
ABSTRACT
Interleukin-32 (IL-32) is a newly described cytokine which is expected to have an important role in autoimmune disorders. It was shown that chronic obstructive pulmonary disease (COPD) has a component of autoimmunity, though the role of IL-32 in its pathogenesis is not known. The aim of this study was to estimate IL-32 concentrations in serum, induced sputum (IS) supernatant and bronchoalveolar lavage (BAL) fluid from patients with COPD, and to compare asthma patients with and healthy subjects. Outpatients with COPD (63.7 ± 8.4 years, n = 51), asthma (58.3 ± 12.4 years, n = 31), and healthy subjects (59.8 ± 8.2 years, n = 9) were studied. The levels of IL-32 in serum, BAL fluid, and IS supernatant samples were analyzed by ELISA. Concentrations of IL-32 were higher in all the studied materials from patients with COPD (BAL 22.46 ± 2.48 pg/ml, IS 19.66 ± 1.69 pg/ml, serum 26.77 ± 2.56 pg/ml) in comparison with patients with asthma (BAL 6.25 ± 1.08 pg/ml, IS 5.82 ± 1.15 pg/ml, serum 6.09 ± 1.16 pg/ml, p < 0.05 respectively) as well as healthy subjects (BAL 4.21 ± 1.13 pg/ml, IS 3.59 ± 0.66 pg/ml, serum 4.63 ± 1.03 pg/ml, p < 0.05 respectively). Moreover, the level of IL-32 was higher in COPD smokers than in COPD ex-smokers in investigated respiratory tissue compartments and serum, and correlated with smoking history. Increased level of IL-32 in serum, IS supernatant, and BAL fluid from patients with COPD in comparison with asthma patients and healthy subjects suggest that IL-32 may play an important role in the pathogenesis of COPD, which depends on the smoking history.
KEYWORDS:
Introduction
Both chronic obstructive pulmonary disease (COPD) and asthma are chronic inflammatory airway diseases with increasing prevalence worldwide, especially in industrialized and developing countries (Citation1–3). These obstructive pulmonary diseases are gene-by-environment diseases, with multifactor pathogenesis, which has not been well established yet. Airway obstruction is a predominant feature for both diseases, although it is reversible and variable in asthma, but irreversible and progressive in COPD. Airway inflammation is associated with various inflammatory cells, cytokines and other inflammatory markers, influenced by many environmental factors (Citation4–8). Certainly, smoking is one of the major agent which leads to the development of COPD (Citation9–11). It was also shown that tobacco smoke is associated with asthma attacks and their severity Citation(12).
A number of recent studies have demonstrated that autoimmune component may play a role in the pathogenesis of COPD (Citation13, Citation14). Many cytokines and chemokines that are secreted in both asthma and COPD are regulated by the transcription factor nuclear factor-ĸB (NF− ĸB), which is activated in the airway epithelial cells and macrophages in both diseases, and may have an important role in increasing airway inflammation (Citation15, Citation16). IL-32, a newly discovered proinflammatory cytokine, is an important player in innate and adaptive immune response (Citation17, Citation18), it is implicated in autoimmune inflammatory disorders and some oncological diseases (Citation17, Citation19, Citation20). It is known that T-lymphocytes, natural killers, monocytes, and epithelial cell lines may produce IL-32 when stimulated by IL-2 or IFN-γ (Citation17, Citation19–22). This proinflammatory cytokine strongly stimulates other cytokines such as TNF-α, IL-1β, IL-6, and macrophage inflammatory protein-2 (MIP-2) (Citation19, Citation20, Citation23). Recently, two studies have shown the regulation of IL-32 expression in primary nasal epithelial cells by inflammatory cytokines (Citation24, Citation25). Several studies demonstrated increased expression of IL-32 in the lung tissue of patients with COPD Citation(21) and in nasal mucosa of patients with allergic rhinitis (Citation25, Citation26), thus suggesting that IL-32 is involved in the pathogenesis of these diseases. However, the role of IL-32 in chronic pulmonary diseases such as COPD and asthma is not well established and a possible relationship with a risk factor such as smoking is not studied properly.
Therefore, we aimed to analyze the level of IL-32 in serum, BAL fluid, and IS supernatant from patients with COPD and to compare those with asthma patients and healthy subjects. Furthermore, we investigated possible relationship between IL-32 level and smoking intensity.
Methods
Study population
A total of 91 adults were recruited for the study: outpatients with stable COPD Citation(9) were divided into smokers (n = 30) and ex-smokers (n = 21). Patients with stable mild-to-moderate asthma were divided into two subgroups: asthma smokers (n = 11) and asthma nonsmokers (n = 20). Nine healthy subjects, smokers (n = 4) and nonsmokers (n = 5), comprised the control group. The subjects were recruited from the outpatient division of the Department of Pulmonology and Immunology, Hospital of the Lithuanian University of Health Sciences, Kaunas. The study protocol was approved by the Regional Ethics Committee for Biomedical Research, Lithuanian University of Health Sciences, and each participant gave his/her informed written consent. The study is registered with Clinical Trials.gov (Identification: NCT01378039).
COPD was diagnosed according to the Global Initiative for Chronic Obstructive Lung Disease criteria Citation(9). The inclusion criteria for COPD patients were as follows: postbronchodilator FEV1/FVC, <0.70; FEV1, <80% of predicted; β2-agonist reversibility, less than 12% and/or 200 mL; smoking history, more than 10 pack/years; and no history of asthma, bronchiectasis, lung cancer, or other significant respiratory or autoimmune disease. All COPD patients had not been treated with systemic steroids and/or antibiotics for at least 1 month before the study. Patients with asthma diagnosed according to Global initiative for Asthma Citation(27) had a clinical history of the disease for ≥1 year, showed positive reversibility to β2-agonist and/or bronchial hyperresponsibility to metacholine, baseline FEV1 >80% of predicted. All patients were instructed to refrain from using inhaled or nasal as well as oral steroids at least 1 month before visits. Control group subjects required to have a baseline FEV1 >80% of predicted, have no symptoms of any respiratory disease, and without any other disease that may influence the results of the study.
Lung function testing
Pulmonary function was tested using a pneumotachometric spirometer “CustovitM” (Custo Med, Germany) with subjects in a sitting position, and the recorded highest value of forced expiratory volume in 1 sec (FEV1) and forced vital capacity (FVC) from at least three technically satisfactory maneuvers differed by less than 5%. Normal values were characterized according to Quanjer and colleagues Citation(28). Subjects had to avoid using short-acting β2-agonists at least 12 h prior the test and long-acting β2 agonists at least 48 h prior the lung function test.
Sputum induction and processing
Subjects inhaled 10 mL of sterile hypertonic saline solution (3%, 4%, or 5% NaCl (Ivex Pharmaceuticals, USA)) at room temperature (RT) from an ultrasonic nebulizer (DeVilbiss Health Care, USA). The duration of each inhalation was 7 min and was stopped after expectoration of an adequate amount of sputum. Spirometry was performed after each inhalation, in order to detect a possible decrease of FEV1. Sputum was poured into a Petri dish and separated from saliva. A four-fold volume of freshly prepared 0.1% dithiothreitol (DTT; Sigma-Aldrich) was added. The mixture was vortexed and placed on a bench rocker for 15 min. at RT. Next, an equal volume of phosphate-buffered saline (PBS; Sigma- Aldrich) solution was added to the DTT. The cell pellet was separated using 40 μm cell stainer (Becton Dickinson, USA). The mixture was centrifuged for 10 min at 4°C; the supernatant was aspirated and stored at –70°C for later assay. The total cell counts, percentage of epithelial cells, and cell viability were investigated using a Neubauer hemocytometer (Heinz-Herenz; Germany) under a microscope (B5 Professional, Motic, China), using Trypan blue exclusion method. Cytospin samples of IS were prepared using a cytofuge instrument (Shandon Southern Instruments, USA).
Bronchoscopy and BAL fluid processing
Bronchoscopy was performed in a week after sputum induction procedure. The subjects were not allowed to drink or eat at least 4 h. To perform BAL fluid test, the local upper airways anesthesia with 5 mL of 2% lidocaine (Grindex, Latvia) was used. All bronchoscopic examinations were performed in the morning. The bronchoscope (Olympus, USA) was wedged into the segmental bronchus of the middle lobe and 20 mL × 7, a total 140 mL, of sterile saline solution (0.9% NaCl) was infused. The fluid was gently aspirated immediately after the infusion has been completed and was collected into a sterile container. The fluid was immediately filtered using 40 μm cell stainer (Becton Dickinson, USA) and centrifuged at 4°C for 10 min. The supernatants were used for enzyme-linked immunosorbent assay (ELISA). The preparation of BAL fluid cytospins was as same as the preparation of IS samples described earlier.
Serum processing
Peripheral blood was collected into the sterile tubes without additives (2 × 5 mL) and stored at room temperature for the formation of surface clot (about 30 min). Then the tubes were centrifuged at 1000 g for 10 min at room temperature. In the upper layer of the sample, serum was vacuumed into sterile cold-resistant Eppendorf tubes and stored at –70°C for further ELISA.
IS and BAL fluid cell analysis
Prepared IS and BAL fluid cytospins were stained by the May-Grünwald-Giemsa method for differential counting of blood cell Citation(29). Cell differentiation was determined by counting approximately 400 cells in random fields viewed under the light microscope, excluding squamous epithelial cells. The cells were identified using standard morphological criteria, by nuclear morphology and cytoplasmic granulation. Cell counts were expressed as percentages of total cells and absolute values (106/L).
Detection of IL-32 in serum, BAL fluid, and induced sputum supernatant
The concentration of IL-32 in serum, BAL fluid, and IS supernatant was estimated by ELISA. Firstly, sterile 96-well plates were coated with monoclonal capture human IL− 32α specific antibody (BioLegend, USA) and incubated for 18 h at 4°C. After washing, nonspecific reactions were blocked with an assay diluent (10%BSA/PBS). Serial dilutions of IL-32α, negative control, and samples in duplicates were added to the wells and incubated for 2 hours. After washing, biotinylated anti-human IL-32α was used as a detection antibody. The sealed plates were incubated for 1 h and then washed. Then Avidin-HRP solution was added to each well and plate incubated 30 minutes. After washing, plates were filled with tetramethylbenzidine (TMB) substrate solution for 15 min in the dark. Immunoenzymatic reaction was stopped by adding 2 N of sulfuric acid. The optical density of samples was evaluated by a microplate reader (Murex Diagnostics, Germany), at 450 nm wave length. The concentration of IL-32α in pg/ml was calculated from the standart curve. The minimum detectable concentration for IL-32 was 4 pg/mL.
Statistical analysis
All statistical analyses were performed using Prism version 6 for Windows (GraphPad Software, San Diego, California, USA). The normality assumption of data was verified with Shapiro-Wilk test. All the data that were normally distributed are presented as mean and standard error of the mean (SEM). The data that did not follow a normal distribution were expressed as median and range (min–max).
Due to a skewed distribution of the variable, nonparametric tests were used. The Kruskal-Wallis test was used to evaluate statistical differences between both groups of patients and control group. If significant differences were detected, differences between two independent groups were determined by the Mann-Whitney U test. Spearmen's rank test was used to assess the relationships between measurements. Statistical significance was assumed at a P value of <0.05.
Results
Characteristics of studied subjects
A total of 91 adults (45 men and 46 women; mean age, 60 ± 8.5 years) were examined. The demographic and clinical characteristics of the subjects are presented in . There were no significant differences of age and gender comparing the groups. FEV1 (%) was significantly lower in the patients with COPD when comparing those with asthma patients and healthy subjects.
Table 1. Demographic and clinical characteristics of the subjects.
Levels of IL-32 in respiratory compartments (BAL fluid, IS supernatant)
Levels of IL-32 in BAL fluid and in IS samples were significantly higher in patients with COPD when compared with asthma and healthy subjects (). Whereas the levels of IL-32 did not differ in the BAL fluid and IS from patients with asthma and healthy subjects.
Figure 1. Levels of IL-32 (pg/ml) in BALF and IS from patients with COPD patients, asthma and healthy subjects. Data are shown as median (range). *P < 0.05, compared with patients with asthma #P < 0.05, compared with healthy subjects.
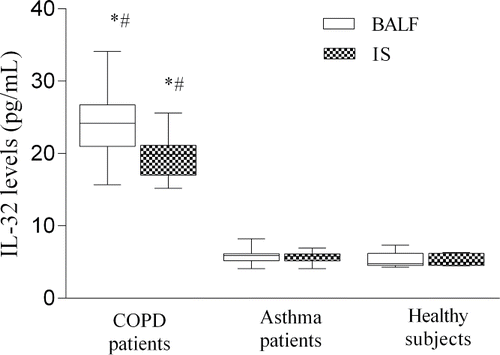
An increased levels of IL-32 in BAL fluid was documented in COPD smokers compared with COPD ex-smokers (p < 0.05) (). However, the level of IL-32 did not differ between asthma and control groups, and did not depend on smoking status in these groups.
Table 2. Distribution of IL-32 concentration according to the smoking status.
Levels of IL-32 in serum
The tendency of variation in IL-32 concentration in serum was almost the same as in BAL fluid and IS supernatant. The level of IL-32 in serum was higher in COPD patients than in the serum of asthmatics as well as healthy subjects (26.77 ± 2.56 pg/ml v.s. 6.09 ± 1.16 pg/ml, 4.63 ± 1.03 pg/ml p < 0.05 respectively). We did not found significant difference in serum IL-32 level comparing patients with asthma and healthy subjects. Also, the level of IL-32 in patients with COPD was dependant on smoking status – level of IL-32 in serum was higher in COPD smokers compared with COPD ex-smokers ().
Relation of IL-32 with airway inflammatory cells
The level of IL-32 in BAL fluid significantly correlated with the number of macrophages in COPD and asthma groups (), but there was no correlation between these parameters in healthy subjects. We did not find correlation between IL-32 level in BAL fluid and other inflammatory cells. As well as we have not found correlation between IL-32 levels and lung function parameters. While examining cellular composition of the respiratory compartments (BAL fluid, IS supernatant), we found that the macrophage count in the BAL fluid was higher in COPD smokers compared with COPD ex-smokers (). However, we did not find any significant difference in differential cell counts of IS supernatant when comparing different groups ().
Figure 2. Association between BAL fluid IL-32 levels and number of macrophages in patients with COPD (A) and asthma (B).
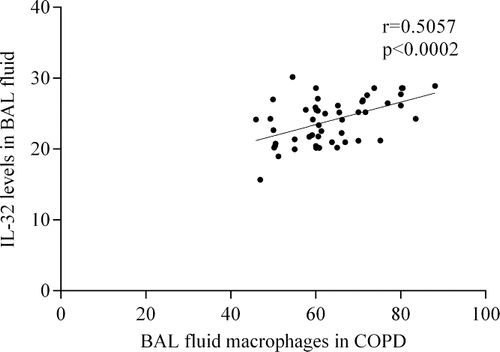
Table 3. Differential cell counts in IS supernatant and BAL fluid.
Discussion
The goal of this study was to investigate one of the newest cytokine, interleukin 32, in local (BAL fluid and IS supernatant) and systemic (serum) compartments of patients with COPD and to compare that with those of asthma patients and healthy subjects. According to the latest scientific data, an autoimmune mechanism-driven chronic inflammation plays an important role in the pathogenesis of COPD Citation(30). Previous studies have shown that IL-32 is an orchestrating cytokine in the autoimmune response (Citation21, Citation31). That is why we aimed to study this cytokine in COPD. The results of our study showed that patients with COPD have significantly increased levels of IL-32 in comparison with patients with asthma and healthy subjects. It suggests that IL-32 could be an important player in COPD pathogenesis. The inflammation in COPD is described by the activation of type 1 helper T lymphocytes (Th1) and production of various cytokines such as IFN-γ and others, therefore inducing lung tissue infiltration by inflammatory cells (Citation32, Citation33). It was shown in vitro that IL-32 is induced probably by IFN-γ Citation(34), and depletion of IL-32 decreases IFN-γ production Citation(20). It was also described that IL-32 may induce production of various cytokines through the activation of transfer factor, thus promoting autoimmune inflammation Citation(34). Whereas in most cases of asthma, lung inflammation is characterized by allergen-induced activation of mast cells. These cells release pro-inflammatory cytokines and mediators, which cause activation of type 2 helper T lymphocytes (Th2) and macrophages, recruitment and degranulation of eosinophils Citation(35), thus leading to acute bronchoconstriction and airway obstruction.
We designed our study in order to investigate the newly described pro-inflammatory cytokine IL-32 levels consistently in local and systemic levels. The compartments of respiratory tract (BAL fluid and IS supernatant) represent local and venous blood serum stands for systemic inflammation. Moreover, differential cell counts were examined in the IS supernatant and BAL fluid in order to evaluate the inflammatory cells in proximal and distal airways. The use of induced sputum has previously been shown to be an appropriate and feasible method to assess airway inflammation in chronic lung diseases (Citation36, Citation37). However, induced sputum mainly represents larger airways Citation(38) and may be contaminated with resident mucus Citation(39). Despite the fact that the use of bronchoscopy and BAL in research is limited due to its invasiveness, this method is still very important for evaluating the inflammatory pattern in distal airways.
The results of our study (increased level of IL-32 in all investigated compartments in COPD group) once again prove that COPD is a systemic disease and inflammatory processes touch not only respiratory tract. To our knowledge, this is the first study to investigate IL-32 level in such a variety of human biological substances (serum, BAL fluid, IS supernatant). Our data are consistent with other authors who found increased IL-32 level in vivo and in vitro in IS supernatant and increased IL-32 expression in the lung tissue of COPD patients (Citation17, Citation21). Whereas Green M. et al. stated that IL-32 level did not differ in COPD and healthy subjects Citation(40). In their research, the studied number of COPD patients was lower than that in our study, also we noticed that the majority of their COPD patients were ex-smokers, though their healthy subject group was considerably bigger. Citation(40). It was also shown that IL-32 induces the production of other cytokines that cause airway inflammation leading to impaired lung function Citation(18). However, in our study correlation between IL-32 concentration and lung function was not obtained.
Our research group found no differences between IL-32 concentrations when analysing asthma group subjects and healthy subjects. These results may be due to relatively small group of asthma patients and a very small number of healthy subjects. Although, N. Meyer et al. in their study revealed that serum levels of IL-32 were significantly higher in asthmatic patients compared with healthy control subjects Citation(41). Other research groups showed that IL-32 might play a role in allergic diseases. For example, H. J. Jeong et al. showed that patients with allergic rhinitis differ significantly from healthy control individuals in having increased expression of IL-32 in the adenoid tissue, nasal mucosa tissue, and serum Citation(26). IL-32 was expressed in lesional skin of atopic dermatitis, whereas it was not detected in skin biopsy specimens from healthy persons Citation(19). Accordingly, the role of IL-32 in asthmatic inflammation can be explained by the participation of Th1-cell-driven mechanism in one endotype according to the classification Citation(42) and therefore may influence angiogenesis Citation(41). However, in our study we did not differentiate patients with asthma according to their endotypes, because our studied asthma group was relatively small, that is why we cannot claim that IL-32 does not play a role in the case of asthma. But we can say that IL-32 plays definitely a more significant role in COPD than in asthma.
Active exposure to cigarette smoke causes the vast majority of COPD cases and contributes to increased incidence of related pulmonary diseases such as asthma. Cigarette smoking is the main risk factor for COPD, which initiates an inflammatory cascade (production of various mediators, ROS) in the peripheral airways and lung parenchyma (Citation29, Citation43). To assess the potential influence of smoking on inflammatory markers, we investigated patients with different smoking experience – smokers and ex-smokers. Our study on human respiratory compartments and serum shows that tobacco smoke may have an impact on the production of IL-32 in COPD smokers as well as in ex-smokers with COPD. The level of IL-32 differed in all investigated biological substances: BAL fluid, IS supernatant, and serum in patients with COPD according to their smoking status. The level of IL-32 was significantly higher in all investigated materials in COPD smokers compared with ex-smokers. Thus we can assume that IL-32 is increasing in COPD patients and depends on smoking status. Our results are consistent with data from human and animal studies performed by other researchers (Citation22, Citation44). However when analysing asthmatics according to their smoking status, we did not see the same tendency. These findings let us suggest that in asthma pathogenesis other factors (not only smoking) play an important role (e.g. allergens). We should also assume the relatively small number of investigated asthma patients in our study.
The macrophages are one of the main cells participating in innate immune response and in smoking-induced inflammation in COPD patients. In the case of COPD, there is an increasing evidence that lung macrophages orchestrate inflammation through the release of chemokines and cytokines that attract inflammatory cells and the release of proteases Citation(43) Citation(29). The macrophages are the major source of IL-32, which have been shown to produce this cytokine in vitro Citation(20) Citation(45). On the other hand, in asthma, it seems that alveolar macrophages are inappropriately activated and are implicated in the development and progression of the disease. These cells in asthma patients' airway may be activated by allergens through low-affinity IgE receptors to release inflammatory mediators that amplify inflammatory response Citation(46). While examining the cellular composition of BAL fluid, we found that macrophage cell count was significantly higher in COPD smokers compared with COPD ex-smokers, but this difference was not confirmed in IS supernatant. We have identified the correlation between IL-32 level and macrophage cell count in BAL fluid from COPD patients; nevertheless, we did not find relation between IS IL-32 level and IS macrophage cell count. We also found a positive correlation between number of BAL fluid macrophages and the level of IL-32 in BAL fluid from patients with asthma, but not healthy subject group. This data could suggest us that inflammation process is more pronounced in distal airways that in proximal, as BAL fluid better reflects processes occurring in distal airways (Citation47, Citation48).
We believe that our study has several strengths. We studied IL-32 level in three different human compartments (BAL, IS, serum), whereas previous studies analysed only surgically resected lung tissue Citation(21) or serum Citation(40) and IS Citation(49) from patients with COPD. To our knowledge, this is the first study to compare IL-32 level in COPD and asthmatic patients. Also, some limitations need to be taken into account when interpreting our results. Our groups were limited in subject numbers due to invasiveness of procedure (bronchoscopy with BAL) and ethical considerations, especially in asthmatics (n = 31) and healthy subjects (n = 9). This may affect the evaluation of correlation between data groups.
Conclusions
To sum up, we investigated the levels of IL-32 in serum, BAL fluid, and induced sputum samples of patients with COPD, asthma as well as healthy subjects. As we have found increased levels of IL-32 in all investigated compartments of patients with COPD in comparison with asthma, and this increase was dependent on subjects' smoking status, it may be suggested that IL-32 plays an important role in the pathogenesis of COPD and therefore could be a marker of inflammation in this disease. Moreover, IL-32 is detectable with less invasive methods, such as BAL and IS while investigating local inflammatory pattern.
Declarations of interest
The authors declare that they have no competing financial interests.
References
- Barnes PJ. Chronic obstructive pulmonary disease: A growing but neglected global epidemic. PLoS Med 2007; 4(5):e112.
- Mannino DM, Buist AS. Global burden of COPD: Risk factors, prevalence, and future trends. Lancet 2007; 370(9589):765–773.
- Pearce N, Ait-Khaled N, Beasley R, Mallol J, Keil U, Mitchell E, et al. Worldwide trends in the prevalence of asthma symptoms: Phase III of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax 2007; 62(9):758–766.
- Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: A systematic review and a meta-analysis. Thorax 2004; 59(7):574–580.
- Cockayne DA, Cheng DT, Waschki B, Sridhar S, Ravindran P, Hilton H, et al. Systemic biomarkers of neutrophilic inflammation, tissue injury and repair in COPD patients with differing levels of disease severity. PloS One 2012; 7(6):e38629.
- Barbu C, Iordache M, Man MG. Inflammation in COPD: Pathogenesis, local and systemic effects. Rom J Morphol Embryol 2011; 52(1):21–27.
- Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 2014; 35(1):71–86.
- Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 2000; 161(5):1720–1745.
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, Committee GS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001; 163(5):1256–1276.
- Rom O, Avezov K, Aizenbud D, Reznick AZ. Cigarette smoking and inflammation revisited. Respir Physiol Neurobiol. 2013; 187(1):5–10.
- Pelegrino NR, Tanni SE, Amaral RA, Angeleli AY, Correa C, Godoy I. Effects of active smoking on airway and systemic inflammation profiles in patients with chronic obstructive pulmonary disease. Am J Med Sci. 2013; 345(6):440–445.
- Siroux V, Pin I, Oryszczyn MP, Le Moual N, Kauffmann F. Relationships of active smoking to asthma and asthma severity in the EGEA study. Epidemiological study on the Genetics and Environment of Asthma. Eur Respir J 2000; 15(3):470–477.
- Shaykhiev R, Crystal RG. Innate immunity and chronic obstructive pulmonary disease: A mini-review. Gerontology 2013; 59(6):481–489.
- Deckers J, Branco Madeira F, Hammad H. Innate immune cells in asthma. Trends Immunol 2013; 34(11):540–547.
- Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med 1998; 158(5 Pt 1):1585–1592.
- Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003; 58(4):348–351.
- Kudo M, Ogawa E, Kinose D, Haruna A, Takahashi T, Tanabe N, et al. Oxidative stress induced interleukin-32 mRNA expression in human bronchial epithelial cells. Respir Res 2012; 13:19.
- Hong J, Bae S, Kang Y, Yoon D, Bai X, Chan ED, et al. Suppressing IL-32 in monocytes impairs the induction of the proinflammatory cytokines TNFalpha and IL-1beta. Cytokine 2010; 49(2):171–176.
- Meyer N, Zimmermann M, Burgler S, Bassin C, Woehrl S, Moritz K, et al. IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol 2010; 125(4):858–865e10.
- Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis 2006; 65 Suppl 3:iii61–64.
- Calabrese F, Baraldo S, Bazzan E, Lunardi F, Rea F, Maestrelli P, et al. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 178(9):894–901.
- Deng X, Zhang Z, Gu W, Li Y, Liu M. Budesonide inhibits interleukin-32 expression in a rat model of chronic obstructive pulmonary disease. Exp Lung Res 2012; 38(6):295–301.
- Alsaleh G, Sparsa L, Chatelus E, Ehlinger M, Gottenberg JE, Wachsmann D, et al. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther 2010; 12(4):R135.
- Keswani A, Chustz RT, Suh L, Carter R, Peters AT, Tan BK, et al. Differential expression of interleukin-32 in chronic rhinosinusitis with and without nasal polyps. Allergy 2012; 67(1):25–32.
- Soyka MB, Treis A, Eiwegger T, Menz G, Zhang S, Holzmann D, et al. Regulation and expression of IL-32 in chronic rhinosinusitis. Allergy 2012; 67(6):790–798.
- Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS, Kim HM. IL-32 up-regulation is associated with inflammatory cytokine production in allergic rhinitis. J Pathol 2011; 224(4):553–563.
- Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M, et al. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J 2008; 31(1):143–178.
- Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3–95-year age range: The global lung function 2012 equations. Eur Respir J 2012; 40(6):1324–1343.
- Babusyte A, Stravinskaite K, Jeroch J, Lotvall J, Sakalauskas R, Sitkauskiene B. Patterns of airway inflammation and MMP-12 expression in smokers and ex-smokers with COPD. Respir Res 2007; 8:81.
- Kheradmand F, Shan M, Xu C, Corry DB. Autoimmunity in chronic obstructive pulmonary disease: Clinical and experimental evidence. Expert Rev Clin Immunol 2012; 8(3):285–292.
- Du Y, Wang W, Yang W, He B. Interleukin-32, not reduced by salmeterol/fluticasone propionate in smokers with chronic obstructive pulmonary disease. Chinese Med J-Peking 2014; 127(9):1613–1618.
- Koutsokera A, Kostikas K, Nicod LP, Fitting JW. Pulmonary biomarkers in COPD exacerbations: A systematic review. Respir Res 2013; 14:111.
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008; 8(3):183–192.
- Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: A cytokine and inducer of TNFalpha. Immunity 2005; 22(1):131–142.
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 2008; 118(11):3546–3556.
- Fahy JV, Boushey HA, Lazarus SC, Mauger EA, Cherniack RM, Chinchilli VM, et al. Safety and reproducibility of sputum induction in asthmatic subjects in a multicenter study. Am J Respir Crit Care Med 2001; 163(6):1470–1475.
- Paggiaro PL, Chanez P, Holz O, Ind PW, Djukanovic R, Maestrelli P, et al. Sputum induction. Eur Respir J Suppl 2002; 37:3s–8s.
- Kips JC, Fahy JV, Hargreave FE, Ind PW, in't Veen JC. Methods for sputum induction and analysis of induced sputum: A method for assessing airway inflammation in asthma. Eur Respir J Suppl 1998; 26:9S–12S.
- Keatings VM, Evans DJ, O'Connor BJ, Barnes PJ. Cellular profiles in asthmatic airways: A comparison of induced sputum, bronchial washings, and bronchoalveolar lavage fluid. Thorax 1997; 52(4):372–374.
- Greene CM, Low TB, O'Neill SJ, McElvaney NG. Anti-proline-glycine-proline or antielastin autoantibodies are not evident in chronic inflammatory lung disease. Am J Respir Crit Care Med 2010; 181(1):31–35.
- Meyer N, Christoph J, Makrinioti H, Indermitte P, Rhyner C, Soyka M, et al. Inhibition of angiogenesis by IL-32: Possible role in asthma. J Allergy Clin Immunol 2012; 129(4):964–973e7.
- Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 2011; 127(2):355–360.
- Barnes PJ. Mechanisms in COPD: Differences from asthma. Chest 2000; 117(2 Suppl):10 S–14 S.
- Hacievliyagil SS, Mutlu LC, Temel I. Airway inflammatory markers in chronic obstructive pulmonary disease patients and healthy smokers. Niger J Clin Pract 2013; 16(1):76–81.
- Netea MG, Azam T, Lewis EC, Joosten LA, Wang M, Langenberg D, et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-gamma-dependent mechanism. PLoS Med 2006; 3(8):e277.
- Peters-Golden M. The alveolar macrophage: The forgotten cell in asthma. Am J Respir Cell Mol Biol 2004; 31(1):3–7.
- Balbi B, Pignatti P, Corradi M, Baiardi P, Bianchi L, Brunetti G, et al. Bronchoalveolar lavage, sputum and exhaled clinically relevant inflammatory markers: values in healthy adults. Eur Respir J 2007; 30(4):769–781.
- Sabroe I, Parker LC, Calverley PM, Dower SK, Whyte MK. Pathological networking: A new approach to understanding COPD. Postgrad Med J 2008; 84(991):259–264.
- Rong Y, Xiang XD, Li YM, Peng ZY, Li JX. IL-32 was involved in cigarette smoke-induced pulmonary inflammation in COPD. Clin Respir J 2014. doi:10.1111/crj.12157. [Epub ahead of print]