Abstract
Abnormalities in the expression and functional activity of cell adhesion molecules are implicated in the development and progression of the majority of colorectal cancers (CRC). Cell–cell adhesion molecule E-cadherin regulates cell polarity, differentiation, proliferation and migration through its intimate association to the actin cytoskeletal network. During colorectal carcinogenesis changes in intercellular adhesion and dynamic rearrangements in the actin cytoskeleton result in altered signalling and migration with loss of contact inhibition. The adenomatous polyposis coli (APC) protein, besides its established role in the β catenin/Wnt signalling pathway, can coordinate microtubule and actin organization during cell migration. The actin-bundling protein Fascin promotes cell motility and is overexpressed in CRC. Based on recent molecular and pathological studies, this review focusses on the role of these molecules sharing the common feature of being associated with the cytoskeletal network during colorectal carcinogenesis and metastasis. The potential use of these molecules as prognostic markers and/or therapeutic targets will also be discussed.
INTRODUCTION
Colorectal cancers (CRC) is the third commonest malignancy worldwide and the second cause of cancer related death in western countries. Understanding of the genetics and molecular basis of colorectal cancer has advanced in the last decade and prompted a new era in clinical practice in which molecular pathogenetic events can be translated into benefits for screening, diagnosis and treatment of this malignancy. Adherens junctions provide critical adhesive contacts between adjacent normal epithelial cells. Epithelial (E)-cadherin is the major transmembrane protein of the adherens junctions and represents the prime mediator of calcium-dependent intercellular adhesion in normal epithelial cell (Shiozaki et al., Citation1996). E-cadherin binds directly or indirectly to cytoplasmic molecules of the catenins family and through them regulates the organization of actin cytoskeleton and the establishment and maintenance of cell polarity and tissue architecture (Takeichi, Citation1991). Interactions between E-cadherin molecules and their connection to the actin cytoskeleton are involved in a wide variety of cellular processes (migration, proliferation, apoptosis) important for the maintenance of the colonic epithelium homeostasis (CitationEfstathiou and Pignatelli, 1998; Wijnhoven and Pignatelli, Citation1999). Abnormalities in the E-cadherin/catenin complex and the disruption of the cytoskeletal molecules’ network appear to be an important step in the development and progression of colorectal cancer (CitationGagliardi et al., 1995; CitationBuda and Pignatelli, 2004).
Convergence of E-cadherin, catenin system and the cytoskeletal network in the colorectal cancer jigsaw
E-cadherin, encoded by the CDH1 gene on chromosome 16q22, forms the key functional component of adherens junctions of colonic epithelial cells and is bound via series of membrane proximal proteins, the catenins (α-, β-, γ- and p120), to the cytoskeletal actin. β catenin and γ catenin bind directly to the cytoplasmic tail of E-cadherin whereas α catenin links the bound β catenin to the actin microfilament network of the cellular cytoskeleton (CitationBenjamin et al., 2010).
In CRC E-cadherin/catenin complex does not always show absent or reduced expression, but often there is redistribution from cell membrane to the cytoplasm (CitationJiang and Hiscox, 1997). Failure to localise to the membrane and/or bind to the cytoskeleton in spite of normal expression may be due to post-transcriptional modification of the complex (van Roy and Berx, Citation2008; CitationFujita et al., 2002; Shen et al., Citation2008). Colorectal carcinoma cells in vivo have been shown to express receptors for several growth factors (e.g. epidermal growth factor, hepatocyte growth factor) known to induce a more malignant phenotype (increased invasiveness, higher proliferation). These changes can be explained, at least in part, by the ability of both growth factors to disrupt the association of cytoskeletal components (actin, α-actinin, vinculin) with the E-cadherin/catenin complex via tyrosine phosphorylation of β catenin (Shiozaki et al., Citation1996; Shibamoto et al., Citation1994).
E-cadherin mediated intercellular adhesion limits cell motility and establish cell apical-basal polarity (Takeichi, Citation1991). Loss of E-cadherin expression and disassembly of the E-cadherin/catenin complex on the cell surface induces a transition from a stationary to a motile phenotype and enables tumour cells to disseminate and metastasise (Vleminckx et al., Citation1991).
The role of downregulation of E-cadherin expression and functional activity in tumour progression and metastasis of colorectal cancer is not only related to loss of cell to cell adhesion but also to loss of suppression of nuclear β catenin activity (Wong and Pignatelli, Citation2002).
β catenin is one of the downstream effectors of the Wnt signalling pathway which is critically deregulated in the pathogenesis of CRC (Wong and Pignatelli, Citation2002; CitationClevers, 2006). Disruption of the E-cadherin/β catenin complex releases β catenin from the membranous to the cytoplasmic pool. Cytosolic β-catenin may subsequently be degraded or trans-located into the nucleus. The degradation of β-catenin involves binding of the protein to a complex composed of APC, AXIN and glycogen synthase kinase (GSK)-3β proteins (Yost et al., Citation1996). GSK-3β phosphorylates serine and threonine residues on β-catenin, a crucial step required to target the protein for ubiquitination and proteosomal degradation. Both APC and AXIN promote β-catenin degradation enhancing its phosphorylation. When ‘E-cadherin-free’ cytoplasmic β-catenin is not completely degraded, it can enter into the nucleus inducing the transcription of genes involved tumour progression (CitationCheng et al., 2005; CitationLogan and Nusse, 2004; CitationFagotto et al., 1996). In colon cancer β catenin increases cell proliferation by the induction of the cell cycle regulators C-myc and cyclin D1 (Tetsu and McCormick, 1999; CitationHe et al., 1998).
The α catenin is an actin bundling protein which mediates the connection of the cadherin/catenin complex to the actin cytoskeleton (Rimm et al., Citation1995). Loss of α catenin is frequently described in colorectal cancer, and reduced α catenin expression is significantly related with depth of tumour invasion, the presence of liver and lymph node metastasis and Duke's stage (Raftopoulos et al., Citation1998).
Unlike E-cadherin, α catenin expression has been shown to be an independent factor in predicting survival, especially, in patients with stage II (Duke's B) colon cancer (Ropponen et al., Citation1999). The role of adjuvant therapy in this subgroup of patients is still controversial. α catenin could represent a molecular prognostic and predictive marker able to identify the subjects with greater risk of disease progression who might benefit from early adjuvant therapy.
IQGAP1 is a multifunctional scaffolding protein involved in the remodelling of actin cytoskeleton assembly during cell adhesion and migration and is upregulated at the invasive front of colorectal carcinoma (Nabeshima et al., Citation2002). Recently Hayashi et al. have addressed the clinical and pathological significance of this increased expression in a series of advanced CRC and demonstrated that a diffuse expression pattern of IQGAP1 can predict a shorter overall survival (CitationHayashi et al., 2010). The mechanism proposed for the enhanced cell invasion relies on the negative regulation of IQGAP1 on intercellular adhesion through the dissociation of E-cadherin from α-catenin.
The p120-catenin is a member of the catenin gene family, homologous to β- and γ-catenin which was originally described as a substrate for the Src oncoprotein (Reynolds et al., Citation1994). p120 interacts directly with the cytoplasmic domain of E-cadherin and promotes the stability of the cadherin complex and the local assembly of the underlying actin cytoskeleton. In normal colon, p120 is localised at the cell membrane and in the cytosol of colonocytes, both in the crypt and surface epithelium, with greater expression in the proliferative compartment (Valizadeh et al., Citation1997). Abnormalities in p120 expression are common in adenomatous polyps and colon cancer and correlate with reduced E-cadherin expression and inversely with tumour size (CitationGold et al., 1998). In colorectal carcinoma p-120 regulates E-cadherin function and recently a mutation of the p-120 gene has been shown in a colonic carcinoma cell line unable to establish normal E-cadherin mediated adhesion (CitationIreton et al., 2002).
Expression and inactivation of E-cadherin in colorectal carcinogenesis
Malignant transformation is usually characterised by decrease in cell–cell adhesion and perturbation of the cytoskeletal organisation. Inactivation of the E-cadherin function is associated with loss of differentiation and tissue architecture. Downregulation of E-cadherin facilitates tumour cell invasion (CitationFrixen et al., 1991) whereas restoring E-cadherin binding by genetic manipulation prevents tumour progression and invasiveness (Perl et al., Citation1998; Vleminckx et al., Citation1991). Several clinical and experimental studies have shown reduced expression of E-cadherin in colorectal adenomas and poorly differentiated invasive CRC (Valizadeh et al., Citation1997; CitationKinsella et al., 1994). Indeed, apart from its adhesive properties, E-cadherin is also important for the negative control of cell growth by upregulation of the cyclin-dependent kinase inhibitor (cdk) p27 (St Croix et al., Citation1998).
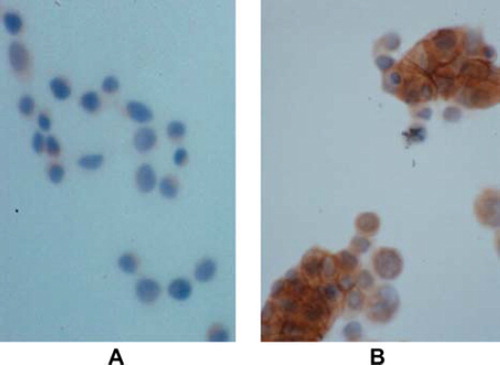
E-cadherin can be downregulated through a variety of mechanisms including mutation, epigenetic silencing and transcriptional repression. E-cadherin-inactivating mutations were first described in diffuse gastric cancer (CitationBecker et al., 1993), and loss of the E-cadherin locus on the long arm of chromosome 16 (16q22) occurs frequently in hepatocellular carcinomas, lobular breast and oesophageal cancer (Strathdee et al., 2002; Strumane et al., Citation2004). However, mutations or loss of heterozygosity (LOH) within the E-cadherin gene are rare in sporadic CRC (More et al., Citation2007; Richards et al., 1999; Vécsey-Semjén et al., Citation2002). Efstathiou et al. have screened a panel of 49 human colorectal carcinoma cell lines and found that only 7% exhibited inactivating E-cadherin mutation (CitationEfstathiou et al., 1999). In the LS174T carcinoma cell line which lacks E-cadherin due to a truncating mutation of CDH1, cells display a poor cohesive growth pattern in both three dimensional gel and in monolayer colture. E-cadherin transfection can reverse LS174T to a non-transformed phenotype () confirming the functional significance of these molecule during colorectal carcinogenesis.
Figure 1. Induction of cell adhesion of E-cadherin transfectans. LS174T cell line lacks E-cadherin expression due to CDH1 gene mutation. These cells show poor differentiation and poor cohesive pattern of growth (A). E-cadherin transfection with mouse full length E-cadherin cDNA reverse LS174T cells to a non-transforming phenotype with cells being more cohesive and tightly connected with each other in monolayer (B); E-cadherin expression at intercellular adhesions is demonstrated in transfected cells by immunocytochemistry (brown immunostaining); A) and B) magnification × 20.
Epigenetic events are alternative mechanisms of gene modulation affecting the expression of a gene without disrupting its sequence. Abnormal methylation of CpG-rich islands in the promoter region of several genes has been studied in CRC. E-cadherin promoter methylation associated with decrease or absence of E-cadherin expression at both mRNA and protein levels has been detected in nearly 60% of sporadic tumours in relatively large series of CRC (CitationGarinis et al., 2002; Wheeler et al., Citation2001) and in normal colonic mucosa neighbouring cancer cells in CRC with microsatellite instability (MSI +) (Ramírez et al., Citation2008).
Different repressors of E-cadherin transcription have been associated with the progression of multiple cancer types. The presence of E-cadherin transcriptional repressors such as Slug, Snail, Twist1 and Zeb-1 in colonic epithelial tumour cells is inversely related with E-cadherin expression and associated with increased metastasis and poorer clinical prognosis (Schmalhofer et al., Citation2009; Thiery et al., Citation2009).
A common polymorphism of the CDH1 promoter at nucleotide–160 relative to the transcription start (CDH1-160C > A) has been reported to influence CDH1 transcription (CitationLi et al., 2000). Pittman et al. have recently shown that CDH1-160C > A polymorphism is a risk factor for CRC and since a high proportion of the European population are carriers of the at-risk allele; this variant is likely to contribute substantially to CRC development (Pittman et al., Citation2009).
Proteolytic processing of E-cadherin in cancer represents an alternative route to inhibit its function. The characterisation of proteins released from tumour cells in vitro, referred as the secretome approach (CitationVolmer et al., 2005), not only enables the discovery of novel cancer biomarkers but also provides the opportunity to characterize the protein secretion pathways. Among non-classical secretion mechanisms, shedding of ectodomains has been recognised as a key mechanism for regulating the function of cell surface proteins including E-cadherin (Rios-Doria et al., Citation2003). Matrix metalloproteinases, including MMP3, MMP7, MMP9 and MMP14, ADAM10 and ADAM15 and several other proteases such as the serine protease kallikrein, are able to cleave E-cadherin ectodomain near the membrane (Noë et al., Citation2001; Najy et al., Citation2008; CitationKlucky et al., 2007). Soluble E-cadherin fragments may function as pseudo-ligands to block E-cadherin adhesion or activate signalling pathways stimulating cell proliferation and migration (Najy et al., Citation2008; CitationJohnson et al., 2007). The role of soluble E-cadherin as a serum biomarker in CRC has been investigated with inconsistent results (Wilmanns et al., Citation2004; Velikova et al., Citation1998). However, recently Weiß et al. (Citation2011) have studied soluble E-cadherin levels in blood samples from CRC, familial adenomatous polyposis (FAP) syndrome and inflammatory bowel disease (IBD) patients, patients with adenomas and healthy controls. The most important findings were the significantly increased soluble E-cadherin levels in FAP and patients with stage III and IV CRC compared to healthy subjects. Interestingly, levels in IBD patients and patients with adenoma did not differ from control, possibly due to the relatively high levels found in the latter. Although soluble E-cadherin like CEA is not an adequate marker for screening purposes, its use as a follow-up marker after surgery or to monitor therapy can be envisaged, particularly in CEA negative patients at diagnosis.
E-cadherin transcriptional silencing and the epithelial-mesenchymal transition program during colorectal cancer invasion and metastasis
The overall process of tumour spreading and metastasis involves the genesis of an invasive and metastatic phenotype with dramatic alterations of epithelial cell structure and function. Increased malignancy is characterized by dedifferentiation, loss of adhesion and increased cellular motility (Sanz-Moreno and Marshall, Citation2010). All these morphogenetic changes mirror the cellular processes occurring during physiological events such as embryogenesis and wound repair and are referred to as epithelial-mesenchymal transition (EMT). Accumulating evidence indicated that the concept of EMT could be extended to cancer progression and metastasis (Thiery, Citation2002). In colon cancer, EMT occurs at the invasive front and generates single migratory cells that lose E-cadherin expression with coexisting deregulation of Wnt signalling pathway and selective loss of basement membrane (Spaderna et al., Citation2006; Ueno et al., Citation2002). The loss of E-cadherin is considered the initial step of the EMT program (CitationIwatsuki et al., 2010). In CRC the disruption of cell–cell adhesion is associated with the gain of mesenchymal markers such as fibronectin vimentin and N-cadherin which is followed by the loss of basolateral polarization of cells and acquisition of front-rear polarization instead (Nelson, Citation2009). Transcriptional repression of E-cadherin by zinc finger proteins (ZEB1, ZEB2), basic-helix-loop-helix (bHLH) proteins (Twist) and snail family proteins (SNAI1) are associated with EMT (CitationBatlle et al., 2000; Yang et al., Citation2004; Vandewalle et al., Citation2004).
SNAI1 correlates with E-cadherin and vitamin D receptor downregulation in CRC and in normal mucosa adjacent to tumour tissue and promotes the metastatic ability of cancer cells (CitationPálmer et al., 2004; Peña et al., Citation2005; Peña et al., Citation2009). Vitamin D treatment could be used in Snail-negative tumours and SNAI1 itself could represent a potential target for distant metastasis chemoprevention (González-Sancho et al., 2006; Wali et al., Citation2008). Shiori et al. have shown that SNAI2 (Slug) is an independent risk factor for poor prognosis in CRC patients (Shioiri et al., Citation2006).
ZEB1 represses E-cadherin transcription and induces EMT in epithelial cells in vitro and in vivo models (CitationComijn et al., 2001; CitationEger et al., 2005). Expression of ZEB1 is regulated by factors known to activate EMT such as TGF-β, NFkB, hypoxia and SNAI1 (Peinado, 2007). Recently ZEB1 has been shown to interact with the chromatin remodelling protein BRG1 to repress E-cadherin promoter in CRC cell lines. BRG1 interacts with DNA-binding protein and activate/repress transcription (Trotter and Archer, Citation2008). Since ZEB 1 was found expressed only in E-cadherin negative cells at the invasive front the ZEB1/BRG1 interaction has been suggested as a new transcriptional mechanism of E-cadherin repression during EMT (Sánchez-Tilló et al., Citation2010). Some of the E-cadherin repressors are also driving the EMT associated chemoresistance (Wang et al., Citation2004; Yang et al., Citation2008; CitationHoshino et al., 2009). The specific inhibition of these transcription repressors by siRNA or by chemical compounds could reduce the tumour aggressiveness and improve CRC management, Immunohistochemical studies have shown that E-cadherin could be increasingly expressed in liver and lymph nodes metastasis compared to primary gastric and colorectal tumours (CitationCai et al., 2001; Mayer et al., Citation1993). This event has been explained by the fact that metastatic E-cadherin positive cells can arise from primary E-cadherin negative ones rather than metastatic colonization of E-cadherin expressing cells escaping the primary tumour. E-cadherin negative breast carcinoma cells have been shown to induce lung metastases from orthotropic primary tumours in mice, and these metastases expressed E-cadherin (CitationChao et al., 2010). A subset of disseminated cancer cells can receive signals from the metastatic site organ to revert the de-differentiating programme activated by EMT and start a mesenchymal-epithelial reverting transition (MErT) (Wells et al., Citation2008). Since E-cadherin adhesion exerts survival signals and prevents anoikis even when it binds to heterotypic cell (Yates et al., Citation2007), it can be speculated that E-cadherin expression in metastastic cancer cells could contribute to a selective growth advantage. Thus, E-cadherin plays a critical role in phenotypic changes noted in both primary and secondary cancer sites, supporting a model of tumour cell plasticity. Recent studies on small non-coding RNAs (miRNAs) have significantly increased our knowledge on the regulation of gene and protein expression in metastasis. Indeed, a new evaluation of EMT has been proposed, identifying one regulated by the miRNA network. The function and dysregulation of miRNAs in colorectal cancer has been reviewed elsewhere (Wu et al., Citation2011). MiR-21 has an oncogenic function and in vitro studies have demonstrated that it is able to promote cell growth and inhibit apoptosis in prostate and hepatocellular carcinoma cells (Si et al., Citation2007; Meng et al., Citation2007; CitationLi et al., 2009). In colorectal cancer miR-21 expression is increased compared to normal and adenomatous tissue and PTEN and programmed cell death 4 (PDCD4) tumour suppressors gene have been identified as its potential targets (CitationChang et al., 2011). Furthermore, miR-21 levels inversely correlate with advanced stage, metastasis and prognosis of CRC patients (Shibuya et al., Citation2010) supporting the potential clinical significance of miR-21 as a diagnostic and prognostic marker (Schetter et al., Citation2008).
The cross-modulation between E-cadherin repressors and specific miRNAs has also potential implications on the EMT changes linked to tumour metastases. Members of the miR-200 and miR-205 family can repress ZEB1 and ZEB2 protein translation, increasing E-cadherin expression (CitationKorpal et al., 2008; CitationGregory et al., 2008). Expression analysis of miRNAs showed that those targeting the ZEB family are particularly abundant in normal epithelial tissue and downregulated in de-differentiated cancer (Sempere et al., Citation2007; CitationFeber et al., 2008).
During tumour cells invasion loss of E-cadherin is accompanied by induced expression of the mesenchymal (N)-cadherin. E-cadherin repressors (SNAI1/2, ZEB1) can induce N-cadherin during EMT (Cano et al., 2000). This ‘cadherin-switching’ where E-cadherin is replaced by N-cadherin accompanied by de novo vimentin expression appears to be part of the de-differentiating programme during EMT induced invasion and metastasis (Thiery et al., Citation2009). Recently, Pino et al. reported abnormal E-cadherin cytoplasmic localisation and membranous N-cadherin expression associated with poor prognosis and microsatellite stability in sporadic CRC (Pino et al., Citation2010). The molecular mechanisms underlying the potential ability of N-cadherin to promote cancer cell metastasis in vivo remain to be determined, though, it has been hypothesised that N-cadherin may regulate the interactions between cancer cells and either endothelial or stromal cells (CitationHazan et al., 1997; CitationHazan et al., 2000). An N-cadherin antagonist (ADH-1) has been discovered (Williams et al., Citation2000) and in animal models this drug has been shown to inhibit tumour metastasis in pancreatic cancer and melanoma (Shintani et al., Citation2008; CitationAugustine et al., 2008). However, neither of the studies has addressed the effect ADH-1 on tumour vasculature and a recent clinical experience on N-cadherin positive gynaecological malignancies showed significant changes in tumour blood flow only in 20% of patients (Perotti et al., Citation2009). Tumour budding is a morphological finding consisting in transition from glandular structures to single cells or clusters up to four cells at the invasive margin of CRC (Ueno et al., Citation2002). Budding cells in CRC have been recognized to be able to re-differentiate locally or at metastatic site, and tumour budding is an independent risk factor for local spread, lymph node, lung and liver metastasis and recurrence and survival after curative surgery (Nakamura et al., Citation2005; Tanaka et al., Citation2003). Tissue microarray analysis in a series of more than a thousand CRC demonstrated that loss of E-cadherin independently predicts tumour budding (CitationLugli et al., 2007).
Cells undergoing EMT develop resistance to pharmacological treatment allowing rapid progression of the tumour. Anticancer drugs and treatment (e.g. radiation, chemotherapy) themselves can induce EMT (CitationAndarawewa et al, 2007; Tsukamoto et al., Citation2007; Li, 2010). Colon cancer cells under chronic exposure to oxaliplatin became resistant and develop phenotypic changes that resemble EMT, including the capacity to migrate and invade (Yang et al., Citation2006). EMT can also confer resistance to active targeted drugs against molecular effectors of tumour progression and invasion such as Bevacizumab, Cetuximab and Enzastaurin. These agents are used in metastatic CRC and target the vascular endothelial growth factor (VEGF), the epidermal growth factor receptor (EGFR) and the protein kinase C (PKC) β, respectively. Colonic cancer cell lines expressing either mutated K-ras, an EMT inducer (Shao et al., Citation2004; Singh et al., Citation2009), or mesenchymal markers (N-cadherin, vimentin) and E-cadherin repressors, ZEB1 and TWIST, were resistant to Enzastaurin (Serova et al., Citation2010).
APC, the cytoskeleton and cell migration: a novel role in colorectal carcinogenesis
The role of APC tumour suppressor protein in the development and progression of CRC have been extensively studied for more than a decade. Briefly, the APC mutations found in these cancers almost always cause truncation of the central region of the protein containing the 20-amino acid β-catenin-binding site (CitationBéroud and Soussi, 1996; Behrens et al., 2000). The resulting loss of APC protein binding to β-catenin significantly reduces phosphorylation of β-catenin by GSK-3β, thereby, leading to accumulation of cytosolic β-catenin and its subsequent nuclear translocation (CitationBéroud and Soussi, 1996). APC mutation occurs early during tumour formation, and the role of APC as a gatekeeper gene in CRC carcinogenesis is widely accepted (Kinzler et al., 1997). However, with the discovery of its localization at the cell–cell adherens junctions and the association with the microtubule-based and actin-based cytoskeleton, other important functional roles of APC have become apparent (Mimori-Kiyosue et al., Citation2000; McCartney and Näthke, 2008).
A series of reports demonstrated that APC is involved in intercellular adhesion through its interactions with the cadherin/catenin system. APC has a positive effect on E-cadherin localization at the plasma membrane. Stable expression of APC in the colonic cancer cell line SW480 restores their adhesive properties and triggers the translocation of the E-cadherin/β catenin complex to the cell membrane (CitationFaux et al., 2004). In the APC Minβ (multiple intestinal neoplasia) mouse model, an animal model of familial adenomatous polyposis (FAP), colonic cells express a truncated APC allele associated with disruption of E-cadherin/β catenin complex and decreased E-cadherin membranous expression (CitationCarothers et al., 2001). The mechanism linking wild type APC to the redistribution of E-cadherin and β catenin to the adherens junctions is not completely understood.
In interphase cells, APC has been found at the actin-rich cortex of epithelial cells and joining the growth of polymerizing microtubule network (Reilein and Nelson, Citation2005). APC protein contains multiple domains that facilitate the interaction with other tip-binding protein at the end of microtubules such as KAP3 (CitationJimbo et al., 2002), IQGAP1 (Watanabe et al., Citation2004) and Asef (N-terminal Armadillo domain) (CitationKawasaki et al., 2000), EB1 and mDIA (C-terminus basic domain) (Wen et al., Citation2004; Okada et al., Citation2010). APC is able to move along microtubules by the binding with the kinesin adaptor protein KAP3 and accumulates at their plus-ends where it drives the formation of membrane protrusions which direct cell movements. Overexpression of KAP3 increases the concentration of endogenous APC in clusters whereas dominant negative form of KAP3 reduces APC clusters at membrane protrusions (CitationJimbo et al., 2002; Sharma et al., Citation2006).
APC promotes cell migration through its subcellular localisation at the actin-dependent membrane ruffles and lamellopodia. At the plasma membrane IQGAP1, APC and microtubules via the CLIP-170 microtubule binding protein, forms a complex regulated by activated Rac1 and Cdc42 (Watanabe et al., Citation2004). Another important finding that underlines the crucial role of APC in the formation of the actin meshwork at the leading edge of migrating cells was the discovery of the interaction with the Rac-specific guanine-nucleotide-exchange factor, Asef. Asef-APC binding modulates actin activity to decrease intercellular adhesion and promote cell motility (CitationKawasaki et al., 2000). Interestingly, binding of the C-truncated APC mutant form to Asef is able to activate migration, and this gain-of-function displayed by APC mutants could contribute to the abnormal migratory properties of CRC cells (CitationKawasaki et al., 2003). EB1 and mDIA proteins interact with the C-terminus of APC, facilitating microtubules dynamics and actin polymerization (Wen et al., Citation2004; Okada et al., Citation2010). Although these binding proteins are not essential for APC trafficking to membrane clusters which is mediated by N-terminal domains molecules, they could be responsible for the lack of stability and retention of the APC C-truncated form at the leading migratory edge.
Regulation of cell migration and directing intercellular adhesion in epithelial cell represent important non-traditional roles of the APC tumour suppressor protein (CitationKawasaki et al., 2000). A defect in cell migration along the crypt-villous axis has been demonstrated in normal flat mucosa of Apc Min mice, and in adenomas of mice carrying a truncated form of Apc (CitationMahmoud et al., 1997). All this evidence suggests that APC is involved in colorectal development and progression beyond its role in Wnt signalling. Mutation of Apc and the consequential loss of its function as a mobile scaffold coordinating microtubule assembly could determine a defect in the maturation and migration of intestinal epithelial cells leading to an aberrant expansion of the proliferative zone and possible accumulation of other genetic events described during CRC carcinogenesis; loss of the APC C-terminus regulation of the cytoskeletal actin network during cell motility could be involved in tumour progression (Okada et al., Citation2010).
Rearrangement of actin cytoskeleton in the promotion of a migratory and invasive phenotype of colorectal cancer cells: a FASCINating link
Tumour progression and tissue invasiveness require increased cell motility and enhanced plasticity of the actin cytoskeleton. The three-dimensional organisation of actin filaments is regulated by the function of multiple actin-bundling proteins.
Fascin is a cross-linking protein that localises to the core actin bundles of spikes and filopodia at the leading edge of migratory cells (CitationKureishy et al., 2002). Alterations in fascin expression have been implicated in several diseases and a dramatic increase of this protein has been described in cancers. We have first reported an upregulation of fascin in a series of human colorectal adenocarcinomas and demonstrated that fascin overexpression is associated with alterations in the structural assembly of focal adhesions, increased proliferation and invasive phenotype in vitro (CitationJawhari et al., 2003). More recently four other studies have demonstrated that fascin expression correlates with poor prognosis either across combined or within different CRC stages (CitationHashimoto et al., 2006; Puppa et al., Citation2007; Tsai et al., Citation2007; CitationChan et al., 2010). These data clearly show that the expression of fascin represents a phenotypic change in CRC.
To fully understand the role of fascin in CRC carcinogenesis it is important to determine at what stage it becomes deregulated and the potential relation to clinical risk factors associated with malignant progression. Qualtrough et al. have addressed these questions and demonstrated not only that fascin is consistently overexpressed in adenomas compared to normal tissue but also a significant positive correlations between expression of fascin with tumour size, histology and dysplasia (Qualtrough et al., Citation2009). Another important finding of this study was the observation of fascin immunoreactivity focussed around the stalk of the polyps which represents the potential site for malignant progression and where the epithelial cells are in contact with the mesenchyme. This corroborates the role of fascin in malignant progression and also allows one to speculate on the possibility of epithelial-mesenchymal crosstalk regulating fascin expression (Qualtrough, 2009).
CONCLUDING REMARKS
The multiple interactions between adhesion complexes and the cytoskeletal proteins are critical for the maintenance of a normal intestinal epithelial architecture and allow individual cells to respond to different stimuli and signals as an integrated network. In colorectal cancer, gene mutations and functional derangements in a variety of cytoskeletal-associated adhesion molecules have been described (). Results from clinico-pathological studies have shown that in colorectal cancer loss of differentiation and increased invasiveness are associated with loss of expression and function of E-cadherin. These observations clearly demonstrate that E-cadherin acts as an ‘invasion-suppressor’ gene and dissecting the molecular mechanisms that regulate its expression and function has, therefore, become pivotal for understanding tumour progression and metastasis development. The phenotypic conversion of epithelial cells to mesenchymal cells plays an important role in the process of CRC spreading; this event has been proposed to be reversible at the metastatic sites. To the same extent the downregulation or silencing of E-cadherin has emerged as a molecular hallmark of carcinoma EMT, re-expression of E-cadherin elicits the mesenchymal to epithelial reverting transition during the metastatic seeding.
Continued progress on the multiple non-traditional roles of APC including microtubule-organization and control of actin-dependent migration has highlighted its contribution to colorectal carcinogenesis outside the Wnt pathway. An increasing number of studies have shown that the actin-bundling protein Fascin is implicated in the metastatic process of colonic carcinoma cells via its motility-inducing properties. Recently, fascin overexpression and a positive correlation with the degree of dysplasia has been demonstrated in colorectal adenomas, the precursor lesions of the vast majority of colon cancers. These findings suggest that Fascin could have an important role even during malignant progression at early stages of carcinogenesis.
The central role of E-cadherin and the cytoskeletal network in tumour development and progression provides a tantalising area for further research with important implications for the development of novel molecular markers and potential targets for new anticancer drugs.
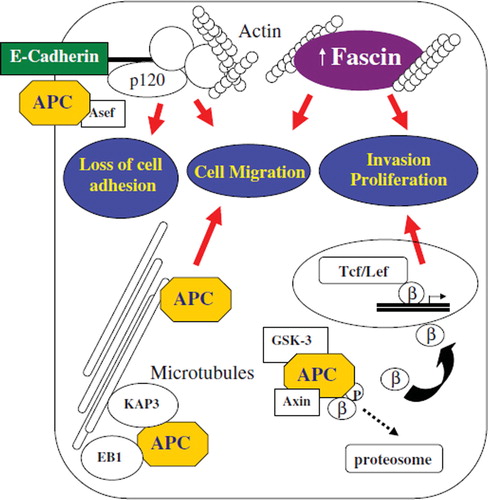
Figure 2. Interactions between cytoskeletal adhesion molecules and potential mechanisms in the development and progression of colorectal cancer. E-cadherin binds catenins to form cytoskeletal complexes with actin cytoskeleton. Abnormalities in E-cadherin expression (genetic mutation, epigenetic silencing, transcriptional repression) result in reduced cell–cell adhesiveness and conversion to a migratory and metastatic phenotype. Fascin forms tight and stable bundles with F-actin. Fascin overexpression is associated with increased proliferation and cell migration, inducing an invasive behaviour of colorectal cancer cells. As a tumour suppressor, APC protein controls β catenin proteosome degradation in association with GSK-3β and Axin. Mutations in APC reduce β catenin degradation and induce nuclear translocation with increased transcription of genes promoting tumour growth/ invasion. Different subcellular localization of APC (e.g. adherens junctions), have prompted the discovery of new non-traditional roles such as cell migration and adhesion. APC is directed along cytoskeletal microtubules by KAP3 to the plasma membrane where it regulates cell migration. APC binding with EB1 at the plus end of microtubule mediate microtubules’ stabilization and polymerization, thus promoting cell migration. APC has also been implicated in cell to cell adhesions. Truncated form of APC can activate Asef, decreasing intercellular adhesions and promote motility. Gain-of-function of migratory properties and loose of adherens junctions by truncated cancer-mutant APC are critical events in tumour formation and spreading. Abbreviations: GSK-3β, glycogen synthase kinase-3β; Tcf/Lef, T-cell/Lymphoid enhancer transcription factors.
ACKNOWLEDGEMENTS
The authors acknowledge the kind gift of E-cadherin cDNA from Dr M. Takeichi.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Andarawewa KL, Erickson AC, Chou WS, . (2007). Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 67: 8662–8670.
- Augustine CK, Yoshimoto Y, Gupta M, Zipfel PA, Selim MA, Febbo P, Pendergast AM, Peters WP, Tyler DS (2008). Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 68(10): 3777–3784.
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2(2): 84–89.
- Becker KF, Atkinson MJ, Reich U, Huang HH, Nekarda H, Siewert JR, Höfler H (1993). Exon skipping in the E-cadherin gene transcript in metastatic human gastric carcinomas. Hum Mol Genet. 2(6): 803–804.
- Behrens J (2000). Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci. 910: 21–33; discussion 33–35.
- Benjamin JM, Kwiatkowski AV, Yang C, Korobova F, Pokutta S, Svitkina T, Weis WI, Nelson WJ (2010). AlphaE-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. J Cell Biol. 189(2): 339–352.
- Béroud C, Soussi T (1996). APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 24(1): 121–124.
- Buda A, Pignatelli M (2004). Cytoskeletal network in colon cancer: from genes to clinical application. Int J Biochem Cell Biol. 36(5): 759–765.
- Cai J, Ikeguchi M, Tsujitani S, Maeta M, Liu J, Kaibara N (2001). Significant correlation between micrometastasis in the lymph nodes and reduced expression of E-cadherin in early gastric cancer. Gastric Cancer. 4(2): 66–74.
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2(2): 76–83.
- Carothers AM, Melstrom KA Jr, Mueller JD, Weyant MJ, Bertagnolli M (2001). Progressive changes in adherens junction structure during intestinal adenoma formation in Apc mutant mice. J Biol Chem. 276(42): 39094–102.
- Chan C, Jankova L, Fung CL, Clarke C, Robertson G, Chapuis PH, Bokey L, Lin BP, Dent OF, Clarke S (2010). Fascin expression predicts survival after potentially curative resection of node-positive colon cancer. Am J Surg Pathol. 34(5): 656–666.
- Chang KH, Miller N, Kheirelseid EA, Ingoldsby H, Hennessy E, Curran CE, Curran S, Smith MJ, Regan M, McAnena OJ, Kerin MJ (2011). MicroRNA-21 and PDCD4 expression in colorectal cancer. Eur J Surg Oncol. 37(7): 597–603.
- Chao YL, Shepard CR, Wells A (2010). Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 9: 179.
- Cheng XX, Wang ZC, Chen XY, Sun Y, Kong QY, Liu J, Gao X, Guan HW, Li H (2005). Frequent loss of membranous E-cadherin in gastric cancers: a cross-talk with Wnt in determining the fate of beta-catenin. Clin Exp Metastasis. 22(1): 85–93.
- Clevers H (2006). Wnt/beta-catenin signaling in development and disease. Cell.127: 469–80.
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001). The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 7(6): 1267–78.
- Efstathiou JA, Liu D, Wheeler JM, Kim HC, Beck NE, Ilyas M, Karayiannakis AJ, Mortensen NJ, Kmiot W, Playford RJ, Pignatelli M, Bodmer WF (1999). Mutated epithelial cadherin is associated with increased tumorigenicity and loss of adhesion and of responsiveness to the motogenic trefoil factor 2 in colon carcinoma cells. Proc Natl Acad Sci U S A. 96(5): 2316–2321.
- Efstathiou JA, Pignatelli M (1998). Modulation of epithelial cell adhesion in gastrointestinal homeostasis. Am J Pathol. 153(2): 341–347.
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R (2005). DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 24(14): 2375–2385.
- Fagotto F, Funayama N, Gluck U, Gumbiner BM (1996). Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol. 132(6): 1105–1114.
- Faux MC, Ross JL, Meeker C, Johns T, Ji H, Simpson RJ, Layton MJ, Burgess AW (2004). Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J Cell Sci. 117 (Pt 3): 427–439.
- Feber A, Xi L, Luketich JD, Pennathur A, Landreneau RJ, Wu M, Swanson SJ, Godfrey TE, Litle VR (2008). MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg. 135(2): 255–260.
- Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Löchner D, Birchmeier W (1991). E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 113(1): 173–185.
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002). Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 4(3): 222–231.
- Gagliardi G, Kandemir O, Liu D, Guida M, Benvestito S, Ruers TG, Benjamin IS, Northover JM, Stamp GW, Talbot IC, . (1995). Changes in E-cadherin immunoreactivity in the adenoma-carcinoma sequence of the large bowel. Virchows Arch. 426(2): 149–154.
- Garinis GA, Menounos PG, Spanakis NE, Papadopoulos K, Karavitis G, Parassi I, Christeli E, Patrinos GP, Manolis EN, Peros G (2002). Hypermethylation-associated transcriptional silencing of E-cadherin in primary sporadic colorectal carcinomas. J Pathol. 198(4): 442–449.
- Gold JS, Reynolds AB, Rimm DL (1998). Loss of p120ctn in human colorectal cancer predicts metastasis and poor survival. Cancer Lett. 132(1–2): 193–201.
- González-SaSancho JM, Larriba MJ, Ordóñez-Morán P, Pálmer HG, Muñoz A (2006). Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer Res. 26(4A): 2669–2681.
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10(5): 593–601.
- Hashimoto Y, Skacel M, Lavery IC, Mukherjee AL, Casey G, Adams JC (2006). Prognostic significance of fascin expression in advanced colorectal cancer: an immunohistochemical study of colorectal adenomas and adenocarcinomas. BMC Cancer. 6: 241.
- Hayashi H, Nabeshima K, Aoki M, Hamasaki M, Enatsu S, Yamauchi Y, Yamashita Y, Iwasaki H (2010). Overexpression of IQGAP1 in advanced colorectal cancer correlates with poor prognosis-critical role in tumor invasion. Int J Cancer. 126(11): 2563–2574.
- Hazan RB, Kang L, Whooley BP (1997). Borgen PIN-cadherin promotes adhesion between invasive breast cancer cells and the stroma. Cell Adhes Commun. 4(6): 399–411.
- Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA (2000). Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 148(4): 779–790.
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, . (1998). Identification of c-MYC as a target of the APC pathway. Science. 281: 1509–1512.
- Hoshino H, Miyoshi N, Nagai K, Tomimaru Y, Nagano H, Sekimoto M, Doki Y, Mori M, Ishii H (2009). Epithelial-mesenchymal transition with expression of SNAI1-induced chemoresistance in colorectal cancer. Biochem Biophys Res Commun. 390(3): 1061–1065.
- Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van Roy F, Reynolds AB (2002). A novel role for p120 catenin in E-cadherin function. J Cell Biol Nov. 159(3): 465–476.
- Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M (2010). Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 101(2): 293–299.
- Jawhari AU, Buda A, Jenkins M, Shehzad K, Sarraf C, Noda M, Farthing MJ, Pignatelli M, Adams JC (2003). Fascin, an actin-bundling protein, modulates colonic epithelial cell invasiveness and differentiation in vitro. Am J Pathol. 162(1): 69–80.
- Jiang W, Hiscox S (1997). beta-catenin-cell adhesion and beyond. Int J Oncol. 11(3): 635–641.
- Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K, Akiyama T (2002). Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat Cell Biol. 4(4): 323–327.
- Johnson SK, Ramani VC, Hennings L, Haun RS Johnson SK (2007). Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer. 109(9): 1811–1820.
- Kawasaki Y, Sato R, Akiyama T (2003). Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol. 5(3): 211–215.
- Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y, Higuchi O, Akiyama T (2000). Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 289(5482): 1194–1197.
- Kinsella AR, Lepts GC, Hill CL, Jones M (1994). Reduced E-cadherin expression correlates with increased invasiveness in colorectal carcinoma cell lines. Clin Exp Metastasis. 12(4): 335–342.
- Kinzler KW, Vogelstein B (1997). Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 386(6627): 761–763.
- Klucky B, Mueller R, Vogt I, Teurich S, Hartenstein B, Breuhahn K, Flechtenmacher C, Angel P, Hess J (2007). Kallikrein 6 induces E-cadherin shedding and promotes cell proliferation, migration, and invasion. Cancer Res. 67(17): 8198–8206.
- Korpal M, Lee ES, Hu G, Kang Y (2008). The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 283(22): 14910–14914.
- Kureishy N, Sapountzi V, Prag S, Anilkumar N, Adams JC (2002). Fascins, and their roles in cell structure and function. Bioessays. 24(4): 350–361.
- Li LC, Chui RM, Sasaki M, Nakajima K, Perinchery G, Au HC, Nojima D, Carroll P, Dahiya R (2000). A single nucleotide polymorphism in the E-cadherin gene promoter alters transcriptional activities. Cancer Res. 60(4): 873–876.
- Li QQ, Chen ZQ, Cao XX, Xu JD, Xu JW, Chen YY, Wang WJ, Chen Q, Tang F, Liu XP, Xu ZD (2011). Involvement of NF-κB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial-mesenchymal transition of breast cancer cells. Cell Death Differ. 18(1): 16–25.
- Li T, Li D, Sha J, Sun P, Huang Y (2009). MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem Biophys Res Commun. 383(3): 280–285.
- Logan CY, Nusse R (2004). The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 20: 781–810.
- Lugli A, Zlobec I, Minoo P, Baker K, Tornillo L, Terracciano L, Jass JR (2007). Prognostic significance of the wnt signalling pathway molecules APC, beta-catenin and E-cadherin in colorectal cancer: a tissue microarray-based analysis. Histopathology. 50(4): 453–464.
- Mahmoud NN, Boolbol SK, Bilinski RT, Martucci C, Chadburn A, Bertagnolli MM (1997). Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 57(22): 5045–5050.
- Mayer B, Johnson JP, Leitl F, Jauch KW, Heiss MM, Schildberg FW, Birchmeier W, Funke I 1993. E-cadherin expression in primary and metastatic gastric cancer: down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 53(7): 1690–1695.
- McCartney BM, Näthke IS (2008). Cell regulation by the Apc protein Apc as master regulator of epithelia. Curr Opin Cell Biol. 20(2): 186–193.
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T 2007. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 133(2): 647–658.
- Mimori-Kiyosue Y, Shiina N, Tsukita S 2000. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J Cell Biol. 148(3): 505–518.
- More H, Humar B, Weber W, Ward R, Christian A, Lintott C, Graziano F, Ruzzo AM, Acosta E, Boman B, Harlan M, Ferreira P, Seruca R, Suriano G, Guilford P 2007. Identification of seven novel germline mutations in the human E-cadherin (CDH1) gene. Hum Mutat. 28(2): 203.
- Nabeshima K, Shimao Y, Inoue T, Koono M 2002. Immunohistochemical analysis of IQGAP1 expression in human colorectal carcinomas: its overexpression in carcinomas and association with invasion fronts. Cancer Lett. 176(1): 101–109.
- Najy AJ, Day KC, Day ML 2008. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem. 283(26): 18393–18401.
- Nakamura T, Mitomi H, Kikuchi S, Ohtani Y, Sato K 2005. Evaluation of the usefulness of tumor budding on the prediction of metastasis to the lung and liver after curative excision of colorectal cancer. Hepatogastroenterol. 52(65): 1432–1435.
- Nelson WJ 2009. Remodeling epithelial cell organization: transitions between front-rear and apical-basal polarity. Cold Spring Harb Perspect Biol. 1(1): a000513.
- Noë V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M 2001. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 114(Pt 1): 111–118.
- Okada K, Bartolini F, Deaconescu AM, Moseley JB, Dogic Z, Grigorieff N, Gundersen GG, Goode BL 2010. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J Cell Biol. 189(7): 1087–1096.
- Pálmer HG, Larriba MJ, García JM, Ordóñez-Morán P, Peña C, Peiró S, Puig I, Rodríguez R, de la Fuente R, Bernad A, Pollán M, Bonilla F, Gamallo C, de Herreros AG, Muñoz A (2004). The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 10(9): 917–919.
- Peinado H, Olmeda D, Cano A 2007. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 7(6): 415–428.
- Peña C, García JM, Larriba MJ, Barderas R, Gómez I, Herrera M, García V, Silva J, Domínguez G, Rodríguez R, Cuevas J, de Herreros AG, Casal JI, Muñoz A, Bonilla F 2009. SNAI1 expression in colon cancer related with CDH1 and VDR downregulation in normal adjacent tissue. Oncogene. 28(49): 4375–4385.
- Peña C, García JM, Silva J, García V, Rodríguez R, Alonso I, Millán I, Salas C, de Herreros AG, Muñoz A, Bonilla F 2005. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 14(22): 3361–3370.
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 392(6672): 190–193.
- Perotti A, Sessa C, Mancuso A, Noberasco C, Cresta S, Locatelli A, Carcangiu ML, Passera K, Braghetti A, Scaramuzza D, Zanaboni F, Fasolo A, Capri G, Miani M, Peters WP, Gianni L 2009. Clinical and pharmacological phase I evaluation of Exherin (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann Oncol. 20(4): 741–745.
- Pino MS, Kikuchi H, Zeng M, Herraiz MT, Sperduti I, Berger D, Park DY, Iafrate AJ, Zukerberg LR, Chung DC 2010. Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterol. 138(4): 1406–1417.
- Pittman AM, Twiss P, Broderick P, Lubbe S, Chandler I, Penegar S, Houlston RS 2009. The CDH1-160C > A polymorphism is a risk factor for colorectal cancer. Int J Cancer. 125(7): 1622–1625.
- Puppa G, Maisonneuve P, Sonzogni A, Masullo M, Chiappa A, Valerio M, Zampino MG, Franceschetti I, Capelli P, Chilosi M, Menestrina F, Viale G, Pelosi G 2007. Independent prognostic value of fascin immunoreactivity in stage III–IV colonic adenocarcinoma. Br J Cancer. 96(7): 1118–1126.
- Qualtrough D, Singh K, Banu N, Paraskeva C, Pignatelli M 2009. The actin-bundling protein fascin is overexpressed in colorectal adenomas and promotes motility in adenoma cells in vitro. Br J Cancer. 101(7): 1124–1169.
- Raftopoulos I, Davaris P, Karatzas G, Karayannacos P, Kouraklis G 1998. Level of alpha-catenin expression in colorectal cancer correlates with invasiveness, metastatic potential, and survival. J Surg Oncol. 68(2): 92–99.
- Ramírez N, Bandrés E, Navarro A, Pons A, Jansa S, Moreno I, Martínez-Rodenas F, Zárate R, Bitarte N, Monzó M, García-Foncillas J 2008. Epigenetic events in normal colonic mucosa surrounding colorectal cancer lesions. Eur J Cancer. 44(17): 2689–2695.
- Reilein A, Nelson WJ 2005. APC is a component of an organizing template for cortical microtubule networks. Nat Cell Biol. 7(5): 463–473.
- Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z 1994. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes, Mol Cell Biol. 14: 8333–8342.
- Richards FM, McKee SA, Rajpar MH, Cole TR, Evans DG, Jankowski JA, McKeown C, Sanders DS, Maher ER 1999. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 8(4): 607–610.
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS 1995. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 92(19): 8813–8817.
- Rios-Doria J, Day KC, Kuefer R, Rashid MG, Chinnaiyan AM, Rubin MA, Day ML 2003. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J Biol Chem. 278(2): 1372–1379.
- Ropponen, K.M., Eskelinen MJ, Lipponen PK, Alhava EM, Kosma VM 1999. Reduced expression of alpha catenin is associated with poor prognosis in colorectal carcinoma. J Clin Pathol. 52(1): 10–16.
- Sánchez-Tilló E, Lázaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A 2010. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 29(24): 3490–3500.
- Sanz-Moreno V, Marshall CJ 2010. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol. 22(5): 690–696.
- Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC 2008. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 299(4): 425–436.
- Schmalhofer O, Brabletz S, Brabletz T 2009. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 28(1–2): 151–166.
- Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, Wells W, Kauppinen S, Cole CN 2007. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 67(24): 11612–11620.
- Serova M, Astorgues-Xerri L, Bieche I, Albert S, Vidaud M, Benhadji KA, Emami S, Vidaud D, Hammel P, Theou-Anton N, Gespach C, Faivre S, Raymond E 2010. Epithelial-to-mesenchymal transition and oncogenic Ras expression in resistance to the protein kinase Cbeta inhibitor enzastaurin in colon cancer cells. Mol Cancer Ther. 9(5): 1308–1317.
- Shao J, Mark Evers B, Sheng H 2004. Roles of Phosphatidylinositol 3-Kinase and Mammalian Target of Rapamycin/p70 Ribosomal Protein S6 Kinase in K-Ras-Mediated Transformation of Intestinal Epithelial Cells. Cancer Res. 64: 229–235.
- Sharma M, Leung L, Brocardo M, Henderson J, Flegg C, Henderson BR 2006. Membrane localization of adenomatous polyposis coli protein at cellular protrusions: targeting sequences and regulation by beta-catenin. J Biol Chem. 281(25): 17140–17149.
- Shen Y, Hirsch DS, Sasiela CA, Wu WJ 2008. Cdc42 regulates E-cadherin ubiquitination and degradation through an epidermal growth factor receptor to Src-mediated pathway. J Biol Chem. 283(8): 5127–5137.
- Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Oku N, Miyazawa K, Kitamura N, Takeichi M, Ito F 1994. Tyrosine phosphorylation of beta-catenin and plakoglobin enhanced by hepatocyte growth factor and epidermal growth factor in human carcinoma cells. Cell Adhes Commun. 1(4): 295–305.
- Shibuya H, Iinuma H, Shimada R, Horiuchi A, Watanabe T 2010. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncol. 79(3–4): 313–320.
- Shintani Y, Fukumoto Y, Chaika N, Grandgenett PM, Hollingsworth MA, Wheelock MJ, Johnson KR 2008. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int J Cancer. 122(1): 71–77.
- Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, Takano S, Miyazaki M 2006. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer. 94(12): 1816–1822.
- Shiozaki H, Oka H, Inoue M, Tamura S, Monden M 1996. E-cadherin mediated adhesion system in cancer cells. Cancer. 77(8 Suppl): 1605–1613.
- Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo HH 2007. MiR-21-mediated tumor growth, Oncogene. 26; pp. 2799–2803.
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J 2009. A gene expression signature associated with ‘K-Ras addiction’ reveals regulators of EMT and tumor cell survival. Cancer Cell. 15(6): 489–500.
- Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T 2006. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterol. 131(3): 830–840.
- St Croix B, Sheehan C, Rak JW, Flørenes VA, Slingerland JM, Kerbel RS 1998. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J Cell Biol. 142(2): 557–571.
- Strathdee, G 2002. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol. 12(5): 373–379.
- Strumane K, Berx G, Van Roy F 2004. Cadherins in cancer. Handb Exp Pharmacol. 165: 69–103.
- Takeichi M 1991. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 251(5000): 1451–1455.
- Tanaka M, Hashiguchi Y, Ueno H, Hase K, Mochizuki H 2003. Tumor budding at the invasive margin can predict patients at high risk of recurrence after curative surgery for stage II, T3 colon cancer. Dis Colon Rectum. 46(8): 1054–1059.
- Tetsu O, McCormick F 1999. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 398: 422–426.
- Thiery JP, Acloque H, Huang RY, Nieto MA 2009. Epithelial-mesenchymal transitions in development and disease. Cell. 139(5): 871–890.
- Thiery JP 2002. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2(6): 442–454.
- Trotter KW, Archer TK 2008. The BRG1 transcriptional coregulator. Nucl Recept Signal. 6.
- Tsai WC, Chao YC, Sheu LF, Chang JL, Nieh S, Jin JS 2007. Overexpression of fascin-1 in advanced colorectal carcinoma: tissue microarray analysis of immunostaining scores with clinicopathological parameters. Dis Markers. 23: 153–160.
- Tsukamoto H, Shibata K, Kajiyama H, Terauchi M, Nawa A, Kikkawa F 2007. Irradiation-induced epithelial-mesenchymal transition (EMT) related to invasive potential in endometrial carcinoma cells. Gynecol Oncol. 107(3): 500–504.
- Ueno H, Murphy J, Jass JR, Mochizuki H, Talbot IC 2002. Tumour ‘budding’ as an index to estimate the potential of aggressiveness in rectal cancer. Histopathol. 40(2): 127–132.
- Valizadeh A, Karayiannakis AJ, el-Hariry I, Kmiot W, Pignatelli M 1997. Expression of E-cadherin-associated molecules (alpha-, beta-, and gamma-catenins and p120) in colorectal polyps. Am J Pathol. 150(6): 1977–1984.
- van Roy F, Berx G 2008. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 65(23): 3756–3788.
- Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G 2004. IP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 33(20): 6566–6578.
- Vécsey-Semjén B, Becker KF, Sinski A, Blennow E, Vietor I, Zatloukal K, Beug H, Wagner E, Huber LA 2002. Novel colon cancer cell lines leading to better understanding of the diversity of respective primary cancers. Oncogene. 21(30): 4646–4662.
- Velikova G, Banks RE, Gearing A, Hemingway I, Forbes MA, Preston SR, Hall NR, Jones M, Wyatt J, Miller K, Ward U, Al-Maskatti J, Singh SM, Finan PJ, Ambrose NS, Primrose JN, Selby PJ 1998. Serum concentrations of soluble adhesion molecules in patients with colorectal cancer. Br J Cancer. 77(11): 1857–1863.
- Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, van Roy F 1991. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 66(1): 107–119.
- Volmer MW, Stühler K, Zapatka M, Schöneck A, Klein-Scory S, Schmiegel W, Meyer HE, Schwarte-Waldhoff (2005). Differential proteome analysis of conditioned media to detect Smad4 regulated secreted biomarkers in colon cancer. Proteomics. 5(10): 2587–2601.
- Wali RK, Kunte DP, Koetsier JL, Bissonnette M, Roy HK 2008. Polyethylene glycol-mediated colorectal cancer chemoprevention: roles of epidermal growth factor receptor and Snail. Mol Cancer Ther. 7(9): 3103–3111.
- Wang X, Ling MT, Guan XY, Tsao SW, Cheung HW, Lee DT, Wong YC 2004. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogen. 23: 474–482.
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K 2004. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 7(6): 871–883.
- Weiß JV, Klein-Scory S, Kübler S, Reinacher-Schick A, Stricker I, Schmiegel W, Schwarte-Waldhoff I 2011. Soluble E-cadherin as a serum biomarker candidate: elevated levels in late stage colorectal carcinoma patients and in FAP. Int J Cancer. 128(6): 1384–1392.
- Wells A, Yates C, Shepard CR 2008. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis. 25(6): 621–628.
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG 2004. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 6(9): 820–830.
- Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF 2001. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 48(3): 367–371.
- Wijnhoven BP, Pignatelli M 1999. E-cadherin-catenin: more than a ‘sticky’ molecular complex. Lancet. 354(9176): 356–357.
- Williams E, Williams G, Gour BJ, Blaschuk OW, Doherty P 2000. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 275(6): 4007–4012.
- Wilmanns C, Grossmann J, Steinhauer S, Manthey G, Weinhold B, Schmitt-Gräff A, von Specht BU 2004. Soluble serum E-cadherin as a marker of tumour progression in colorectal cancer patients. Clin Exp Metastasis. 21(1): 75–78.
- Wong NA, Pignatelli M 2002. Beta-catenin--a linchpin in colorectal carcinogenesis? Am J Pathol. 160(2): 389–401.
- Wu WK, Law PT, Lee CW, Cho CH, Fan D, Wu K, Yu J, Sung JJ 2011. MicroRNA in colorectal cancer: from benchtop to bedside. Carcinogenes. 32(3): 247–253.
- Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ, Ellis L 2006. Mascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 66(1): 46–51.
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 117(7): 927–939.
- Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ 2008. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 10(3): 295–305.
- Yates CC, Shepard CR, Stolz DB, Wells A 2007. Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer. 96(8): 1246–1252.
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT 1996. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 10(12): 1443–1454.