
ABSTRACT
Toll-like receptor 3 (TLR3) is involved in the virus-induced pulmonary inflammatory response, but its role in airway remodeling after viral infection is unclear. We explored the role of TLR3 in poly(I:C)-induced inflammatory cytokines and mucin 5AC (MUC5AC) production in human bronchial epithelial cells by Western blotting, RT-PCR and ELISA. The expression of TLR3, MUC5AC, Matrixmetalloproteinase (MMP9), Transforming growth factor (TGF-β1) and Vascular endothelial growth factor (VEGF) in human bronchial epithelial cells increased in a dose-dependent manner after exposure to poly(I:C), and this effect was inhibited by treatment with TLR3 siRNA. The phosphorylation of epithelial growth factor receptor (EGFR)/ERK/P38 Mitogen-activated protein kinases (MAPK) proteins increased after poly(I:C) treatment, and inhibition of this signaling pathway decreased TLR3 expression and MUC5AC and TGF-β1 production in human bronchial epithelial cells. The TLR3-EGFR signaling pathway is involved in the production of airway remodeling cytokines after virus infection. Inhibiting EGFR and its signaling pathway may be a therapeutic strategy for modifying airway remodeling.
KEYWORDS:
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by incompletely reversible airflow limitation and loss of lung function. Mucin hypersecretion is a characteristic feature of COPD. Excessive mucus production and secretion in the proximal and distal airways are associated with disabling symptoms (cough and sputum), lung function decline, exacerbations and mortality in patients with COPD (Citation1–3). Viral infection is closely associated with the development of COPD. Rhinovirus and respiratory syncytial virus (RSV) were detected in the respiratory secretions of more than 30% of COPD patients with acute exacerbations or in the stable phase Citation(4). Acute exacerbations caused by a viral infection are characterized by more severe symptoms, longer duration, and even worse pulmonary function than exacerbations due to other causes. Viral infections are associated with mucin secretion. Rhinovirus infection of the mucociliary epithelium results in the overexpression of genes associated with bronchial remodeling Citation(5). RSV induces metaplastic changes to the epithelium and increases the number of goblet cells and the expression of mucin 5AC (MUC5AC) and chloride channel calcium-activated 3 protein (GOB5) Citation(6). However, the mechanism by which viral infections affect MUC production is poorly understood. We previously demonstrated that Lipopolysaccharide (LPS) stimulation of NCI-H292 cells and primary human bronchial epithelial cells increases epithelial growth factor receptor (EGFR)-mediated MUC5AC transcription and protein expression. LPS-induced activation of MMP-9 is involved in EGFR transactivation Citation(7). Ichikawa et al. recently demonstrated that TLR3 activation augments MMP production via the generation of reactive nitrogen species in human lung fibroblasts, thus potentiating virus-induced airway remodeling Citation(8). Therefore, we hypothesized that virus-induced MUC secretion may also be associated with the EGFR signaling pathway. In this study, we used dsRNA poly(I:C) as a TLR3 ligand to stimulate bronchial epithelial cells and to explore the effect of virus infection on the production of inflammatory cytokines and MUC5AC and the possible mechanism underlying the observed effect.
Methods
Cell culture
Human bronchial epithelial cells (HBECs) were purchased from ATCC (Manassas, VA, USA). The cells were cultivated in Bronchial Epithelial Cell Medium (ScienCell, San Diego, CA, USA, Cat # 3211) and Bronchial Epithelial Cell Growth Supplement (ScienCell, San Diego, CA, USA, Cat # 3262), which contain growth factors, hormones and proteins necessary for the culture of normal bronchial epithelial cells, at 37°C in a humidified 5% CO2 water-jacketed incubator.
Treatment of cells with poly(I:C) and inhibitors
For poly(I:C) treatment, HBECs were cultured without the Bronchial Epithelial Cell Growth Supplement for 24 hours after reaching 70% confluence. Then, the HBECs were incubated with dsRNA poly(I:C) for 24 or 48 hours; untreated cells served as a control. For inhibition studies, the cells were pretreated with inhibitor for 30 minutes before exposure to stimuli. Then, the cells were washed three times with serum-free medium and cultured for another 24 hours in medium with poly(I:C) at the same concentration used in the pretreatment period. To determine the role of TLR3/EGFR in poly(I:C)-induced MUC5AC production, the cells were treated with 10 µM AG1478, an EGFR inhibitor (Sigma, St. Louis, MO, USA). To identify the affected MAPK signaling pathway, the cells were treated with 50 µM PD98059, an Extracellular regulated protein kinases (ERK) 1/2 inhibitor (Gene Operation, Ann Arbor, MI, USA), or with 10 µM SB203580, a P38 inhibitor (Gene Operation, Ann Arbor, MI, USA) for 1 hour.
ELISA assay
Supernatants were obtained from cells cultured for 48 hours. The concentrations of MUC5AC, TGF-β1, MMP9 and VEGF in the cell culture supernatants were determined using ELISA kits (Cloud-Clone Corp., Houston, TX, USA) according to the manufacturer's instructions.
Western blot analysis
Samples containing 100 µg of protein were mixed with loading buffer and boiled for 5 minutes. The proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis in 7.5% tris–glycine gels before electrophoretic transfer to polyvinylidene fluoride membranes. The membranes were blocked in milk and then incubated in Tris-buffered saline (TBS). The following primary antibodies were used: anti-TLR3 (1:1000; Abcam, Cambridge, UK) and anti-EGFR (1:1000), anti-phospho-EGFR (Tyr1068; 1:1000), anti-ERK1/2(1:1000), anti-phospho-ERK1/2 (Thr202/Tyr204; 1:2000), anti-p38 (1:1000), anti-phospho-p38 (Thr180/Tyr182; 1:1000), anti-c-Jun N-terminal kinase (JNK) (1:1000), and anti-phospho-JNK (Thr183/Tyr185; 1:1000) (Cell Signaling, Danvers, MA, USA). The membranes were then washed twice with TBS and incubated with a horseradish peroxidase-linked secondary antibody at room temperature for 1 hour. The immunoreactive bands were detected using an enhanced chemiluminescence Western blotting system (ECL Western Blotting Substrate, Pierce, USA, Cat # 32106).
RNA isolation and real-time reverse transcription PCR
Total RNA was isolated from the cultured cells using TRIzol reagent (Amresco, Radnor, PA, USA, ET111-01) following the manufacturer's instructions, and the RNA was quantified by spectrophotometry. cDNA was prepared using a cDNA synthesis kit (TIANGEN, KR104) and random hexamers. Real-time PCR was performed using a real-time PCR kit (Amresco, Radnor, PA, USA). The TLR3 and EGFR primers were designed and synthesized by Sangon Biotech Co. Ltd. (Shanghai, China). The primer sequences were as follows:
TLR3: (forward) 5′- ATTTGCCACACACTTCCCTGA- 3′,
(reverse) 5′-TGCACACAGCATCCCAAAGG- 3′.
EGFR: (forward) 5′- TTGCCGCAAAGTGTGTAACG- 3′,
(reverse) 5′- GAGATCGCCACTGATGGAGG- 3′.
The PCR mixture was subjected to 45 cycles of denaturation at 95°C for 10 seconds and annealing and extension at 60°C for 1 minute. The fold change in MUC5AC and TLR3 mRNA expression relative to β-actin was calculated based on the threshold cycle.
TLR3 siRNA transfection
TLR3 siRNA duplexes were purchased from Integrated Biotech Solutions Co. Ltd. (Shanghai, China). The selected 21-nt sequences were as follows:
TLR3-siRNA1: (sense) 5′ - GGUAUAGCCAGCUAACUAGTT - 3′,
(antisense) 5′ - CUAGUUAGCUGGCUAUACCTT - 3′.
TLR3-siRNA2: (sense) 5′ - GGUACAUCAUGCAGUUCAATT - 3′,
(antisense) 5′ - UUGAACUGCAUGAUGUACCTT - 3′.
TLR3-normal control (NC) was used as a transfection control. The siRNA was transfected into cells using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacturer's instructions. Successful target knockdown was confirmed by Western blotting. After treatment with TLR3-siRNA1 or TLR3-siRNA2 for various lengths of time (24, 48 or 72 hours), TLR3 protein expression was quantified by Western blotting.
Statistical analysis
Data are expressed as the mean ± SD. Differences between groups were analyzed by one-way analysis of variance (ANOVA). When statistical significance was identified by ANOVA, the Student–Newman–Keuls test was used for multiple comparisons. Statistically significant differences were accepted at p < 0.05.
Results
Poly(I:C) stimulation induces the expression of TLR3 in HBECs
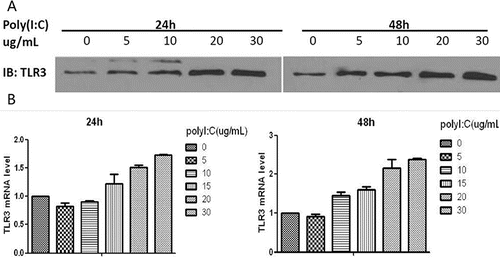
To evaluate concentration-dependent changes in TLR3, cells were exposed to a range of concentrations of poly(I:C) (0, 5, 10, 20 or 30 µg/ml) for either 24 or 48 hours and analyzed by Western blotting. RT-PCR was performed for cell extracts that had been pretreated with various doses of poly(I:C) (0, 5, 10, 15, 20 or 30 µg/ml) for 24 or 48 hours. Poly(I:C) induced the expression of the TLR3 protein and TLR3 mRNA in a dose-dependent manner: the highest level was observed at 30 µg/ml. At the same concentration of poly(I:C), the increase in TLR3 expression was greater after exposure for 48 hours than that after 24 hours (). The mean ± SD of triplicate wells from a representative experiment (n = 3) is shown.
Figure 1. Western blotting and RT-PCR analysis of TLR3 expression after exposure to poly(I:C). (A) Maximal TLR3 protein expression was observed after 48 hours of treatment with 30 µg/ml poly(I:C). (B) Maximal TLR3 mRNA expression also occurred at 48 hours after treatment with 30 µg/ml. The mean ± SD of triplicate wells from a representative experiment (n = 3) is shown.
Poly(I:C) treatment increases the production of MUC5AC and inflammatory cytokines in HBECs
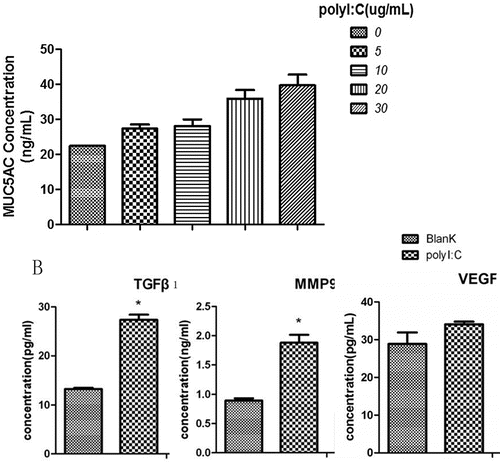
We quantified MUC5AC expression using an ELISA following exposure to various doses of poly(I:C) (0, 5, 10, 20 and 30 µg/ml). Poly(I:C) treatment increased MUC5AC levels in a dose-dependent manner. Moreover, the expression levels of TGF-β1, MMP9 and VEGF in the culture supernatants were higher after treatment of the cells with poly(I:C) (25 µg/ml) ().
Figure 2. ELISA analysis of MUC5AC, TGF-β1, MMP9 and VEGF levels after treatment with poly(I:C). (A) The MUC5AC level increased with increasing doses of poly(I:C). (B) TGF-β1, MMP9 and VEGF expression levels increased after exposure to poly(I:C). The mean ± SD of triplicate wells from a representative experiment (n = 3) is shown.
Poly(I:C) activates the EGFR-ERK/P38 MAPK signaling pathway in a TLR3-dependent manner
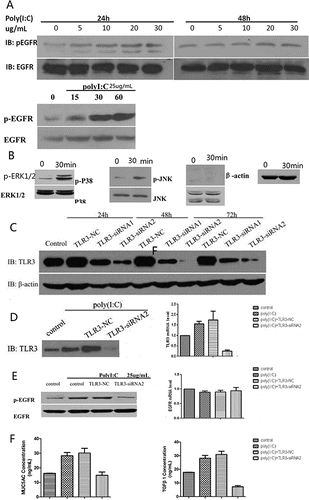
We analyzed cells that had been exposed to poly(I:C) (0, 5, 10, 20 or 30 µg/ml) for either 24 or 48 hours by Western blotting. p-EGFR expression increased as the concentration of poly(I:C) increased. When cells were exposed to 25 µg/ml poly(I:C) for different lengths of time (0, 15, 30 or 60 minutes), EGFR expression remained unchanged, whereas the p-EGFR level was significantly increased at 30 minutes and 60 minutes. To identify the MAPK kinase signaling pathway downstream of EGFR activation, cells were exposed to 25 µg/ml poly(I:C), and the expression of p-ERK1/2, ERK1/2, p-p38, p38, p-JNK and JNK was examined by Western blotting. After poly(I:C) stimulation for 30 minutes, p-p38 and p-ERK1/2 were activated, whereas p-JNK was not activated.
After treatment with TLR3-siRNA1 or TLR3-siRNA2 for various intervals (24, 48 or 72 hours), the expression of TLR3 was quantified by Western blotting. Interference by TLR3-siRNA2 for 48 hours suppressed TLR3 expression most effectively compared to the control, TLR3-NC and TLR3-siRNA1. We examined TLR3, p-EGFR and EGFR expression and transcription by Western blotting and RT-PCR following treatment with TLR3-siRNA2. TLR3 production and transcription were remarkably suppressed after TLR3-siRNA2 treatment. Pretreatment with TLR3-siRNA2 decreased p-EGFR expression but not EGFR expression and transcription. ELISA analysis revealed that treatment with TLR3-siRNA2 reduced MUC5AC and TGF-β1 expression levels ().
Figure 3. Western blotting analysis of p-EGFR, p-p38, p-ERK1/2 and p-JNK after exposure to poly(I:C) and of TLR3 after treatment with TLR3-NC, TLR3-siRNA1 or TLR3-siRNA2. Western blotting and RT-PCR analysis of TLR3, p-EGFR and EGFR; and ELISA analysis of MUC5AC and TGF-β1 after exposure to TLR3-siRNA2. (A) p-EGFR expression increased with increasing poly(I:C) concentration. (B) p-p38 and p-ERK1/2 were activated, whereas p-JNK was not activated. (C) Treatment with TLR3-siRNA2 for 48 hours was more effective at suppressing TLR3 than TLR3-NC or TLR3-siRNA1. (D) TLR3 production and transcription were remarkably suppressed after TLR3-siRNA2 treatment. (E) TLR3-siRNA2 decresed p-EGFR expression, but did not attenuate EGFR expression and transcription. (F) MUC5AC and TGF-β1 expression levels were diminished by treatment with TLR3-siRNA2. The mean ± SD of triplicate wells from a representative experiment (n = 3) is shown.
EGFR-ERK/P38 MAPK inhibition suppresses the production of MUC5AC and inflammatory cytokines
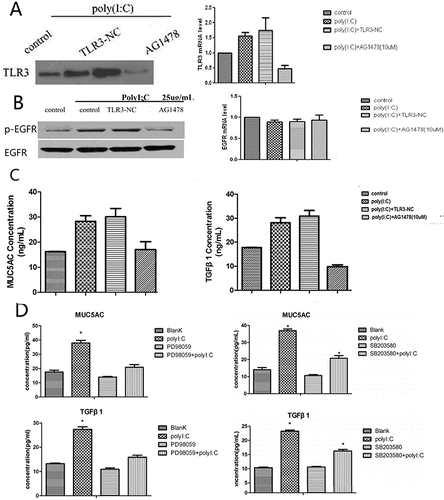
We examined the effects of the EGFR inhibitor AG1478 on TLR3, p-EGFR and EGFR expression and transcription by Western blotting and RT-PCR. TLR3 expression and transcription were remarkably suppressed by AG1478 treatment. Pretreatment with AG1478 decreased p-EGFR expression but not EGFR expression and transcription. ELISA analysis revealed that MUC5AC and TGF-β1 expression levels were reduced by treatment of cells with AG1478. We also evaluated the levels of these proteins in the culture supernatants by ELISA. The poly(I:C)-induced increases in MUC5AC and TGF-β1 expression were inhibited by pretreatment with PD98059 or SB203580 ().
Figure 4. Western blotting and RT-PCR analysis of TLR3, p-EGFR and EGFR; ELISA analysis of MUC5AC and TGF-β1 after treatment with AG1478; and ELISA analysis of MUC5AC and TGF-β1 after pretreatment with PD98059 or SB203580. (A) TLR3 production and transcription were remarkably suppressed by AG1478. (B) Pretreatment with AG1478 decreased p-EGFR expression but did not attenuate EGFR expression and transcription. (C) MUC5AC and TGF-β1 expression levels were diminished by AG1478. (D) The poly(I:C)-induced increases in MUC5AC and TGF-β1 expression were inhibited by pretreatment with PD98059 or SB203580. The mean ± SD of triplicate wells from a representative experiment (n = 3) is shown.
Discussion
In the present study, we demonstrated that TLR3 expression in HBECs increased after stimulation with poly(I:C). This increase in TLR3 expression triggered the phosphorylation of EGFR, leading to the secretion of MUC5AC, TGF-β1, MMP9 and VEGF. EGFR-ERK/P38 MAPK inhibition repressed the production of MUC5AC and inflammatory cytokines. These findings suggest that TLR3 is involved in virus-induced inflammatory cytokine production in HBECs via the EGFR-ERK/P38 MAPK signaling pathway.
TLR3 plays an important role in defensive antiviral immunity after respiratory virus infections. Guillot et al. reported that TLR3 contributes directly to the immune response of respiratory epithelial cells to influenza A virus (IAV) and dsRNA Citation(9). Le Goffic R et al. demonstrated that inhibition of TLR3 impairs the expression of proinflammatory cytokines in epithelial cells infected by IAV Citation(10). More recently, Stowell et al. showed that TLR3 knockout mice are protected from poly(I:C)-induced pathological changes and consequent lung function impairment Citation(11). In this study, we found that TLR3 expression in HBECs was increased in a dose- and time-dependent manner after poly(I:C) treatment. The increased expression of TLR3 was accompanied by elevated levels of MUC5AC, TGF-β1, MMP9 and VEGF, all of which contribute to airway remodeling in chronic airway diseases.
The mechanisms of TLR3 activation by viral infection are complex. Koff observed that Interleukin (IL)-8 and VEGF production were increased in HBECs after treatment with poly(I:C), and these increases were inhibited by the EGFR inhibitor AG1478 Citation(12). Kenneth Liu observed elevated IL-8 and Intercellular adhesion molecule (ICAM)-1 expression in rhinovirus-infected HBECs, and these responses were also completely blocked by treatment with AG1478 Citation(13). Thus, we speculated that the EGFR pathway might be associated with the virus-induced TLR3 expression. In the present study, EGFR phosphorylation increased after poly(I:C) treatment. The phosphorylation of EGFR was inhibited by treatment with TLR3 siRNA. The expression of the inflammatory cytokines MUC5AC and TGF-β1 was diminished by AG1478. Interestingly, we observed that TLR3 production and transcription were remarkably suppressed after blockade of the EGFR signaling pathway by AG1478. EGFR inhibitors can partially block the phosphorylation of TLR3 Citation(14). EGFR kinase activity is required for Tyr858 phosphorylation of TLR3, which is followed by assembly of the signaling complex, activation of transcription factors, gene induction and antiviral effects Citation(14).
Johnsen et al. observed that TLR3 induces signaling mechanisms involving TRIF, p38 MAPK and MK2 to enhance the stabilization of IFN-β mRNA, thus contributing to enhanced IFN-β levels during pathogen infection Citation(15). To determine which EGFR downstream signaling pathways is involved in airway mucus secretion and airway remodeling after virus infection, we evaluated changes in MUC5AC and TGF-β1 expression after treatment with ERK1/2, p38 and JNK inhibitors. TLR3, MUC5AC and TGF-β1 expression was inhibited by the ERK inhibitor PD98059 and the P38 inhibitor SB203580. These results indicate that TLR3 expression in bronchial epithelial cells after poly(I:C) treatment involves the TLR3-dependent EGFR-ERK/P38 MAPK signaling pathway.
COPD induces a series of airway remodeling events, including goblet cell and submucosal gland hyperplasia, smooth muscle cell and fibroblast hyperplasia, collagen deposition and microvascular changes. Zhu et al. determined that rhinovirus induces MUC5AC production in HBECs, which results in hypersecretion of MUC by airway goblet cells and submucosal glands Citation(16). Sugiura et al. observed that poly(I:C) augments the release of TGF-β1 in human fetal lung fibroblasts, thus affecting the differentiation of fibroblasts to myofibroblasts and enhancing the production of extracellular matrix proteins Citation(17). Merkle et al. demonstrated that TLR3 induces the upregulation of MMP-9 in human mesothelial cells after virus infection, thus influencing the synthesis and degradation of extracellular matrix components Citation(18). Thuringer et al. revealed that the interaction between heat-shock protein 27 and TLR3 activates Nuclear factor (NF)κB, leading to VEGF-mediated cell migration and angiogenesis in human microvascular endothelial cells Citation(19). These studies suggest that airway remodeling factors, including MUC5AC, TGF-β1, MMP-9 and VEGF, are associated with viral infection in HBECs. In the present study, we confirmed that the expression of MUC5AC, TGF-β1, MMP-9 and VEGF increases after exposure to poly(I:C). The expression of MUC5AC and TGF-β1 was inhibited by TLR3 siRNA and inhibitors of EGFR, ERK and P38.
Conclusions
In conclusion, the present study demonstrates that the TLR3-EGFR-ERK/p38 MAPK signaling pathway is involved in the production of airway remodeling cytokines after viral infection. MUC5AC and TGF-β1 expression was inhibited by treatment with an EGFR, ERK or P38 inhibitor. These findings provide new insights suggesting that the TLR3-EGFR signaling pathway is a potential target for virus infection-related airway inflammation.
Funding
This work was funded by research grants from the Zhejiang Provincial Natural Science Foundation of China (no. LY15H010003) and the Major Project of the Science Technology Department of Zhejiang Province (no. 2012C13022-2).
References
- Corhay JL, Vincken W, Schlesser M, Bossuyt P, Imschoot J. Chronic bronchitis in COPD patients is associated with increased risk of exacerbations: a cross-sectional multicentre study. Int J Clin Pract 2013; 67(12):1294–1301.
- Burgel P, Nesme-Meyer P, Chanez P, Caillaud D, Carre P, Perez T. Cough and sputum production are associated with frequent exacerbations and hospitalizations in COPD subjects. Chest 2009; 135(4):975–982.
- Hogg JC, Chu FSF, Tan WC, Sin DD, Patel SA, Pare PD, et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am J Respir Crit Care Med 2007; 176(5):454–459.
- Sikkel MB, Quint JK, Mallia P, Wedzicha JA, Johnston SL. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatr Infect Dis J 2008; 27(10 Suppl):S63–S70.
- Jakiela B, Gielicz A, Plutecka H, Hubalewska-Mazgaj M, Mastalerz L, Bochenek G, et al. Th2-type cytokine-induced mucus metaplasia decreases susceptibility of human bronchial epithelium to rhinovirus infection. Am J Respir Cell Mol Biol 2014; 51(2):229–241.
- Mata M, Sarrion I, Armengot M, Carda C, Martinez I, Melero JA, et al. Respiratory syncytial virus inhibits ciliagenesis in differentiated normal human bronchial epithelial cells: effectiveness of N-acetylcysteine. PLoS One 2012; 7(10):e48037.
- Wang Y, Shen Y, Li K, Zhang P, Wang G, Gao L, et al. Role of matrix metalloproteinase-9 in lipopolysaccharide-induced mucin production in human airway epithelial cells. Arch Biochem Biophys 2009; 486(2):111–118.
- Ichikawa T, Sugiura H, Koarai A, Minakata Y, Kikuchi T, Morishita Y, et al. TLR3 activation augments matrix metalloproteinase production through reactive nitrogen species generation in human lung fibroblasts. J Immunol 2014; 192(11):4977–4988.
- Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 2005; 280(7):5571–5580.
- Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 2007; 178(6):3368–3372.
- Stowell NC, Seideman J, Raymond HA, Smalley KA, Lamb RJ, Egenolf DD, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res 2009; 10:43.
- Koff JL, Shao MXG, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol 2008; 294(6):L1068–L1075.
- Liu K, Gualano RC, Hibbs ML, Anderson GP, Bozinovski S. Epidermal growth factor receptor signaling to ERK1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 2008; 283(15):9977–9985.
- Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. Epidermal growth factor receptor is essential for toll-like receptor 3 signaling. Sci Signal 2012; 5(233):ra50.
- Johnsen IB, Nguyen TT, Bergstrøm B, Lien E, Anthonsen MW. Toll-like receptor 3-elicited MAPK activation induces stabilization of interferon-β mRNA. Cytokine 2012; 57(3):337–346.
- Zhu L, Lee P, Lee W, Zhao Y, Yu D, Chen Y. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am J Respir Cell Mol Biol 2009; 40(5):610–619.
- Sugiura H, Ichikawa T, Koarai A, Yanagisawa S, Minakata Y, Matsunaga K, et al. Activation of toll-like receptor 3 augments myofibroblast differentiation. Am J Respir Cell Mol Biol 2009; 40(6):654–662.
- Merkle M, Ribeiro A, Sauter M, Ladurner R, Mussack T, Sitter T, et al. Effect of activation of viral receptors on the gelatinases MMP-2 and MMP-9 in human mesothelial cells. Matrix Biol 2010; 29(3):202–208.
- Thuringer D, Jego G, Wettstein G, Terrier O, Cronier L, Yousfi N, et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J 2013; 27(10):4169–4183.