group. Lung homogenate from guinea pigs exposed to LPS pretreated with Montelukast (30 mg/kg) showed a significant increase in HDAC activity. In contrast, Montelukast (10 mg/kg) and dexamethasone did not improve to any significant extent the decrease in HDAC activity induced by LPS. Increase of HDAC activity by high-dose Montelukast may enhance anti-inflammatory gene transcription that it may “unlock” the glucocorticoid resistance of COPD and potentiate its suppressive effects. This suggests that high-dose Montelukast may restore the anti-inflammatory effect of glucocorticoids and thus controls the underlying disease process in COPD.
Tahan et al. (Citation41) reported that Montelukast at concentrations of 0.01–10 μM resulted in substantial suppression of histone acetyl transferase (HAT) activity following activation of a monocyte/macrophage cell line with TNF-α. The precise molecular mechanism underlying these inhibitory effects of Montelukast on HAT was not established, but may involve interference with the activation of transcriptional coactivator proteins, a prerequisite for histone acetylation, chromatin unwinding, and gene transcription. Suppression of HAT activity may explain the increase in HDAC in the present study.
Conclusion
Montelukast improved some aspects of the pathological and functional changes induced by chronic exposure to LPS. The HDAC-targeted anti-inflammatory activity of this agent demonstrated in the current study may contribute to the beneficial effects of this agent. In addition, Montelukast attenuated the production of LTB4 representing an additional therapeutic activity that may contribute to the control of corticosteroid-insensitive neutrophil-mediated inflammation. High-dose Montelukast might provide a way of managing this common and globally increasing disease. Nevertheless, this needs to be confirmed in clinical studies in COPD patients.
Acknowledgments
The author wishes to thank Dr. Safaa Mohammed Shaker Mohammed, Assistant Professor of Histology, Faculty of Medicine–Ain Shams University for her help and support in the histopathological part of this study.
Declaration of Interest Statement
The authors have declared that no competing interests exist. The authors alone are responsible for the content and writing of the paper.
References
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet 2009; 373:1905–1917.
- Wang W, Li JJ, Foster PS, et al. Potential therapeutic targets for steroid-resistant asthma. Curr Drug Targets 2010; 11(8):957–970.
- Milara J, Navarro A, Almudéver P, et al. Oxidative stress-induced glucocorticoid resistance is prevented by dual PDE3/PDE4 inhibition in human alveolar macrophages. Clin Exp Allergy 2011; 41(4):535–546.
- Barnes PJ. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thoracic Soc 2009; 6:693–696.
- Montuschi P, Peters-Golden ML. Leukotriene modifiers for asthma treatment. Clin Exp Allergy 2010; 40(12):1732–1741.
- Montuschi P, Santini G, Valente S, et al. Liquid chromatography-mass spectrometry measurement of leukotrienes in asthma and other respiratory diseases. J Chromatogr B Analyt Technol Biomed Life Sci. 2014; 964(1):12–25.
- Thompson AB, Daughton D, Robbins RA, et al. Intraluminal airway inflammation in chronic bronchitis—characterization and correlation with clinical parameters. Am Rev Respir Dis 1989; 140:1527–1537.
- Claesson HE, Odlander B, Jakobsson PJ. 1992. Leukotriene-B4 in the immune-system. Int J Immunopharmacol 14:441–449.
- Chaney MO, Froelich LL, Gapinski DM, Mallett BE, Jackson WT. Characterization of the spatial arrangement of the 2 acid-binding sites on the human neutrophil ltb (4) receptor. Receptor 1992; 2:169–179.
- Conklyn MJ, AlpertRB, Wright KF, Showell HJ. Inhibition of spontaneous apoptosis in neutrophils by 5-lipoxygenase metabolites of arachidonic acid: potential involvement of the low affinity LTB4 receptor. J Leukoc Biol 1997; 55:13.
- Montuschi P. Leukotrienes, antileukotrienes and asthma. Mini Rev Med Chem 2008; 8(7):647–656.
- Bateman ED, Hurd SS, Barnes PJ, et al. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J 2008; 31,143–178.
- Tintinger GR, Feldman C, Theron AJ. 2010. Montelukast: more than a cysteinyl leukotriene receptor antagonist? Review. Scient World J 2010; 14;10:2403–2413.
- Tsuchida H, Takahashi S, Nosaka E, et al. Novel triple neurokinin receptor antagonist CS-003 inhibits respiratory disease models in guinea pigs. Eur J Pharmacol 2008; 596(1–3):153–159.
- Wu Y, Zhou C, Tao J, Li S. Montelukast prevents the decrease of interleukin-10 and inhibits NF-kappaB activation in inflammatory airway of asthmatic guinea pigs. Can J Physiol Pharmacol 2006; 84(5):531–537.
- Toward TJ, Broadler KJ. Chronic lipopolysaccharide exposure on airway function, cell infiltration, and nitric oxide generation in conscious guinea pigs: effect of rolipram and dexamethasone. J Pharmacol Exp Ther 2001; 298:298–306.
- Kaneko Y, Takashima K, Suzuki N, Yamana K. Effects of theophylline on chronic inflammatory lung injury induced by LPS exposure in guinea pigs. Allergol Int 2007; 56(4):445–456.
- Pennock BE, Cox CP, Rogers RM, et al. A non-invasive technique for measurement of changes in specific airway resistance. J Appl Physiol 1979; 46:399–406.
- Tulić MK, Wale JL, Holt PG, Sly PD. Modification of the inflammatory response to allergen challenge after exposure to bacterial lipopolysaccharide. Am J Respir Cell Mol Biol 2000; 22(5):604–612.
- Kasahara DI, Perini A, Lopes FD, et al. Effect of salbutamol on pulmonary responsiveness in chronic pulmonary allergic inflammation in guinea pigs. Braz J Med Biol Res 2005; 38(5):723–730.
- Underwood DC, Osborn RR, Bochnowicz, S, et al. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol 2000; 279(5):L895–902.
- Buege JA, Aust SD. Microsomal lipid peroxidation. Meth Enzymol 1978; 52:302–310.
- Wegener D, Wirsching F, Riester D, Schwienhorst A. 2003. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol 2003; 10:61–68.
- Lefort J, Singer M, Leduc D, et al. Systemic administration of endotoxin induces bronchopulmonary hyperreactivity dissociated from TNF-alpha formation and neutrophil sequestration into the murine lungs. J Immunol 1998; 161(1):474–480.
- Pauwels RA, Kips JC, Peleman RA, Van Der Straeten ME. The effect of endotoxin inhalation on airway responsiveness and cellular influx in rats. Am Rev Respir Dis 1990; 141(3):540–545.
- Whelan CJ. Inhibition of PAF-, LPS-, and cytokine-induced granulocyte accumulation in guinea pig lung by dexamethasone: evidence that inhibition of IL-5 release is responsible for the selective inhibition of eosinophilia by glucocorticoids in guinea pigs. Inflamm Res 1996; 45: 166–170.
- Belvisi MG. Regulation of inflammatory cell function by corticosteroids. Proc Am Thorac Soc 2004; 1(3):207–214.
- Whelan CJ, Hughes SC, Wren GP. Inhibition of some aspects of acute inflammation of guinea-pig lung by intraperitoneal dexamethasone and mifepristone: demonstration of agonist activity of mifepristone in the guinea-pig. Inflamm Res 1995; 44(3):131–138.
- Montuschi P, Barnes PJ. New perspectives in pharmacological treatment of mild persistent asthma. Drug Discov Today 2011; 16(23–24):1084–1091.
- Montuschi P, Malerba M, Santini G, Miravitlles M. Pharmacological treatment of chronic obstructive pulmonary disease: from evidence-based medicine to phenotyping. Drug Discov Today 2014; 19(12):1928–1935.
- Zhu J, Qiu YS, Figueroa DJ, et al. Localization and upregulation of cysteinyl leukotriene-1 receptor in asthmatic bronchial mucosa. Am J Respir Cell Mol Biol 2005; 33(6):531–540.
- Anderson R, Theron AJ, Gravett CM, et al. Montelukast inhibits neutrophil pro-inflammatory activity by a cyclic AMP-dependent mechanism. Br J Pharmacol 2009; 156, 105–115.
- Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153(2):530–534.
- Basyigit I, Sahin M, Sahin D, et al. Anti-inflammatory effects of Montelukast on smoke-induced lung injury in rats. Multidiscip Respir Med 2010; 5(2):92–98.
- Maeba S, Ichiyama T, Ueno Y, et al. Effect of Montelukast on nuclear factor kappa B activation and proinflammatory molecules. Ann Allergy Asthma Immunol 2005; 94(6):670–674.
- Can M, Yüksel B, Demirtaş S, Tomaç N. 2006. The effect of Montelukast on soluble interleukin-2 receptor and tumor necrosis factor alpha in pediatric asthma. Allergy Asthma Proc 27(4):383–386.
- Barnes PJ. New concepts in chronic obstructive pulmonary disease. Ann Rev Med 2003; 54:113–129.
- Cuciureanu M, Caruntu ID, Paduraru O, et al. The protective effect of Montelukast sodium on carbon tetrachloride induced hepatopathy in rat. Prostaglandins Other Lipid Mediat 2009; 88:82–88.
- Sener G, Sehirli O, Ogunc AV, et al. 2006. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol Res 54:65–71.
- Du J, Cui D, Guo Y. Detection of ET-1, TNF-alpha, oxidative radicals in sera of asthmatics and in sera and lung tissue of asthmatic guinea pigs. Zhonghua Jie He He Hu Xi Za Zhi 1998; 21(5):293–296.
- Tahan F, Jazrawi T, Rovati GE, Adcock IM. Montelukast inhibits tumour necrosis factor-α-mediated interleukin-8 expression through inhibition of nuclear factor-κB p65-associated histone acetyltransferase activity. Clin Exp Allergy 2008; 38:805–811.