Abstract
Inflammation is a central feature of asthma and chronic obstructive pulmonary disease (COPD). Despite recent advances in the knowledge of the pathogenesis of asthma and COPD, much more research on the molecular mechanisms of asthma and COPD are needed to aid the logical development of new therapies for these common and important diseases, particularly in COPD where no effective treatments currently exist. In the future the role of the activation/repression of different transcription factors and the genetic regulation of their expression in asthma and COPD may be an increasingly important aspect of research, as this may be one of the critical mechanisms regulating the expression of different clinical phenotypes and their responsiveness to therapy, particularly to anti-inflammatory drugs.
INTRODUCTION
Inflammation is a central feature of asthma and chronic obstructive pulmonary disease (COPD). The precise nature of the inflammatory response in each disease and the predominant localization within the airways of inflammation differ, but both involve activation and alteration in the structural cells of the airways and the lungs (airway and lung remodelling) and the activation and/or recruitment of infiltrating inflammatory cells (CitationBarnes, 2008a; CitationCaramori et al., 2005; GINA, 2011; GOLD, Citation2011). Asthma and COPD are both characterized by enhanced expression of pro-inflammatory proteins including cytokines, chemokines, growth factors, neuropeptides, enzymes, receptors, and adhesion molecules and reduced expression of anti-inflammatory mediators, such as interleukin (IL)-10 (CitationBarnes et al., 1998; CitationBarnes, 2004a, Citation2008b, Citation2009, Citation2011a, Citation2011b). This altered expression is generally associated with concomitant changes in gene expression profiles in a cell-specific manner during the inflammatory process (CitationBarnes, 2006a, Citation2006b).
Changes in gene transcription are controlled by transcription factors, which are proteins that regulate and activate the transcriptional response in a DNA-dependent manner. Some transcription factors in addition to their regulation of homeostatic genes control the expression of many inflammatory genes and may therefore play a key role in the pathogenesis of asthma and COPD (CitationBarnes et al., 1998; CitationBarnes, 2004b, Citation2008b, Citation2009, Citation2011a, Citation2011b).
In addition, individual transcription factors may also amplify and perpetuate the inflammatory process by modulating either the DNA-binding capacity of other transcription factors or their ability to activate the basal transcription machinery and may thereby contribute to determine disease severity and response to treatment.
Increased understanding of the role of transcription factors and of the signaling pathways leading to their activation in the pathogenesis of asthma and COPD has also provided an opportunity for the development of new potential anti-inflammatory drugs (CitationBarnes, 2012a, Citation2012b). Several new compounds targeting these pathways and/or transcription factors are now in development for the treatment of asthma and COPD. Furthermore, some drugs already in clinical use, such as glucocorticoids, act through their own transcription factor, the glucocorticoid receptor (GR), to control the expression of inflammatory and anti-inflammatory genes (CitationAdcock et al., 2012; CitationBarnes, 2012a, Citation2012b). Glucocorticoids may also have a beneficial effect on COPD exacerbations, although this is less marked than in asthma, highlighting the importance of how distinct cell-and context-specific patterns of transcription factor action and subsequent gene expression profiles control asthma and COPD inflammation.
One concern about this approach is the specificity of such drugs, but it is clear that transcription factors have selective effects on gene expression profiles and this may make it possible to be more selective. In addition, there are cell-specific transcription factors that may be targeted for inhibition, which could provide selectivity of drug action. One such example is GATA-3, which has been reported to have a restricted cellular distribution (CitationCaramori et al., 2001). In asthma and COPD it may be possible to selectively target drugs to the airways by inhalation, as is currently performed for inhaled glucocorticoids, to decrease the potential for systemic effects.
Despite the fact that many transcription factors have now been discovered there is still a paucity of data concerning the regulation of transcription factors in the human lung and airway cells in asthma and COPD. This review briefly summarizes the physiological function of the transcription factors in the normal cells and the role of some transcription factors that may be relevant in the pathogenesis of asthma and COPD.
TRANSCRIPTION FACTORS
Transcription factors generally bind to DNA-regulatory sequences (enhancers and silencers), localized in the 5’-upstream region of target genes, to modulate the rate of gene transcription.
However, genome-wide analysis using chromatin immunoprecipitation-deep sequencing (ChIP-Seq) approaches has indicated that some transcription factors bind at very distal 3’ and 5’ to transcriptional start sites or even within coding regions (CitationLanglais et al., 2012). This results in increased or decreased gene transcription and usually altered cellular function. Many transcription factors have now been identified and a large proportion of the human genome appears to code for these proteins (CitationMaher, 2012).
Transcription factors are classified into several classes and members of each class share structural characteristics [for a detailed description please refer to the “transcription factor encylopedia” (CitationYusuf et al., 2012) at http://www.cisreg.ca/tfe; and to the “transcription factor catalogue” (CitationFulton et al., 2009) at http://www.tfcat.ca]. These groups include (with many families and members inside of each group):
helix-turn-helix group (e.g. Oct-1),
winged helix-turn-helix (e.g. Foxp3 and interferon regulatory factors),
zinc-coordinating group (e.g. glucocorticoid receptors, GATA proteins),
zipper-type group [e.g. cyclic AMP response element-binding factor (CREB), activator protein-1 (AP-1)],
other alpha-helix group (e.g. SOX proteins),
beta-sheet group [e.g. nuclear factor-κB (NF-κB)],
beta-hairpin-ribbon group (e.g. the T box proteins),
other groups (such as the STATs proteins),
enzyme group (DNA polymerase-beta family), and
unclassified structure group.
Many transcription factors are common to all inflammatory cell types (ubiquitous), such as AP-1 and NF-κB, and may play a general role in the regulation of inflammatory genes, whereas others are much more cell-specific and may determine the phenotypic characteristics of a cell, such as FoxP3. Activation of transcription factors generally involves kinase activation pathways including protein kinase A (PKA), mitogen-activated protein (MAP) kinases (MAPK)s, Janus kinases (JAKs), and protein kinases C family members (PKCs) stimulated by cell-surface receptors (CitationGilmore, 2006; CitationSaklatvala, 2004) ( and ).
Figure 1. Multiple pathways mediating transcription factor modulation of inflammatory genes. (A) Inflammatory mediator signal transduction activation. The binding of cytokines, growth factors or chemokines to their respective receptors sets in train the activation of a number of signal transduction pathways, including the receptor tyrosine kinases, mitogen-activated protein kinases (MAPKs, including MEKK1 and JNK), Janus kinases (JAKs) and other kinase pathways involved in NF-κB activation. Activation of nuclear factor-κB involves phosphorylation of the inhibitory protein IκB by specific kinases(s), with subsequent ubiquitination and proteolytic degradation by the proteasome. The free NF-κB then translocates to the nucleus, where it binds to κB sites in the promoter regions of inflammatory genes. Activation of the IκB gene results in increased synthesis of IκB to terminate the activation of NF-κB. (B) JAK-STAT pathways. Cytokine binding to its receptor results in activation of JAK which phosphorylates intracellular domains of the receptor, resulting in phosphorylation of signal transduction-activated transcription factors (STATs). Activated STATs dimerize and translocate to the nucleus where they bind to recognition elements on certain genes. (C and D) Nuclear factor of activated T-cells (NF-AT) is activated via dephosphorylation by calcineurin (CaN) and translocates to the nucleus where it interacts with AP-1 to induce gene transcription. (E) Classical mechanism of steroid action. Glucocorticoids are lipophilic molecules which diffuse readily through cell membranes to interact with cytoplasmic receptors. Upon ligand binding receptors are activated and translocate into the nucleus where they bind to specific DNA elements. The foregoing pathways can interact so that the final signal may be amplified or altered depending upon the exact combination of stimuli. The final response to each stimulus or combination of stimuli by a particular cell depends upon the receptors present in a particular cell along with the exact intracellular transduction pathway activated.
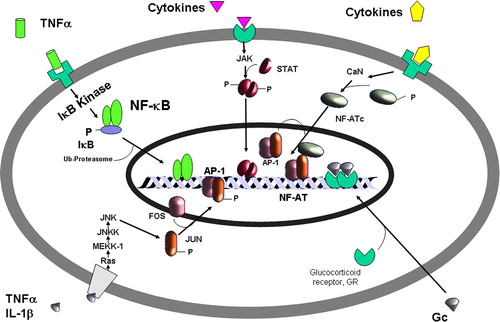
Figure 2. Mechanism of activation of the signal transduction-activated transcription factors (STATs) pathway. Different STATs are activated by different pro-inflammatory cytokines. Cytokine binding to its receptor results in activation of different Janus kinases (JAKs) that phosphorylate intracellular domains of the receptor, resulting in phosphorylation of STATs. Activated STATs dimerize and translocate to the nucleus where they bind to recognition elements on certain responsive genes (please refer to the text for a more detailed description). GM-CSF: granulocyte-macrophage colony stimulating factor; IL: interleukin; IFN: interferon; mRNA: messenger ribonucleic acid.
Transcription factors may also be directly activated by their ligands such as members of the nuclear hormone receptor family including glucocorticoids, estrogens, progesterone, thyroid hormones and vitamins A and D (CitationIto et al., 2006a).
The activity of most transcription factors is regulated by post-translational modifications including phosphorylation, acetylation, methylation and nitration. These can alter either transcription factor DNA-binding capacity or the ability to interact with other proteins and thereby modulate transcriptional activity (CitationPerkins, 2006). This is exemplified by how the myriad of post-translational modifications that occur in the subunit p65 of the nuclear factor (NF)-κB, including phosphorylation and acetylation, which alter its association with transcriptional coactivator and repressor complexes resulting in changes in gene expression profile (CitationChen et al., 2001; CitationOgawa et al., 2005; CitationZhong et al., 2002).
These modifications may occur within the nucleus, often with the transcription factor already bound to DNA, or within the cytoplasm, resulting, for example, in exposure of nuclear localization signals and targeting to the nucleus (CitationIto et al., 2006a).
The drivers for these post-translational modifications are generally extracellular signals such as cytokines and chemokines or stressors such as oxidative/nitrosative stress.
Transcription factors therefore convert cellular exposure to transient environmental signals at the cell surface into long-term changes in gene transcription and alterations in cell function.
Cross-talk between transcription factors
Most transcription factors do not act in isolation and physically interact either with each other to form homodimers or with other transcription factors to form heterodimers. This results in inhibition or enhancement of transcriptional activity. This may occur at the DNA-binding site of one of the heterodimers partners with DNA association at a distinct site from the consensus target for the other transcription factor through a process known as tethering, in a cell- and/or context-dependent manner () (CitationAdcock & Caramori, 2001; CitationLanglais et al., 2012). This cross-talk between different transcription factors results in the complex regulation of gene expression patterns by signal transduction pathways. Generally it is necessary to have coincident activation of several transcription factors in order to achieve maximal gene expression. This may explain how ubiquitous transcription factors only may regulate particular subsets of genes in each specific cell type (CitationBarnes, 2006a, Citation2006b; CitationPerkins, 2006).
This complexity in the activation pathways and their ability to engage in cross-talk at the many levels enables cells to overcome inhibition of one pathway and retain a capacity to activate specific transcription factors and cell specific gene expression profiles.
This cross-talk and redundancy may hinder the search for novel anti-inflammatory agents targeted toward specific transcription factor pathways.
Binding of transcription factors to their specific binding motifs in the regulatory regions of a gene may alter transcription by interacting directly with components of the basal transcription apparatus or via cofactors that link the transcription factor to the basal transcription apparatus (CitationLi et al., 2007). Large proteins that bind to the basal transcription apparatus bind many transcription factors and thus act as integrators of gene transcription. These coactivator molecules include CREB-binding protein (CBP), and the related p300, thus allowing further complex interactions between different signaling pathways to exist (CitationLi et al., 2007).
Histone acetylation and deacetylation in the regulation of gene transcription
The 2–3 m of DNA contained in each cell must be packaged 50,000 times to enable them to fit inside the nucleus. It does this by being wrapped around histone proteins to form nucleosomes and the chromatin fibre in chromosomes (CitationKouzarides, 2007).
This however, then presents problems about how to access the tightly packaged DNA to read the genetic information that it contains.
It has long been recognized at a microscopic level that chromatin may become dense or opaque owing to the winding or unwinding of DNA around the histone core (CitationAllfrey et al., 1964). CBP, p300, and other coactivators, such as p300/CBP-associated factor (PCAF) or K(lysine) acetyltransferase 2A (KAT2A) and histone acetyltransferase 1 (HAT1), have histone acetylase activity (HAT) which is activated by the binding of general pro-inflammatory transcription factors, such as AP-1, NF-κB, and STATs () (CitationKouzarides, 2007; CitationLi et al., 2007).
Figure 3. Histone acetylation by proinflammatory transcription factors. In response to stress and other stimuli, such as cytokines, various second messenger systems are upregulated, leading to activation of signal-dependent transcription factors such as CREB, NF-κB, AP-1 and STAT proteins. Binding of these factors leads to recruitment of CBP and/or other coactivators to signal-dependent promoters and acetylation of histones by an intrinsic acetylase activity (HAT). Induction of histone acetylation allows the formation of a more loosely packed nucleosome structure which enables access to TATA box-binding protein (TBP) and associated factors (TAFs) and the recruitment of further remodeling factors including switch/sucrose nonfermentable (SWI/SNF). Remodeling thereby allows RNA polymerase II recruitment and the activation of inflammatory gene transcription.
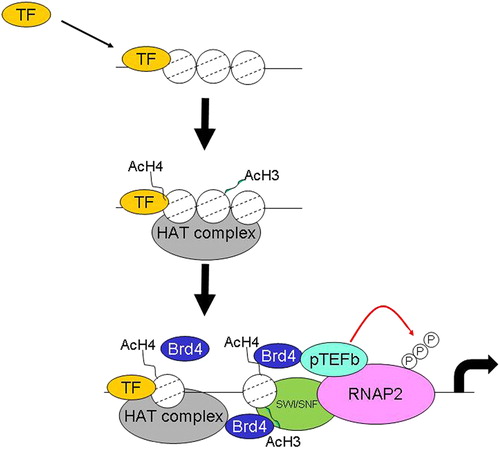
Acetylation of histone residues results in an alteration in the charge between DNA and histones and also leaves a tag that enables the recruitment of proteins that bind acetylated histones (bromodomain-containing proteins). Bromodomains proteins read the acetylated histone tag and enable the recruitment of the basal transcriptional apparatus including protein complexes which have DNA remodelling activity.
This leads to local unwinding of DNA coiled around the histone core, thus opening up the chromatin structure, allowing read through of the basal transcription factor complex resulting in increased transcription. These activating marks deposited by histone acetyltransferases (HATs writers) are removed by specific enzymes [histone deacetylases (HDACs), erasers] that reverses this process, leading to gene repression and allow control of gene expression (Citationde Ruijter et al., 2003). Eighteen isoforms of human HDACs (1–18) have been identified (CitationPan et al., 2012). In general, histone acetylation is associated with increased gene transcription whereas, in contrast, hypoacetylation induced by histone deacetylases (HDACs) is associated with reduced transcription or gene silencing. Many other post-translational modifications of histones exist which control gene expression, such as methylation and phosphorylation but they will not be discussed here. Interested readers are directed to excellent reviews in these areas (CitationDawson et al., 2012; CitationKouzarides, 2007).
Role of the transcription factors in the pathogenesis of bronchial asthma
Asthma is “a chronic inflammatory disorder of the airways, in which many cells and cellular elements play a role. The chronic inflammation is associated with airway hyperresponsiveness that leads to recurrent episodes of wheezing, breathlessness, chest tightness, and coughing, particularly at night or in the early morning. These episodes are usually associated with widespread, but variable, airflow limitation within the lung that is often reversible, either spontaneously or with treatment“ (GINA, 2011).
Asthma is a complex chronic inflammatory disease of the lower airways that involves the activation of many inflammatory and structural cells, all of which release pro-inflammatory mediators that result in the typical clinico-pathological changes of asthma including reticular basement membrane fibrosis under the bronchial epithelium, increased thickness (mainly through hyperplasia) of lower airway smooth muscle cells, increased numbers of bronchial blood vessels (neo-angiogenesis) and increased number/volume of mucus-secreting cells in the bronchial mucosa (surface epithelium and glands). These changes, often referred to as airway remodelling (CitationBousquet et al., 2000), may not be fully reversible with current treatments. The chronic airway inflammation of asthma is unique in that the lower airways wall is infiltrated by an increased number of T-lymphocytes of the T-helper (Th) type 2 phenotype, eosinophils, macrophages/monocytes, and mast cells. In addition, an “acute-on-chronic” inflammation may be observed during asthmatic exacerbations, with an increase in the number of eosinophils and sometimes also neutrophils (CitationBarnes et al., 1998; CitationBarnes, 2008a; CitationCaramori et al., 2005). Lower airway structural cells also produce inflammatory mediators thus bronchial/bronchiolar epithelial cells, airway and lung endothelial cells, airway and lung fibroblasts and bronchial/bronchiolar smooth muscle cells show an altered phenotype in asthma and express multiple inflammatory mediators (CitationBarnes et al., 1998; CitationHolgate, 2011).
The role of transcription factors in differentiation of th1/th2 cells
CD4+ T-helper (Th) cells can be divided into three major subsets, termed Th1, Th2, and Th0 based on the pattern of cytokines they produce (CitationNeurath et al., 2002). Th1 cells produce predominantly IL-2 and interferon gamma (IFN-γ) and predominantly promote cell-mediated immune response. Th2 cells, which produce mainly interleukins (IL-4, IL-5, and IL-13), augment certain B-cell responses. IL-4 in particular is the major inducer of B-cell switching to immunoglobulin E (IgE) production, and therefore plays a crucial role in allergic reactions involving IgE and mast cells. Th cells producing cytokines typical of both Th1 and Th2 clones have also been described and they have been named Th0 (CitationCaramori & Adcock, 2003).
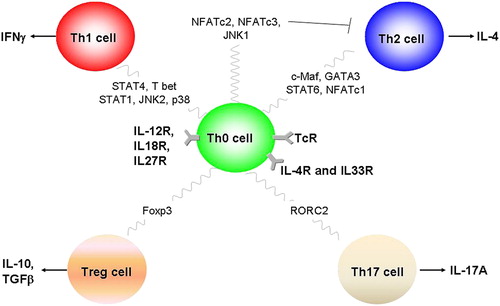
Evidence indicates that Th1 and Th2 cells differentiate from a common precursor, naive T-cells. This appears to be a multistep process, in which naive T-cells pass through an intermediate stage (Th0) at which both Th1 and Th2 cytokines are produced ().
Figure 4. Differentiation of T-cell subtypes depends upon the action of a number of transcription factors. Th0 cells can differentiate into Th1 and Th2 cells following IL-12 or IL-4 stimulation. Stimulation through the IL-12 receptor (IL-12R) activates the transcription factor STAT4 and the MAPK pathways JNK2 and p38 and drives Th1 cell differentiation, leading to cells capable of releasing IFN-γ. In contrast, activation of the IL-4R in concert with T-cell receptor (TCR) stimulation leads to activation of the transcription factors STAT6, GATA3, c-Maf and NFATc1, and drives Th2 cell differentiation. Th2 cells can release a number of cytokines including interleukins 4, 5, 6, 9, 10 and 13. In addition, activation of JNK1, NFATc2 and NFATc3 can prevent Th2 cell differentiation.
Many transcription factors, including GATA-3, T-bet, c-maf, and STAT6, are involved in this process by which Th1 and Th2 cells differentially express the Th1 and Th2 cytokines (CitationZhang et al., 2000; CitationZheng & Flavell, 1997). However, most of these studies were conducted on murine T-cells, and the situation in human T-cells, in physiological and pathological conditions (e.g. bronchial asthma), remains largely unknown.
The publication of the Encode database (http://www.genome.gov/10005107) and recent analysis of transcription factor DNA binding by ChIP-seq in primary human CD4 + T-cells has clearly indicated that transcription factor DNA binding and histone acetylation is associated with the expression of distinct sets of genes (CitationZhang et al., 2012).
A key role for regulatory T-cells (e.g. Treg and Th-17) has also been proposed in asthma (CitationCosmi et al., 2011; CitationMaddur et al., 2012; CitationRay et al., 2010; CitationSilverpil & Lindén, 2012; CitationStrickland & Holt, 2011). Production of the Treg phenotype is associated with the expression of the transcription factor Foxp3 (CitationMarson et al., 2007; CitationRomagnani, 2006) and whose activity is also dependent upon NF-AT (CitationWu et al., 2006). Human Th17 cells are characterized by the expression of the transcription factor RAR-related orphan receptor C (RORC) (CitationIvanov et al., 2006).
TRANSCRIPTION FACTORS AND DETERMINATION OF ASTHMA SEVERITY
Role of the transcription factor NF-κB in the pathogenesis of bronchial asthma
Activation of NF-κB leads to the coordinated induction of multiple genes that are expressed in inflammatory and immune responses. Many of these genes are induced in inflammatory and structural cells and play an important role in the inflammatory process.
While NF-κB is not the only transcription factor involved in regulation of the expression of these genes, it often appears to have a decisive regulatory role. NF-κB often functions in cooperation with other transcription factors, such as AP-1 and C/EBP, which are also involved in regulation of inflammatory and immune genes (CitationBaldwin, 2001; CitationPerkins, 2006). Genes induced by NF-κB include those for the proinflammatory cytokines IL-1β, tumor necrosis factor alpha (TNF-α), and granulocytes-macrophage-colony stimulating factor (GM-CSF) and the chemokines CXCL8 (IL-8), macrophage inflammatory protein-1α (MIP-1α), macrophage chemotactic protein-1 (MCP-1), CCL5 (RANTES), and the eotaxins, that are largely responsible for attracting inflammatory cells into sites of inflammation (CitationBarnes, 2006b; CitationCaramori et al., 2004a). NF-κB also regulates the expression of inflammatory enzymes, including the inducible form of nitric oxide synthase (NOS2) that produces large amounts of nitric oxide (NO). NF-κB also plays an important role in regulating expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), that are expressed on endothelial and epithelial cells at inflammatory sites and play a key role in the initial recruitment of inflammatory cells (CitationBarnes, 2006b; CitationCaramori et al., 2004b). The expression of the receptors for cytokines and chemokines are also under the regulation of NF-κB (CitationPerkins, 2012).
NF-κB may also be activated in vitro by many of the stimuli that exacerbate asthmatic inflammation (e.g. rhinovirus infection, allergen exposure, pro-inflammatory cytokines, and oxidants) (CitationPapi & Johnston, 1999; CitationPoynter et al., 2002; CitationRahman, 2005).
There is also evidence for NF-κB activation in bronchial epithelial and sputum cells of patients with stable asthma (CitationHart et al., 1998), in the peripheral blood mononuclear cells of patients with severe uncontrolled asthma (CitationGagliardo et al., 2003, Citation2011) and in the bronchial and bronchiolar epithelium of patients who died during an asthmatic exacerbation (CitationCaramori et al., 2009). Furthermore rhinovirus infection of asthmatic patients in vivo is associated with enhanced NF-κB p65 nuclear expression in bronchial epithelial cell, increased NF-κB p65 DNA binding in lung tissue and elevated levels of NF-κB-regulated inflammatory mediators (CitationBartlett et al., 2012). Finally, induction of inflammatory mediators by respiratory syncytial virus (RSV) infection of primary human bronchial epithelial cells is also associated with NF-κB activation (CitationHinzey et al., 2011).
Mucoid plugging of the airway lumen is frequently associated with fatal asthma and is associated with increased expression of the mucin MUC5AC in the lung epithelium from patients with fatal asthma (CitationGroneberg et al., 2002). Interestingly, studies suggest an important role for NF-κB-regulated genes in airway epithelium in allergen-induced airway mucus production (CitationBroide et al., 2005). Furthermore, in in vitro experiments both neutrophil elastase and TNF-α induce MUC5AC mucin synthesis via NF-κB activation in airway epithelial cells (CitationCaramori et al., 2009; CitationFujisawa et al., 2009).
NF-κB may be considered, therefore, as an amplifying and perpetuating mechanism that exaggerates the disease-specific inflammatory process through the coordinated expression of multiple inflammatory genes.
The ability of NF-κB to enhance inflammatory gene expression is regulated by post-translational modifications of p65 (CitationPerkins, 2006). However, the degree of NF-κB phosphorylation and acetylation in asthma is unknown as is the specific transcriptome associated with each modified NF-κB (CitationPerkins, 2006).
Inhibitors of the controlling kinase (inhibitor of κB kinase 2, IKK2) are in clinical development for asthma and other inflammatory diseases with the hope of achieving as good an anti-inflammatory effect as glucocorticoids without the side effects (CitationBarnes, 2012a).
Additional benefits of IKK2 inhibition may also be observed independent of NF-κB p65 activation (CitationYan et al., 2013).
However, there are concerns about the potential side effects of long-term therapy with IKK2 inhibitors, such as an increased risk of opportunistic infections. These risks need to be weighed against the potential therapeutic advantages.
In summary, the increased NF-κB activation found in airway cells in patients who have died of status asthmaticus may be involved in the pathogenesis of asthma death through inflammatory cell activation and mucus hypersecretion. This suggests that inhaled inhibitors of NF-κB activation may be beneficial in the treatment of severe exacerbations of asthma (CitationCaramori et al., 2004b).
AP-1
AP-1 is a collection of related transcription factors belonging to the Fos (c-Fos, FosB) and Jun (c-Jun, JunB, JunD) families which dimerize in various combinations through their leucine zipper region. Fos/Jun form heterodimers with high affinity and are the predominant form of AP-1 in most cells, whereas Jun/Jun homodimers bind with low affinity and are less abundant (CitationManning & Davis, 2003). AP-1 may be activated by various cytokines, including TNF-α and IL-1β, via several types of protein tyrosine kinase (PTK) and mitogen-activated protein (MAP) kinases in particular Jun-N-terminal kinase (JNK), which themselves activate a cascade of intracellular kinases (CitationSaklatvala, 2004). AP-1, like NF-κB, regulates many of the inflammatory and immune genes that are overexpressed in asthma. Indeed many of these genes require the simultaneous activation of both transcription factors that work together cooperatively. Interestingly, AP-1 has recently been shown to be involved in glucocorticoid receptor DNA accessibility by regulating chromatin structure (CitationBiddie et al., 2011).
There is evidence for increased expression of c-Fos in bronchial epithelial cells in asthmatic airways (CitationDemoly et al., 1992) and an even greater expression in patients with severe, treatment-insensitive asthma (CitationAdcock & Lane, 2003; CitationLoke et al., 2006), and many of the stimuli relevant to asthma that activate NF-κB will also activate AP-1. Inhibitors of JNK are being developed (CitationManning & Davis, 2003) and these show good effectiveness against bronchial hyperresponsiveness, bronchoalveolar lavage (BAL) inflammatory cells, and airway remodeling in animal models of asthma (CitationNath et al., 2005). Interestingly, JNKs are also involved in T-cell class switching and their inhibitors may also have a profound immunomodulatory role (CitationManning & Davis, 2003).
STATs in the pathogenesis of bronchial asthma
In general, signal transduction and activator of transcription (STAT) family members control T-cell differentiation through regulation of the enhancer landscape (CitationVahedi et al., 2012).
Signal transduction-activated transcription factor-6 (STAT6) also provides a target as a potential treatment for allergic asthma (CitationGoenka & Kaplan, 2011; CitationKuperman & Schleimer, 2008). STAT6 knockout mice have no response to IL-4, do not develop Th2 cells in response to IL-4, and fail to produce IgE, bronchial hyperresponsiveness, or BAL eosinophilia and Treg induction after allergen sensitization indicating the critical role of STAT6 in allergic responses (CitationChapoval et al., 2011; CitationTomkinson et al., 1999). However there are mechanisms of eosinophil mobilization from the bone marrow that are STAT6-independent (CitationWang et al., 2011).
Pendrin-induced expression of MUC5AC, a major mucin in asthma, in bronchial epithelial cells is also STAT6-dependent following activation of the IL-14/IL13 receptor complex (CitationNakao et al., 2008; CitationNofziger et al., 2011). The suppressor of cytokine signaling (SOCS) family of proteins down-regulate specific cytokine signals (CitationInoue et al., 2007). SOCS2 inhibits Th2 development, and its absence enhances STAT5 and STAT6 activation favoring Th2 polarization (CitationKnosp et al., 2011).
In animal models of allergen-induced airway inflammation the expression of the chemokines CCL11, CCL17, and CCL22 involves regulation by STAT6 (CitationFulkerson et al., 2004).
STAT6 has been reported to be overexpressed in bronchial biopsies from asthmatic patients in some (CitationMullings et al., 2001), but not all (CitationTomita et al., 2012) studies. In addition, STAT6 gene polymorphisms are associated with asthma-related symptoms but not with the risk of developing asthma (CitationBeghé et al., 2010; CitationKavalar et al., 2012).
Genistein, a flavonoid in legumes and some herbal medicines, decrease airway inflammation in animal models of allergic asthma and this is associated with a down-regulation of GATA-3 and STAT6 and the up-regulation of T-bet (CitationGao et al., 2012).
Selective STAT6 blockers are also under preclinical development (CitationDubey et al., 2011; CitationMartinez-Nunez et al., 2011; CitationMatsunaga et al., 2011; CitationNagashima et al., 2008; CitationOh et al., 2010; CitationTian et al., 2011; CitationZhou et al., 2012) and some of them are effective in animal models of asthma (CitationChiba et al., 2009a; CitationCortes et al., 2009; CitationDarcan-Nicolaisen et al., 2009; CitationMcCusker et al., 2007; CitationWang et al., 2011), but there is still a complete absence of studies on STAT6 blockers published on human asthmatics.
JAK/STATs inhibitors have proved effective in clinical trials for rheumatoid arthritis and inflammatory bowel disease (CitationFleischmann et al., 2012; CitationSandborn et al., 2012; Citationvan Vollenhoven et al., 2012) but no data are available as yet in asthmatics.
STAT1 has complex and contrasting effects on inflammatory responses which may participate in the pathogenesis of asthma. In vitro in the eosinophil granulocytes stimulation of the cysteinyl leukotriene (CysLT) receptor 1 activates STAT1 through PKC and extracellular- signal-regulated kinases (ERK)1/2 activation, and causes enhanced ICAM-1 surface expression and eosinophil adhesion (CitationProfita et al., 2008). Furthermore, the signal peptide of eosinophil cationic protein (ECPsp) function as a regulator for enhancing macrophage migration through the upregulation of the transcriptional factors STAT1 and STAT2 (CitationLiu et al., 2012). In vitro CCL5 expression in airway epithelial cells is STAT1 dependent (CitationHomma et al., 2010) and IL-4 and IL-13 activate STAT1 in bronchial smooth muscle cells, lung epithelium, endothelium and fibroblasts (CitationChiba et al., 2009b; CitationWang et al., 2004). Thymic stromal lymphopoietin (TSLP) is a cytokine with may be involved in the pathogenesis of bronchial asthma. The TSLP receptor (TSLPR) mediates gene regulation not only through STAT5 and STAT3 but also via STAT1 (CitationWohlmann et al., 2010). In animal models of severe asthma neurokinin-2 receptor (NK2R) expression is STAT1 dependent (CitationKobayashi et al., 2012).
However, in vitro the STAT1-activating cytokines IL-27 and IFN-gamma amplify TGF-β1-induced FoxP3 expression and Treg cells activation (CitationOuaked et al., 2009). Also STAT1 is a critical intracellular signaling molecule for the production of type I interferons (IFNs) (α/β) and IFN-γ and resistance to viral respiratory infections (CitationHashimoto et al., 2005; CitationStrengell et al., 2003).
Suppressor of cytokine signaling (SOCS) 1 gene polymorphisms associated with an increased susceptibility to adult asthma enhance the transcriptional level of SOCS1 in human airway epithelial cells, and induce higher levels of protein expression of SOCS1 and lower phosphorylation of STAT1 stimulated with IFN-beta (CitationHarada et al., 2007).
STAT1 expression is elevated in animal models of asthma (CitationChiba et al., 2011) and in the lower airways of stable asthmatics, but not of COPD patients (CitationDiez et al., 2012; CitationSampath et al., 1999). Other studies, however, did not reproduce these data and were unable to find any significant activation of STAT1 in the bronchial biopsies of patients with stable steroid naïve asthma when compared with healthy controls (CitationLim et al., 2004).
In contrast, baseline phospho-STAT1 and phospho- STAT6 levels are increased in blood CD4 + CD161 + central/effector-memory T cells from steroid naïve asthmatic patients. Systemic glucocorticoids treatment only decreased STAT6, but not STAT1, activation in these cells (CitationGernez et al., 2007).
In animal models of allergen-induced airway inflammation the expression of the chemokines CXCL9 and CXCL10 involves regulation by STAT1 (CitationFulkerson et al., 2004).
Indeed, decoy oligonucleotides against STAT1 are effective in animal models of asthma (CitationLührmann et al., 2010; CitationQuarcoo et al., 2004) and multiple dosing of AVT-01 (Avontec Gmbh), a STAT1 decoy oligonucleotides has been reported to have entered phase II studies in asthma although no results have been published. Orazipone [OR-1384; 3-[4-(methylsulfonyl)benzylidene]pentane-2,4-dione] is a sulfhydryl-modulating compound that has anti-inflammatory properties in experimental models of asthma and in vitro decreases activation of the inflammatory transcription factors NF-κB and STAT1 (CitationSareila et al., 2008).
Interestingly, glucocorticoids can also suppress STAT1 activation (CitationBhattacharyya et al., 2011; CitationDiez et al., 2012).
STAT5 is represented by two similar but distinct proteins: STAT5A and STAT5B (CitationGrimley et al., 1999). In vitro STAT5 activation is necessary and sufficient for IL-2-mediated function in Th2 differentiation, independent of IL-4 signaling; does not up-regulate GATA-3 expression, but does require the presence of GATA-3 for its action (CitationZhu et al., 2006). IL-5 drives bone marrow progenitor cells into IL-4-producing eosinophils by activating STAT5 (CitationZhu et al., 2004) and in human bronchial epithelial cells nitric oxide (NO)-mediated STAT5 activation inhibits both Th1 and Th2 cell proliferation (CitationEriksson et al., 2005). STAT5A is important for the development and proliferation of mast cells (CitationShelburne et al., 2003), whereas STAT5B is a regulator of cytokine production by the same cells (CitationPullen et al., 2012).
There is evidence for a role of granulocyte/macrophage colony stimulating factor (GM-CSF)-activated STAT5 in delaying apoptosis of lung granulocytes in an animal model of asthma (CitationTurlej et al., 2001). Furthermore in another animal model of allergic asthma STAT5 activation is reported in spleen lymphocytes (CitationLi et al., 2004). Finally, in animal models of chronic asthma recombinant human phospholipase D2 suppress the chronic inflammatory reaction through down-regulating PKC expression and STAT1/STAT5a activity in the lungs (CitationCai et al., 2012) but there is still a complete absence of studies on STAT5 blockers published on human asthmatics.
C/EBP (CCAAT/Enhancer binding protein) transcription factor in asthma
Interleukin (IL)-6 is an important inflammatory mediator and targeting this cytokine has proved a clinically effective strategy in rheumatoid arthritis for example (CitationNeurath & Finotto, 2011; CitationO’Shea & Plenge, 2012). IL-6 is thought to be important in asthma pathophysiology and its expression is increased in animal models of asthma (CitationRincon & Irvin, 2012). Levels of sputum IL-6 and BAL soluble IL-6 receptor are increased in patients with severe persistent asthma (CitationHawkins et al., 2012; CitationMorjaria et al., 2011). Importantly, IL-6 activates a number of key transcription factors such as STATS (see above) and members of the CCAAT/enhancer binding protein (C/EBP) family (CitationNeurath & Finotto, 2011).
There is a selective decrease in C/EBPα expression in asthma, particularly in airway smooth muscle cells (CitationRoth et al., 2002, Citation2004), whereas C/EBPδ is decreased in COPD lungs, but with no change in C/EBPα expression (CitationBorger et al., 2007). The reduction in C/EBPα in asthmatic airway smooth muscle cells affects cell proliferation and glucocorticoid responsiveness (CitationRoth et al., 2002, Citation2004).
In contrast, C/EBPβ is associated with the induction of inflammatory genes in both human airway epithelial cells and macrophages (CitationYang et al., 2012). C/EBPβ is bound to the native CXCL8 promoter to a much greater extent in airway smooth muscle cells from asthmatic subjects compared to cells from non-asthmatic control subjects (CitationJohn et al., 2009). This, at least in part, accounts for the hyperproduction of CXCL8 in these cells. C/EBPβ is particularly important in the induction of host defence genes (CitationZhang et al., 2007). These genes, unlike inflammatory genes regulated by NF-κB – either alone or in conjunction with C/EBPβ, are not suppressed by glucocorticoids (CitationJohn et al., 2009; CitationZhang et al., 2007).
NRF2 transcription factor in asthma
The transcription of many antioxidant genes is regulated by the expression and DNA binding of nuclear erythroid 2 p45-related factor 2 [NRF2; also termed nuclear erythroid 2-related factor 2 (NFE2L2)] (CitationRahman & MacNee, 2012). NRF2 is a redox-sensitive basic leucine zipper transcription factor whose expression was initially reported to be reduced in the lungs following allergen exposure in mice and NRF2 knockout mice displayed enhanced allergen-driven airway inflammation and hyperresponsiveness post ovalbumin exposure (CitationRangasamy et al., 2005). Altered oxidant responses have been seen particularly in severe asthma and this has been related to post-translational modifications of NRF2 which altered its ability to switch on anti-oxidant genes (CitationFitzpatrick et al., 2011). NRF2 activation is also involved in inflammatory gene expression following bacterial lipopolysaccharide (LPS)-stimulation of airway epithelial cells and macrophages (CitationYang et al., 2012), controls eotaxin/CCL11 expression and release from airway fibroblasts (CitationFourtounis et al., 2012) and proliferation of airway smooth cells (CitationMichaeloudes et al., 2011).
Other transcription factors in the pathogenesis of bronchial asthma
Cyclosporin A has been used as an immunomodulator for many years although its side effect profile limits its utility in asthma (CitationCaramori & Adcock, 2003). Its target is cyclophilin which controls the dephosphorylation of nuclear factor of activated T-cells (NFAT) and thereby the expression of key Th2 cytokines including IL-13 (CitationBarnes, 2006b; CitationCaramori et al., 2004a).
Sp1 is a ubiquitous transcription factor that binds to GC-boxes and related motifs, which are frequently occurring DNA elements present in many promoters and enhancers. Modification of these sites within the IL-10 and IL-4 and the 5-lipoxygenase promoters has been associated with altered expression of these genes in distinct patient groups (CitationHobbs et al., 1998; CitationLim et al., 1998).
Transcription factors and glucocorticoid action
Glucocorticoids act through their receptor (GR), which belongs to the family of nuclear hormone receptors and are important anti-inflammatory agents used in the treatment of chronic inflammatory diseases such as bronchial asthma (CitationBarnes, 2012b). Functionally they act by suppressing airway hyperresponsiveness, reducing airway edema and the infiltration of inflammatory cells from the blood to the airway and thereby reducing the lower airway inflammatory response (CitationCaramori & Adcock, 2003). Glucocorticoid receptors (GR) are predominantly localized to the airway epithelium, alveolar macrophages and endothelium (CitationAdcock et al., 2012), which are, therefore, probably important sites for the anti-inflammatory action of steroids, especially those delivered by the inhaled route. Airway epithelial cells act as important regulators of the inflammatory reaction, responding to various inflammatory mediators, such as cytokines, by the production of a wide range of cytokines, chemokines, and other inflammatory mediators (CitationIto et al., 2006a).
Glucocorticoids bind to and activate the cytosolic GR which then translocates into the nucleus. Within the nucleus GR can act as a positive transcription factor by forming homodimers of two GR subunits which bind to specific DNA elements (glucocorticoid response elements or GREs) in the promoter regions of glucocorticoid-responsive genes (CitationAdcock et al., 2012). Several genes are upregulated by glucocorticoids, including the β2-adrenergic receptor, MAPK phosphatase (MKP1) and serum leukoprotease inhibitor (SLPI). However, in inflammation the major role of glucocorticoids appears to be gene repression. The major anti-inflammatory action of glucocorticoids is the repression of the pro-inflammatory actions of NF-κB resulting in a reduction in inflammatory gene transcription (CitationIto et al., 2006a).
This requires the action of a GR monomer which either tethers to pro-inflammatory transcription factors such as NF-κB and recruiting transcriptional co-repressors (CitationUhlenhaut et al., 2013) or by binding to negative GREs (CitationSurjit et al., 2011) forming a complex that is structurally and spatially unable to dimerize (CitationHudson et al., 2013).
Although the major effect of glucocorticoids is on gene transcription, some effects on messenger ribonucleic acid (mRNA) stability do occur with specific genes under the control of MKP-1 (CitationAbraham et al., 2006). Several molecular mechanisms for the suppressive actions of the activated GR on NF-κB-mediated gene transcription have been proposed, but it is likely that inhibition of inflammatory mediator-induced histone H3 and H4 acetylation leads to changes in the ability of RNA polymerase 2 to transcribe mRNA (CitationBilodeau et al., 2006; CitationIto et al., 2000, Citation2006a; CitationLuecke & Yamamoto, 2005). This effect on histone acetylation is achieved either by a direct inhibition of CBP-associated histone acetylation via recruitment of HDAC2 to the activated DNA-bound NF-κB complex (CitationIto et al., 2000; CitationUhlenhaut et al., 2013) () or by modulation of RNA polymerase 2 phosphorylation and transcription re-initiation.
Figure 5. The actions of the dexamethasone/GR complex on inhibition of IL-1β stimulated histone acetylation. DNA-bound p65 induces histone acetylation via activation of CBP and a CBP-associated HAT complex to occur. This causes local unwinding of DNA and increased gene transcription. GR, acting as a monomer, interacts with CBP causing an inhibition of CBP-mediated HAT activity. In addition, GR also recruits HDAC2 to the p65/CBP complex, further reducing local HAT activity, leading to enhanced nucleosome compaction and repression of transcription.
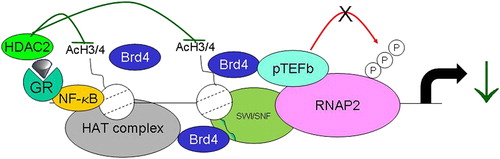
Oxidative stress down-regulates HDAC activity and expression and this is linked to a reduction in the ability of GR to suppress the expression of some inflammatory mediators and reduce glucocorticoid responsiveness (CitationIto et al., 2001). We have previously reported that the expression of HDAC2 is reduced in the lung of patients with stable COPD (CitationIto et al., 2005) and that overexpression of HDAC2 restores glucocorticoid sensitivity in BAL macrophages from these patients (CitationIto et al., 2006b). Furthermore, the failure of glucocorticoids to regulate pro-opiomelanocortin (POMC) expression in Cushing's disease has also been related to a lack of HDAC2 (CitationBilodeau et al., 2006) and targeting of these enzymes may provide another novel target for future drug discovery.
Glucocorticoids are able to activate the expression of a number of anti-inflammatory genes due to GR binding to specific GREs in the promoter regions of responsive genes (CitationIto et al., 2006a). This is often in conjunction with other transcription factors and activation of CAAT/enhancer-binding protein (C/EBPα) by long-acting β2-agonists in airway smooth muscle cells has been proposed to account, at least in part, for the enhanced effect of this combination treatment in asthmatic patients (CitationRoth et al., 2004). Loss of C/EBPα expression in some asthmatic patients may account for a reduction in glucocorticoid effectiveness in these subjects (CitationRoth et al., 2004).
A very small number of asthmatic patients are steroid-resistant and fail to respond to even high doses of oral glucocorticoids (so called steroid-resistant asthma). This defect is also present in their peripheral blood mononuclear cells (PBMCs) and T-lymphocytes and smooth muscle cells (CitationAdcock & Lane, 2003; CitationChang et al., 2012). Although ligand binding data show no change in GR number in these patients, immunocytochemistry indicates a reduced GR nuclear translocation which may account for the reduced binding to DNA seen in these patients (CitationMatthews et al., 2004).
Altered nuclear translocation is affected by GR phosphorylation status (CitationGalliher-Beckley et al., 2011; CitationIrusen et al., 2002), which may also be a target for long-acting β-agonists acting via modulation of protein phosphatase 2A activity which may be reduced in severe asthma (CitationIto et al., 2006a; CitationKobayashi et al., 2011; CitationKobayashi et al., 2012). In the same patients there is a reduced inhibitory effect of glucocorticoids on AP-1 DNA interaction, but not on that of NF-κB. Furthermore, there is an increase in the baseline activity of AP-1 which appears to be due to excessive activation of JNK (CitationKobayashi et al., 2011; CitationLane et al., 1998; CitationSousa et al., 1999) possibly resulting in an alteration in chromatin accessibility at glucocorticoid responsive genes (CitationBiddie et al., 2011). This resistance will be seen at the site of inflammation where AP-1 is activated but not at uninflamed sites. This may explain why patients with steroid-resistant asthma are not resistant to the endocrine and metabolic effects of glucocorticoids, and thus develop the drug's systemic side effects. The situation is probably more complex as different stimuli, including infections and T-cell co-stimulation, can induce a relative glucocorticoid insensitivity in a cell- and context-dependent manner (CitationGalliher-Beckley et al., 2011; CitationGoleva et al., 2002; CitationHinzey et al., 2011; CitationTsitoura & Rothman, 2004) via distinct mechanisms.
TRANSCRIPTION FACTORS IN THE PATHOGENESIS OF COPD
Chronic obstructive pulmonary disease (COPD) is “a common preventable and treatable disease, characterized by persistent airflow limitation that is usually progressive and associated with an enhanced chronic inflammatory response in the airways and the lung to noxious particles or gases. Exacerbations and comorbidities contribute to the overall severity in individual patients “(GOLD, Citation2011).
In contrast to the enormous increase in our understanding of the pathogenesis of asthma, very little is known about the molecular pathogenesis of inflammation in the lower airways of COPD. However, there are accumulating data indicating that the selective activation of different transcription factors and the subsequent induction of distinct transcriptomic patterns play a fundamental role in the pathogenesis of both stable COPD and COPD exacerbations.
Transcription factors and disease susceptibility
The etiology of COPD appears to be due to interactions between environmental factors (particularly cigarette smoking) and genetic factors. Chronic heavy cigarette smoking is currently the cause of more than 90% of cases of COPD in Westernized countries, so environmental factors are clearly very important. However, it is important to identify the factors that determine why only around 25% of chronic heavy cigarette smokers develop at the age of 80 years a symptomatic COPD (CitationGershon et al., 2011). So far, this is little understood, although it is likely that genetic factors are important. There is convincing evidence that several genes influence the development of COPD. In a complex polygene disease such as COPD, multiple genes are operating and the influence of each gene in isolation is probably relatively weak. The susceptibility to develop COPD with smoking is likely to depend on the coincidence of several gene polymorphisms that act together (CitationBarnes & Kleinert, 2004).
The only clearly established, but rare, genetic risk factor for COPD is α1-antitrypsin deficiency (α1-AT) (CitationKalsheker & Morgan, 1994). Approximately 95% of cases of clinical α1-AT deficiency are caused by a single amino acid substitution at position 342 (lysine for glutamic acid) in the coding region of the α1-AT gene (Z allele). This genotype is uncommon, but a Taq1 polymorphism in the 3’-flanking region of the α1-AT gene has been reported to be present in 18% of COPD patients but in only 5% of the general population in the United Kingdom (CitationKalsheker & Morgan, 1994). Although this mutation is not associated with an abnormal α1-AT protein expression, this genetic variant is within an enhancer sequence (which contains C/EBP-binding sites) and may impair the acute-phase increase in α1-AT gene expression in response to IL-6 (CitationMorgan et al., 1997).
In patients with COPD and symptoms of chronic bronchitis there is an increased prevalence of a common polymorphism in the 5’-flanking region of the TNF-α gene, which was previously found to be associated with high baseline and induced TNF-α expression (CitationHuang et al., 1997). Unfortunately, in controlled clinical trials anti-TNFα therapy is ineffective and dangerous (increased risk of infections and neoplasms) in patients with stable COPD (CitationRennard et al., 2007).
Transcription factors and clinical manifestations of COPD
The chronic airflow obstruction in tobacco smoking-related COPD results from a combination of airway inflammation and remodelling, which particularly affects small airways (CitationCaramori et al., 2011), and loss of lung elasticity because of destruction of the lung parenchyma (CitationHogg & Timens, 2009). However, pulmonary emphysema is usually only appearing with increasing severity of the COPD and can also be present in subjects without airflow obstruction (CitationGalbán et al., 2012).
The modulation of the inflammatory response in COPD by genetic factors associated with transcription factors may, in part, cause the different clinical phenotypes of patients with COPD (CitationBarnes, 2006b); however, it is clear that COPD patients have marked inflammation, which increases with disease severity (CitationHogg et al., 2004). Patients with COPD are reported to have increased activation of NF-κB in sputum and lung tissue macrophages and bronchial epithelial cells (CitationCaramori et al., 2003; CitationDi Stefano et al., 2002; CitationSzulakowski et al., 2006). The activation status of NF-κB and its repression by glucocorticoids may also be affected by altered expression of HDAC2 (CitationIto et al., 2005, Citation2006b) and of the protein deacetylase sirtuin (SIRT1) (CitationRajendrasozhan et al., 2008).
Hogg and colleagues (CitationHogg et al., 2004) have shown that the mRNA expression of the transcription factors early growth response (Egr)-1 and c-Fos are increased in bronchial biopsies and that exposure of fibroblasts to cigarette smoke enhanced Egr-1 expression (CitationNing et al., 2004). This results in subsequent induction of matrix metalloprotease (MMP)2 expression in vitro and in vivo (CitationNing et al., 2007). Egr-1 has also been reported to drive autophagy in cigarette smoke-induce COPD (CitationChen et al., 2008).
In contrast to the tissue destruction seen in the alveoli in COPD patients with concomitant pulmonary emphysema, the small airways are characterized by fibrosis and increased extracellular matrix and smooth muscle hypertrophy/hyperplasia. Activation of SMAD family of transcription factors by growth factors, such as TGFβ, may be important in this process and Smad3 knockout mice develop pulmonary emphysema (CitationChen et al., 2005). In addition, expression of the inhibitory SMAD7 is reduced in bronchiolar epithelial cells of stable COPD (Global Initiative for Chronic Obstructive Lung Disease, GOLD stage II) patients whereas basal mRNA expression of SMAD2,3,4 and 7 is not significantly different in the COPD pulmonary fibroblasts compared with controls (CitationZandvoort et al., 2006, Citation2008).
In contrast to asthma, where C/EBPα levels are enhanced, Borger and colleagues have reported a reduction of C/EBPδ in patients with COPD (CitationBorger et al., 2007). Activation of STAT4 is critical for the differentiation of Th1/Tc1 cells T-cells and the production of IFN-γ (CitationDi Stefano et al., 2004). We have reported increased expression of activated phospho-STAT4 in bronchial biopsies and BAL lymphocytes of COPD patients and that this correlated with the degree of airflow obstruction and the number of IFN-γ+ cells. In contrast, T-bet expression was not altered (CitationDi Stefano et al., 2004) as well as the number of FoxP3 immunoreactive cells was not significantly different in the bronchial mucosal of stable COPD patients of different severity compared with controls (CitationCappello et al., 2011).
Both STAT1 and STAT2 are involved in the regulation of the transcription of antiviral interferon genes (CitationSchneider et al., 2010). Interestingly, bronchial epithelial cells obtained from stable COPD patients have increased expression, of STAT1 and STAT2 mRNA expression compared to control non-smoking subjects. In addition, cells from COPD patients express more mRNA for IL28A/IFNλ2, IL29/IFNλ1, IRF7, STAT1, and STAT2 after infection with rhinovirus in vitro (CitationSchneider et al., 2010).
NRF2 transcription factor in COPD
NRF2 is transcription factor that induces a multitude of antioxidant genes that in vitro and in animal models protect cells from the oxidative stress and inflammation induced by cigarette smoke (CitationAdair-Kirk et al., 2008; CitationBlake et al., 2010; CitationBoutten et al., 2011; CitationRangasamy et al., 2004).
Several studies have shown the presence of decreased activity and/or expression of NRF2 in the airway and lung cells of patients with stable COPD (CitationMalhotra et al., 2008; CitationSuzuki et al., 2008) or pulmonary emphysema (CitationGoven et al., 2008). In alveolar macrophages from patients with COPD, S-nitrosylation of HDAC2 is increased and this abolishes its GR-transrepression activity promoting glucocorticoid resistance (CitationMalhotra et al., 2011). Treatment with sulforaphane, a small-molecule activator of NRF2 is able to denitrosylate HDAC2, restoring glucocorticoid sensitivity in alveolar macrophages obtained from patients with stable COPD (CitationMalhotra et al., 2011), suggesting the potential role of NRF2 activators for the treatment of COPD (CitationBarnes, 2012b; CitationBoutten et al., 2011). However, large genetic studies have been unable to find a significant association between polymorphisms in the NRF2 pathway and the rate of decline of lung function (CitationSandford et al., 2012) and the antibody used in the previous studies performed using COPD tissues does not appear to be specific for NRF2 (CitationLau et al., 2013) suggesting the necessity of further human studies in this area using validated specific antibodies.
Theophylline action in the treatment of COPD
Glucocorticoid suppression of inflammatory genes requires recruitment of histone deacetylases (HDACs) to the activation complex by the glucocorticoid receptor (GR). HDACs are not effective in switching off inflammatory genes unless recruited to the active inflammatory site by activated glucocorticoid receptors. Increased oxidative stress in COPD reduces HDAC2 expression and activity thus potentially limiting glucocorticoid effectiveness in suppressing inflammation, as evidenced in vitro studies and clinically in patients with stable COPD (CitationIto et al., 2005, Citation2006b). Theophylline, at concentrations that do not inhibit phosphodiesterase (PDE)4 activity, enhances HDAC2 activity and functionally this enhances glucocorticoid effects (CitationCosio & Soriano, 2009; CitationIto et al., 2002). Theophylline activates different subtypes of HDAC, under conditions of oxidative stress, with equal activation of HDAC1 and HDAC2. There appears to be a relatively selective effect on Class I HDACs, with less effect on Class II HDACs (CitationBarnes, 2003, Citation2005). In vitro low dose theophylline in oxidant stressed cells strongly potentiate the anti-inflammatory actions of glucocorticoids (CitationMarwick et al., 2008). Hyperphosphorylation of HDAC2 impairs its function, and phosphoinositide-3-kinase-delta (PI3K-δ) signaling is, in part, responsible for the hyperphosphorylation and inactivation of HDAC2 and consequent reduction in glucocorticoid responsiveness (CitationGalasinski et al., 2002; CitationMarwick et al., 2009). Low dose theophylline in patients with stable COPD is a potent selective inhibitor of PI3K-δ, which is up-regulated in peripheral lung tissue of patients with stable COPD compared to control subjects (CitationMarwick et al., 2010; CitationTo et al., 2010). The treatment with low dose theophylline during COPD exacerbations potentiate the anti-inflammatory actions of glucocorticoids and increase sputum HDAC activity (CitationCosio & Soriano, 2009). In a small clinical study of 30 patients with stable COPD treatment with low dose slow release theophylline plus low dose inhaled fluticasone significantly reduced sputum neutrophils and improved lung function and this was associated with an increase in HDAC2 activity in circulating peripheral blood monocytes (CitationFord et al., 2010). Further studies in this area are required however.
Despite most current COPD guidelines stating that theophylline is of limited value in the routine management of COPD, many short-term controlled clinical trials support the utility of theophylline in the management of stable COPD patients (CitationRam et al., 2002) and further larger controlled clinical trials are ongoing for investigating the long-term anti-inflammatory and clinical efficacy of low dose theophylline. Bronchial biopsy studies are required to evaluate whether low dose theophylline has, as demonstrated in bronchial asthma, a significant in vivo anti-inflammatory activity (CitationCaramori & Adcock, 2003).
CONCLUSION
In the future the role of the activation/repression of different transcription factors and the genetic regulation of their expression in asthma and COPD may be an increasingly important aspect of research, as this may be one of the critical mechanisms regulating the expression of different clinical phenotypes and their responsiveness to therapy, particularly to anti-inflammatory drugs. Despite recent advances in the knowledge of the pathogenesis of asthma and COPD, much more research on the molecular mechanisms of asthma and COPD are needed to aid the logical development of new therapies for these common and important diseases, particularly in COPD where no effective pharmacological treatments currently exist.
ACKNOWLEDGMENTS
None.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, Saklatvala J, Clark AR (2006). Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 273: 1883–1889.
- Adair-Kirk TL, Atkinson JJ, Griffin GL, Watson MA, Kelley DG, DeMello D, Senior RM, Betsuyaku T (2008). Distal airways in mice exposed to cigarette smoke: Nrf2-regulated genes are increased in Clara cells. Am J Respir Cell Mol Biol. 39: 400–411.
- Adcock IM, Caramori G (2001). Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 79: 376–384.
- Adcock IM, Lane SJ (2003). Corticosteroid-insensitive asthma: Molecular mechanisms. J Endocrinol. 178: 347–355.
- Adcock IM, Caramori G, Kirkham PA (2012). Strategies for improving the efficacy and therapeutic ratio of glucocorticoids. Curr Opin Pharmacol. 12: 246–251.
- Allfrey VG, Faulkner R, Mirsky AE (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 51: 786–794.
- Baldwin AS Jr (2001). Series introduction: The transcription factor NF-kappaB and human disease. J Clin Invest. 107: 3–6.
- Barnes PJ, Chung KF, Page CP (1998). Inflammatory mediators of asthma: an update. Pharmacol Rev. 50: 515–596.
- Barnes PJ (2003). Theophylline: new perspectives for an old drug. Am J Respir Crit Care Med. 167: 813–818.
- Barnes PJ (2004a). Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 56: 515–548.
- Barnes PJ (2004b). Distribution of receptor targets in the lungs. Proc Am Thorac Soc. 1: 345–351.
- Barnes PJ, Kleinert S (2004). COPD – a neglected disease. Lancet. 364: 564–565.
- Barnes PJ (2005). Theophylline in chronic obstructive pulmonary disease: new horizons. Proc Am Thorac Soc. 2: 334–339; discussion 340–341.
- Barnes PJ (2006a). Reduced histone deacetylase in COPD: clinical implications. Chest. 129: 151–155.
- Barnes PJ (2006b). Transcription factors in airway diseases. Lab Invest. 86: 867–872.
- Barnes PJ (2008a). Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 8: 183–192.
- Barnes PJ (2008b). The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 118: 3546–3556.
- Barnes PJ (2009). The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 41: 631–638.
- Barnes PJ (2011a). Pathophysiology of allergic inflammation. Immunol Rev. 242: 31–50.
- Barnes PJ (2011b). Biochemical basis of asthma therapy. J Biol Chem. 286: 32899–32905.
- Barnes PJ (2012a). Development of new drugs for COPD. Curr Med Chem. [Epub ahead of print].
- Barnes PJ (2012b). New drugs for asthma. Semin Respir Crit Care Med. 33: 685–694.
- Bartlett NW, Slater L, Glanville N, Haas JJ, Caramori G, Casolari P, Clarke DL, Message SD, Aniscenko J, Kebadze T, Zhu J, Mallia P, Mizgerd JP, Belvisi M, Papi A, Kotenko SV, Johnston SL, Edwards MR (2012). Defining critical roles for NF-κB p65 and type I interferon in innate immunity to rhinovirus. EMBO Mol Med. 4: 1244–1260.
- Beghé B, Hall IP, Parker SG, Moffatt MF, Wardlaw A, Connolly MJ, Fabbri LM, Ruse C, Sayers I (2010). Polymorphisms in IL13 pathway genes in asthma and chronic obstructive pulmonary disease. Allergy. 65: 474–481.
- Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ (2011). Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc Natl Acad Sci USA. 108: 9554–9559.
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, Stamatoyannopoulos JA, Hager GL (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 43: 145–155.
- Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, Meij B, Drouin J (2006). Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 20: 2871–2886.
- Blake DJ, Singh A, Kombairaju P, Malhotra D, Mariani TJ, Tuder RM, Gabrielson E, Biswal S (2010). Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol. 42: 524–536.
- Borger P, Matsumoto H, Boustany S, Gencay MM, Burgess JK, King GG, Black JL, Tamm M, Roth M (2007). Disease-specific expression and regulation of CCAAT/enhancer-binding proteins in asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 119: 98–105.
- Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM (2000). Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 161: 1720–1745.
- Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M (2011). NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med. 17: 363–371.
- Broide DH, Lawrence T, Doherty T, Cho JY, Miller M, McElwain K, McElwain S, Karin M (2005). Allergen-induced peribronchial fibrosis and mucus production mediated by IkappaB kinase beta-dependent genes in airway epithelium. Proc Natl Acad Sci USA. 102: 17723–17728.
- Cai LQ, Zhang JY, Yu CX, Zhu L (2012). rhPLD2 suppresses chronic inflammation reactions in a guinea pig asthma model. Immunopharmacol Immunotoxicol. 34: 74–78.
- Cappello F, Caramori G, Campanella C, Vicari C, Gnemmi I, Zanini A, Spanevello A, Capelli A, La Rocca G, Anzalone R, Bucchieri F, D’Anna SE, Ricciardolo FL, Brun P, Balbi B, Carone M, Zummo G, Conway de Macario E, Macario AJ, Di Stefano A (2011). Convergent sets of data from in vivo and in vitro methods point to an active role of Hsp60 in chronic obstructive pulmonary disease pathogenesis. PLoS One. 6: e28200.
- Caramori G, Lim S, Ito K, Tomita K, Oates T, Jazrawi E, Chung KF, Barnes PJ, Adcock IM (2001). Expression of GATA family of transcription factors in T-cells, monocytes and bronchial biopsies. Eur Respir J. 18: 466–473.
- Caramori G, Adcock I (2003). Pharmacology of airway inflammation in asthma and COPD. Pulm Pharmacol Ther. 16: 247–277.
- Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, Chung KF, Barnes PJ, Adcock IM, Ciaccia A, Fabbri LM, Papi A (2003). Nuclear localization of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 58: 348–351.
- Caramori G, Ito K, Adcock IM (2004a). Transcription factors in asthma and COPD. IDrugs. 7: 764–770.
- Caramori G, Adcock IM, Ito K (2004b). Anti-inflammatory inhibitors of IkappaB kinase in asthma and COPD. Curr Opin Investig Drugs. 5: 1141–1147.
- Caramori G, Pandit A, Papi A (2005). Is there a difference between chronic airway inflammation in chronic severe asthma and chronic obstructive pulmonary disease? Curr Opin Allergy Clin Immunol. 5: 77–83.
- Caramori G, Oates T, Nicholson AG, Casolari P, Ito K, Barnes PJ, Papi A, Adcock IM, Chung KF (2009). Activation of NF-kappaB transcription factor in asthma death. Histopathology. 54: 507–509.
- Caramori G, Casolari P, Giuffrè S, Barczyk A, Adcock I, Papi A (2011). COPD pathology in the small airways. Panminerva Med. 53: 51–70.
- Chang PJ, Bhavsar PK, Michaeloudes C, Khorasani N, Chung KF (2012). Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma. J Allergy Clin Immunol. 130: 877–885.
- Chapoval SP, Dasgupta P, Smith EP, DeTolla LJ, Lipsky MM, Kelly-Welch AE, Keegan AD (2011). STAT6 expression in multiple cell types mediates the cooperative development of allergic airway disease. J Immunol. 186: 2571–2583.
- Chen H, Sun J, Buckley S, Chen C, Warburton D, Wang XF, Shi W (2005). Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am J Physiol Lung Cell Mol Physiol. 288: L683–691.
- Chen Lf, Fischle W, Verdin E, Greene WC (2001). Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 293: 1653–1657.
- Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM (2008). Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 3: e3316.
- Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M (2009a). A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol. 41: 516–524.
- Chiba Y, Todoroki M, Misawa M (2009b). Activation of signal transducer and activator of transcription factor 1 by interleukins-13 and -4 in cultured human bronchial smooth muscle cells. J Smooth Muscle Res. 45: 279–288.
- Chiba Y, Todoroki M, Misawa M (2011). Antigen exposure causes activations of signal transducer and activator of transcription 6 (STAT6) and STAT1, but not STAT3, in lungs of sensitized mice. Immunopharmacol Immunotoxicol. 33: 43–48.
- Cortes JR, Rivas MD, Molina-Infante J, Gonzalez-Nuñez MA, Perez-G M, Masa JF, Sanchez JF, Zamorano J (2009). Omeprazole inhibits IL-4 and IL-13 signaling signal transducer and activator of transcription 6 activation and reduces lung inflammation in murine asthma. J Allergy Clin Immunol. 124: 607–610.
- Cosio BG, Soriano JB (2009). Theophylline again? Reasons for believing. Eur Respir J. 34: 5–6.
- Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F (2011). Th17 cells: new players in asthma pathogenesis. Allergy. 66: 989–998.
- Darcan-Nicolaisen Y, Meinicke H, Fels G, Hegend O, Haberland A, Kühl A, Loddenkemper C, Witzenrath M, Kube S, Henke W, Hamelmann E (2009). Small interfering RNA against transcription factor STAT6 inhibits allergic airway inflammation and hyperreactivity in mice. J Immunol. 182: 7501–7508.
- Dawson MA, Kouzarides T, Huntly BJ (2012). Targeting epigenetic readers in cancer. N Engl J Med. 367: 647–657.
- Demoly P, Basset-Seguin N, Chanez P, Campbell AM, Gauthier-Rouvière C, Godard P, Michel FB, Bousquet J (1992).c-fos proto-oncogene expression in bronchial biopsies of asthmatics. Am J Respir Cell Mol Biol. 7: 128–133.
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003). Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 370: 737–749.
- Diez D, Goto S, Fahy JV, Erle DJ, Woodruff PG, Wheelock ÅM, Wheelock CE (2012). Network analysis identifies a putative role for the PPAR and type 1 interferon pathways in glucocorticoid actions in asthmatics. BMC Med Genomics. 5: 27.
- Di Stefano A, Caramori G, Oates T, Capelli A, Lusuardi M, Gnemmi I, Ioli F, Chung KF, Donner CF, Barnes PJ, Adcock IM (2002). Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 20: 556–563.
- Di Stefano A, Caramori G, Capelli A, Gnemmi I, Ricciardolo FL, Oates T, Donner CF, Chung KF, Barnes PJ, Adcock IM (2004). STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 24: 78–85.
- Dubey R, Chhabra R, Saini N (2011). Small interfering RNA against transcription factor STAT6 leads to increased cholesterol synthesis in lung cancer cell lines. PLoS One. 6: e28509.
- Eriksson U, Egermann U, Bihl MP, Gambazzi F, Tamm M, Holt PG, Bingisser RM (2005). Human bronchial epithelium controls TH2 responses by TH1-induced, nitric oxide-mediated STAT5 dephosphorylation: implications for the pathogenesis of asthma. J Immunol. 175: 2715–2720.
- Fitzpatrick AM, Stephenson ST, Hadley GR, Burwell L, Penugonda M, Simon DM, Hansen J, Jones DP, Brown LA (2011). Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J Allergy Clin Immunol. 127: 1604–1611.
- Fleischmann R, Cutolo M, Genovese MC, Lee EB, Kanik KS, Sadis S, Connell CA, Gruben D, Krishnaswami S, Wallenstein G, Wilkinson BE, Zwillich SH (2012). Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 64: 617–629.
- Ford PA, Durham AL, Russell RE, Gordon F, Adcock IM, Barnes PJ (2010). Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 137: 1338–1344.
- Fourtounis J, Wang IM, Mathieu MC, Claveau D, Loo T, Jackson AL, Peters MA, Therien AG, Boie Y, Crackower MA (2012). Gene expression profiling following NRF2 and KEAP1 siRNA knockdown in human lung fibroblasts identifies CCL11/Eotaxin-1 as a novel NRF2 regulated gene. Respir Res. 13: 92.
- Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, Wu R (2009). Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol. 183: 6236–6243.
- Fulkerson PC, Zimmermann N, Hassman LM, Finkelman FD, Rothenberg ME (2004). Pulmonary chemokine expression is coordinately regulated by STAT1, STAT6, and IFN-gamma. J Immunol. 173: 7565–7574.
- Fulton DL, Sundararajan S, Badis G, Hughes TR, Wasserman WW, Roach JC, Sladek R (2009). TFCat: the curated catalog of mouse and human transcription factors. Genome Biol. 10: R29.
- Gagliardo R, Chanez P, Mathieu M, Bruno A, Costanzo G, Gougat C, Vachier I, Bousquet J, Bonsignore G, Vignola AM (2003). Persistent activation of nuclear factor-kappaB signaling pathway in severe uncontrolled asthma. Am J Respir Crit Care Med. 168: 1190–1198.
- Gagliardo R, Chanez P, Profita M, Bonanno A, Albano GD, Montalbano AM, Pompeo F, Gagliardo C, Merendino AM, Gjomarkaj M (2011). IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 128: 635–645.
- Galasinski SC, Resing KA, Goodrich JA, Ahn NG (2002). Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem. 277: 19618–19626.
- Galbán CJ, Han MK, Boes JL, Chughtai KA, Meyer CR, Johnson TD, Galbán S, Rehemtulla A, Kazerooni EA, Martinez FJ, Ross BD (2012). Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med. 18: 1711–1715.
- Galliher-Beckley AJ, Williams JG, Cidlowski JA (2011). Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 31: 4663–4675.
- Gao F, Wei D, Bian T, Xie P, Zou J, Mu H, Zhang B, Zhou X (2012). Genistein attenuated allergic airway inflammation by modulating the transcription factors T-bet, GATA-3 and STAT-6 in a murine model of asthma. Pharmacology. 89: 229–236.
- Gernez Y, Tirouvanziam R, Nguyen KD, Herzenberg LA, Krensky AM, Nadeau KC (2007). Altered phosphorylated signal transducer and activator of transcription profile of CD4 + CD161 + T cells in asthma: modulation by allergic status and oral corticosteroids. J Allergy Clin Immunol. 120: 1441–1448.
- Gershon AS, Warner L, Cascagnette P, Victor JC, To T (2011). Lifetime risk of developing chronic obstructive pulmonary disease: a longitudinal population study. Lancet. 378: 991–996.
- Gilmore TD (2006). Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene. 25: 6680–6684.
- Global Initiative for Asthma. Global strategy for Asthma Management and Prevention. NHLBI/WHO Workshop report 2002. NHI Publication 02–3659. Last update 2011. http://www.ginasthma.com (accessed January 2012).
- Global Initiative for Chronic Obstructive Lung Disease (GOLD): global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHLBI/WHO workshop report. NIH Publication No 2701A. Last update 2011. http://www.goldcopd.com/ (accessed 28 May 2012).
- Goenka S, Kaplan MH (2011). Transcriptional regulation by STAT6. Immunol Res. 50: 87–96.
- Goleva E, Kisich KO, Leung DY (2002). A role for STAT5 in the pathogenesis of IL-2-induced glucocorticoid resistance. J Immunol. 169: 5934–5940.
- Goven D, Boutten A, Leçon-Malas V, Marchal-Sommé J, Amara N, Crestani B, Fournier M, Lesèche G, Soler P, Boczkowski J, Bonay M (2008). Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 63: 916–924.
- Grimley PM, Dong F, Rui H (1999). Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 10: 131–157.
- Groneberg DA, Eynott PR, Lim S, Oates T, Wu R, Carlstedt I, Roberts P, McCann B, Nicholson AG, Harrison BD, Chung KF (2002). Expression of respiratory mucins in fatal status asthmaticus and mild asthma. Histopathology. 40: 367–373.
- Harada M, Nakashima K, Hirota T, Shimizu M, Doi S, Fujita K, Shirakawa T, Enomoto T, Yoshikawa M, Moriyama H, Matsumoto K, Saito H, Suzuki Y, Nakamura Y, Tamari M (2007). Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am J Respir Cell Mol Biol. 36: 491–496.
- Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF (1998). Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med. 158: 1585–1592.
- Hashimoto K, Durbin JE, Zhou W, Collins RD, Ho SB, Kolls JK, Dubin PJ, Sheller JR, Goleniewska K, O’Neal JF, Olson SJ, Mitchell D, Graham BS, Peebles RS Jr (2005). Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J Allergy Clin Immunol. 116: 550–557.
- Hawkins GA, Robinson MB, Hastie AT, Li X, Li H, Moore WC, Howard TD, Busse WW, Erzurum SC, Wenzel SE, Peters SP, Meyers DA, Bleecker ER; National Heart, Lung, and Blood Institute–sponsored Severe Asthma Research Program (SARP) (2012). The IL6R variation Asp(358)Ala is a potential modifier of lung function in subjects with asthma. J Allergy Clin Immunol. 130: 510–515.
- Hinzey A, Alexander J, Corry J, Adams KM, Claggett AM, Traylor ZP, Davis IC, Webster Marketon JI (2011). Respiratory syncytial virus represses glucocorticoid receptor-mediated gene activation. Endocrinology. 152: 483–494.
- Hobbs K, Negri J, Klinnert M, Rosenwasser LJ, Borish L (1998). Interleukin-10 and transforming growth factor-beta promoter polymorphisms in allergies and asthma. Am J Respir Crit Care Med. 158: 1958–1962.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Paré PD (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 350: 2645–2653.
- Hogg JC, Timens W (2009). The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 4: 435–459.
- Holgate ST (2011). The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 242: 205–219.
- Homma T, Matsukura S, Hirose T, Ohnishi T, Kimura T, Kurokawa M, Ieki K, Odaka M, Suzuki S, Watanabe S, Sato M, Kawaguchi M, Schleimer RP, Adachi M (2010). Cooperative activation of CCL5 expression by TLR3 and tumor necrosis factor-alpha or interferon-gamma through nuclear factor-kappaB or STAT-1 in airway epithelial cells. Int Arch Allergy Immunol. 152: 9–17.
- Huang SL, Su CH, Chang SC (1997). Tumor necrosis factor-alpha gene polymorphism in chronic bronchitis. Am J Respir Crit Care Med. 156: 1436–1439.
- Hudson WH, Youn C, Ortlund EA (2013). The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. 20: 53–58.
- Inoue H, Fukuyama S, Matsumoto K, Kubo M, Yoshimura A (2007). Role of endogenous inhibitors of cytokine signaling in allergic asthma. Curr Med Chem. 14: 181–189.
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM (2002). p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J Allergy Clin Immunol. 109: 649–657.
- Ito K, Barnes PJ, Adcock IM (2000). Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 20: 6891–6903.
- Ito K, Lim S, Caramori G, Chung KF, Barnes PJ, Adcock IM (2001). Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 15: 1110–1112.
- Ito K, Lim S, Caramori G, Cosio B, Chung KF, Adcock IM, Barnes PJ (2002). A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci USA. 99: 8921–8926.
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ (2005). Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 352: 1967–1976.
- Ito K, Chung KF, Adcock IM (2006a). Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 117: 522–543.
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM (2006b). Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 203: 7–13.
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell. 126: 1121–1133.
- John AE, Zhu YM, Brightling CE, Pang L, Knox AJ (2009). Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF-kappa B p65, C/EBP beta, and RNA polymerase II binding to the CXCL8 promoter. J Immunol. 183: 4682–4692.
- Kalsheker NA, Morgan K (1994). Regulation of the alpha 1-antitrypsin gene and a disease-associated mutation in a related enhancer sequence. Am J Respir Crit Care Med. 150: S183–S189.
- Kavalar MS, Balantic M, Silar M, Kosnik M, Korosec P, Rijavec M (2012). Association of ORMDL3, STAT6 and TBXA2R gene polymorphisms with asthma. Int J Immunogenet. 39: 20–25.
- Knosp CA, Carroll HP, Elliott J, Saunders SP, Nel HJ, Amu S, Pratt JC, Spence S, Doran E, Cooke N, Jackson R, Swift J, Fitzgerald DC, Heaney LG, Fallon PG, Kissenpfennig A, Johnston JA (2011). SOCS2 regulates T helper type 2 differentiation and the generation of type 2 allergic responses. J Exp Med. 208: 1523–1531.
- Kobayashi M, Ashino S, Shiohama Y, Wakita D, Kitamura H, Nishimura T (2012). IFN-γ elevates airway hyper-responsiveness via up-regulation of neurokinin A/neurokinin-2 receptor signaling in a severe asthma model. Eur J Immunol. 42: 393–402.
- Kobayashi Y, Mercado N, Barnes PJ, Ito K (2011). Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS One. 6: e27627.
- Kobayashi Y, Mercado N, Miller-Larsson A, Barnes PJ, Ito K (2012). Increased corticosteroid sensitivity by a long acting β2 agonist formoterol via β2 adrenoceptor independent protein phosphatase 2A activation. Pulm Pharmacol Ther. 25: 201–207.
- Kouzarides T (2007). Chromatin modifications and their function. Cell. 128: 693–705.
- Kuperman DA, Schleimer RP (2008). Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 8: 384–392.
- Lane SJ, Adcock IM, Richards D, Hawrylowicz C, Barnes PJ, Lee TH (1998). Corticosteroid-resistant bronchial asthma is associated with increased c-fos expression in monocytes and T lymphocytes. J Clin Invest. 102: 2156–2164.
- Langlais D, Couture C, Balsalobre A, Drouin J (2012). The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell. 47: 38–49.
- Lau A, Tian W, Whitman SA, Zhang DD (2013). The predicted molecular weight of nrf2: it is what it is not. Antioxid Redox Signal. 18: 91–93.
- Li B, Carey M, Workman JL (2007). The Role of Chromatin during Transcription. Cell. 128: 707–719.
- Li G, Liu Z, Ran P, Qiu J, Zhong N (2004). Activation of signal transducer and activator of transcription 5 (STAT5) in splenocyte proliferation of asthma mice induced by ovalbumin. Cell Mol Immunol. 1: 471–474.
- Lim S, Crawley E, Woo P, Barnes PJ (1998). Haplotype associated with low interleukin-10 production in patients with severe asthma. Lancet. 352: 113.
- Lim S, Caramori G, Tomita K, Jazrawi E, Oates T, Chung KF, Barnes PJ, Adcock IM (2004). Differential expression of IL-10 receptor by epithelial cells and alveolar macrophages. Allergy. 59: 505–514.
- Liu YS, Tsai PW, Wang Y, Fan TC, Hsieh CH, Chang MD, Pai TW, Huang CF, Lan CY, Chang HT (2012). Chemoattraction of macrophages by secretory molecules derived from cells expressing the signal peptide of eosinophil cationic protein. BMC Syst Biol. 6: 105.
- Loke TK, Mallett KH, Ratoff J, O’Connor BJ, Ying S, Meng Q, Soh C, Lee TH, Corrigan CJ (2006). Systemic glucocorticoid reduces bronchial mucosal activation of activator protein 1 components in glucocorticoid-sensitive but not glucocorticoid-resistant asthmatic patients. J Allergy Clin Immunol. 118: 368–375.
- Luecke HF, Yamamoto KR (2005). The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. 19: 1116–1127.
- Lührmann A, Tschernig T, von der Leyen H, Hecker M, Pabst R, Wagner AH (2010). Decoy oligodeoxynucleotide against STAT transcription factors decreases allergic inflammation in a rat asthma model. Exp Lung Res. 36: 85–93.
- McCusker CT, Wang Y, Shan J, Kinyanjui MW, Villeneuve A, Michael H, Fixman ED (2007). Inhibition of experimental allergic airways disease by local application of a cell-penetrating dominant-negative STAT-6 peptide. J Immunol. 179: 2556–2564.
- Maddur MS, Miossec P, Kaveri SV, Bayry J (2012). Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 181: 8–18.
- Maher B (2012). ENCODE: The human encyclopaedia. Nature. 489: 46–48.
- Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S (2008). Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 178: 592–604.
- Malhotra D, Thimmulappa RK, Mercado N, Ito K, Kombairaju P, Kumar S, Ma J, Feller-Kopman D, Wise R, Barnes P, Biswal S (2011). Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 121: 4289–4302.
- Manning AM, Davis RJ (2003). Targeting JNK for therapeutic benefit: From junk to gold? Nat Rev Drug Discov. 2: 554–565.
- Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, Young RA (2007). Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 445: 931–935.
- Martinez-Nunez RT, Louafi F, Sanchez-Elsner T (2011). The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. 286: 1786–1794.
- Marwick JA, Wallis G, Meja K, Kuster B, Bouwmeester T, Chakravarty P, Fletcher D, Whittaker PA, Barnes PJ, Ito K, Adcock IM, Kirkham PA (2008). Oxidative stress modulates theophylline effects on steroid responsiveness. Biochem Biophys Res Commun. 377: 797–802.
- Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, Ito K, Adcock IM, Kirkham PA, Papi A (2009). Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 179: 542–548.
- Marwick JA, Caramori G, Casolari P, Mazzoni F, Kirkham PA, Adcock IM, Chung KF, Papi A (2010). A role for phosphoinositol 3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 125: 1146–1153.
- Matsunaga Y, Inoue H, Fukuyama S, Yoshida H, Moriwaki A, Matsumoto T, Matsumoto K, Asai Y, Kubo M, Yoshimura A, Nakanishi Y (2011). Effects of a Janus kinase inhibitor, pyridone 6, on airway responses in a murine model of asthma. Biochem Biophys Res Commun. 404: 261–267.
- Matthews JG, Ito K, Barnes PJ, Adcock IM (2004). Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy Clin Immunol. 113: 1100–1108.
- Michaeloudes C, Chang PJ, Petrou M, Chung KF (2011). Transforming growth factor-β and nuclear factor E2–related factor 2 regulate antioxidant responses in airway smooth muscle cells: role in asthma. Am J Respir Crit Care Med. 184: 894–903.
- Morgan K, Scobie G, Marsters P, Kalsheker NA (1997). Mutation in an alpha1-antitrypsin enhancer results in an interleukin-6 deficient acute-phase response due to loss of cooperativity between transcription factors. Biochim Biophys Acta. 1362: 67–76.
- Morjaria JB, Babu KS, Vijayanand P, Chauhan AJ, Davies DE, Holgate ST (2011). Sputum IL-6 concentrations in severe asthma and its relationship with FEV1. Thorax. 66: 537.
- Mullings RE, Wilson SJ, Puddicombe SM, Lordan JL, Bucchieri F, Djukanović R, Howarth PH, Harper S, Holgate ST, Davies DE (2001). Signal transducer and activator of transcription 6 (STAT-6) expression and function in asthmatic bronchial epithelium. J Allergy Clin Immunol. 108: 832–838.
- Nagashima S, Nagata H, Iwata M, Yokota M, Moritomo H, Orita M, Kuromitsu S, Koakutsu A, Ohga K, Takeuchi M, Ohta M, Tsukamoto S (2008). Identification of 4-benzylamino-2-[(4-morpholin-4-ylphenyl)amino]pyrimidine-5-carboxamide derivatives as potent and orally bioavailable STAT6 inhibitors. Bioorg Med Chem. 16: 6509–6521.
- Nakao I, Kanaji S, Ohta S, Matsushita H, Arima K, Yuyama N, Yamaya M, Nakayama K, Kubo H, Watanabe M, Sagara H, Sugiyama K, Tanaka H, Toda S, Hayashi H, Inoue H, Hoshino T, Shiraki A, Inoue M, Suzuki K, Aizawa H, Okinami S, Nagai H, Hasegawa M, Fukuda T, Green ED, Izuhara K (2008). Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J Immunol. 180: 6262–6269.
- Nath P, Eynott P, Leung SY, Adcock IM, Bennett BL, Chung KF (2005). Potential role of c-Jun NH2-terminal kinase in allergic airway inflammation and remodelling: effects of SP600125. Eur J Pharmacol. 506: 273–283.
- Neurath MF, Finotto S, Glimcher LH (2002). The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 8: 567–573.
- Neurath MF, Finotto S (2011). IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 22: 83–89.
- Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP, Otterbein SL, Song R, Hayashi S, Zhou Z, Pinsky DJ, Watkins SC, Pilewski JM, Sciurba FC, Peters DG, Hogg JC, Choi AM (2004). Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. 101: 14895–14900.
- Ning W, Dong Y, Sun J, Li C, Matthay MA, Feghali-Bostwick CA, Choi AM (2007). Cigarette Smoke Stimulates Matrix Metalloproteinase-2 Activity via EGR-1 in Human Lung Fibroblasts. Am J Respir Cell Mol Biol. 36: 480–490.
- Nofziger C, Vezzoli V, Dossena S, Schönherr T, Studnicka J, Nofziger J, Vanoni S, Stephan S, Silva ME, Meyer G, Paulmichl M (2011). STAT6 links IL-4/IL-13 stimulation with pendrin expression in asthma and chronic obstructive pulmonary disease. Clin Pharmacol Ther. 90: 399–405.
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK (2005). Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 122: 707–721.
- Oh CK, Geba GP, Molfino N (2010). Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 19: 46–54.
- O’Shea JJ, Plenge R (2012). JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 36: 542–550.
- Ouaked N, Mantel PY, Bassin C, Burgler S, Siegmund K, Akdis CA, Schmidt-Weber CB (2009). Regulation of the foxp3 gene by the Th1 cytokines: the role of IL-27-induced STAT1. J Immunol. 182: 1041–1049.
- Pan H, Cao J, Xu W (2012). Selective histone deacetylase inhibitors. Anticancer Agents Med Chem. 12: 247–270.
- Papi A, Johnston SL (1999). Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kappaB-mediated transcription. J Biol Chem. 274: 9707–9720.
- Perkins ND (2006). Post-translational modifications regulating the activity and function of the nuclear factor kappaB pathway. Oncogene. 25: 6717–6730.
- Perkins ND (2012). The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 12: 121–132.
- Poynter ME, Irvin CG, Janssen-Heininger YM (2002). Rapid activation of nuclear factor-kappaB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 160: 1325–1334.
- Profita M, Sala A, Bonanno A, Siena L, Ferraro M, Di Giorgi R, Montalbano AM, Albano GD, Gagliardo R, Gjomarkaj M (2008). Cysteinyl leukotriene-1 receptor activation in a human bronchial epithelial cell line leads to signal transducer and activator of transcription 1-mediated eosinophil adhesion. J Pharmacol Exp Ther. 325: 1024–1030.
- Pullen NA, Barnstein BO, Falanga YT, Wang Z, Suzuki R, Tamang TD, Khurana MC, Harry EA, Draber P, Bunting KD, Mizuno K, Wilson BS, Ryan JJ (2012). Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J Biol Chem. 287: 2045–2054.
- Quarcoo D, Weixler S, Groneberg D, Joachim R, Ahrens B, Wagner AH, Hecker M, Hamelmann E (2004). Inhibition of signal transducer and activator of transcription 1 attenuates allergen-induced airway inflammation and hyperreactivity. J Allergy Clin Immunol. 114: 288–295.
- Rahman I (2005). Redox signaling in the lungs. Antioxid Redox Signal. 7: 1–5.
- Rahman I, MacNee W (2012). Antioxidant pharmacological therapies for COPD. Curr Opin Pharmacol. 12: 256–265.
- Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I (2008). SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 177: 861–870.
- Ram FS, Jones PW, Castro AA, De Brito JA, Atallah AN, Lacasse Y, Mazzini R, Goldstein R, Cendon S (2002). Oral theophylline for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 4: CD003902.
- Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S (2004). Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 114: 1248–1259.
- Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S (2005). Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 202: 47–59.
- Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P (2010). Regulatory T cells in many flavors control asthma. Mucosal Immunol. 3: 216–229.
- Rennard SI, Fogarty C, Kelsen S, Long W, Ramsdell J, Allison J, Mahler D, Saadeh C, Siler T, Snell P, Korenblat P, Smith W, Kaye M, Mandel M, Andrews C, Prabhu R, Donohue JF, Watt R, Lo KH, Schlenker-Herceg R, Barnathan ES, Murray J; COPD Investigators (2007). The Safety and Efficacy of Infliximab in Moderate-To-Severe Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 156: 926–934.
- Rincon M, Irvin CG (2012). Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 8: 1281–1290.
- Romagnani S (2006). Regulation of the T cell response. Clin Exp Allergy. 36: 1357–1366.
- Roth M, Johnson PR, Rüdiger JJ, King GG, Ge Q, Burgess JK, Anderson G, Tamm M, Black JL (2002). Interaction between glucocorticoids and beta2 agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet. 360: 1293–1299.
- Roth M, Johnson PR, Borger P, Bihl MP, Rüdiger JJ, King GG, Ge Q, Hostettler K, Burgess JK, Black JL, Tamm M (2004). Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med. 351: 560–574.
- Saklatvala J (2004). The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 4: 372–377.
- Sampath D, Castro M, Look DC, Holtzman MJ (1999). Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J Clin Invest. 103: 1353–1361.
- Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W; Study A3921063 Investigators (2012). Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 367: 616–624.
- Sandford AJ, Malhotra D, Boezen HM, Siedlinski M, Postma DS, Wong V, Akhabir L, He JQ, Connett JE, Anthonisen NR, Paré PD, Biswal S (2012). NFE2L2 pathway polymorphisms and lung function decline in chronic obstructive pulmonary disease. Physiol Genomics. 44: 754–763.
- Sareila O, Hämäläinen M, Nissinen E, Kankaanranta H, Moilanen E (2008). Orazipone inhibits activation of inflammatory transcription factors nuclear factor-kappa B and signal transducer and activator of transcription 1 and decreases inducible nitric-oxide synthase expression and nitric oxide production in response to inflammatory stimuli. J Pharmacol Exp Ther. 324: 858–866.
- Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, Curtis JL, Martinez FJ, Hershenson MB, Sajjan U (2010). Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 182: 332–340.
- Shelburne CP, McCoy ME, Piekorz R, Sexl V, Roh KH, Jacobs-Helber SM, Gillespie SR, Bailey DP, Mirmonsef P, Mann MN, Kashyap M, Wright HV, Chong HJ, Bouton LA, Barnstein B, Ramirez CD, Bunting KD, Sawyer S, Lantz CS, Ryan JJ (2003). Stat5 expression is critical for mast cell development and survival. Blood. 102: 1290–1297.
- Silverpil E, Lindén A (2012). IL-17 in human asthma. Expert Rev Respir Med. 6: 173–186.
- Sousa AR, Lane SJ, Soh C, Lee TH (1999). In vivo resistance to corticosteroids in bronchial asthma is associated with enhanced phosphorylation of JUN N-terminal kinase and failure of prednisolone to inhibit JUN N-terminal kinase phosphorylation. J Allergy Clin Immunol. 104: 565–574.
- Strengell M, Matikainen S, Sirén J, Lehtonen A, Foster D, Julkunen I, Sareneva T (2003). IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol. 170: 5464–5469.
- Strickland DH, Holt PG (2011). T regulatory cells in childhood asthma. Trends Immunol. 32: 420–427.
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P (2011). Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 145: 224–241.
- Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M (2008). Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 39: 673–682.
- Szulakowski P, Crowther AJ, Jimenez LA, Donaldson K, Mayer R, Leonard TB, MacNee W, Drost EM (2006). The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 174: 41–50.
- Tian XR, Tian XL, Bo JP, Li SG, Liu ZL, Niu B (2011). Inhibition of allergic airway inflammation by antisense-induced blockade of STAT6 expression. Chin Med J (Engl). 124: 26–31.
- To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, Elliott WM, Hogg JC, Adcock IM, Barnes PJ (2010). Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 182: 897–904.
- Tomita K, Caramori G, Ito K, Sano H, Lim S, Oates T, Cosio B, Chung KF, Tohda Y, Barnes PJ, Adcock IM (2012). STAT6 expression in T cells, alveolar macrophages and bronchial biopsies of normal and asthmatic subjects. J Inflamm (Lond). 9: 5.
- Tomkinson A, Kanehiro A, Rabinovitch N, Joetham A, Cieslewicz G, Gelfand EW (1999). The failure of STAT6-deficient mice to develop airway eosinophilia and airway hyperresponsiveness is overcome by interleukin-5. Am J Respir Crit Care Med. 160: 1283–1291.
- Tsitoura DC, Rothman PB (2004). Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4 + T cells. J Clin Invest. 113: 619–627.
- Turlej RK, Fiévez L, Sandersen CF, Dogné S, Kirschvink N, Lekeux P, Bureau F (2001). Enhanced survival of lung granulocytes in an animal model of asthma: evidence for a role of GM-CSF activated STAT5 signalling pathway. Thorax. 56: 696–702.
- Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hübner N, Evans RM (2013). Insights into Negative Regulation by the Glucocorticoid Receptor from Genome-wide Profiling of Inflammatory Cistromes. Mol Cell. 49: 158–171.
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O’Shea JJ (2012). STATs shape the active enhancer landscape of T cell populations. Cell. 151: 981–993.
- van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, Forejtova S, Zwillich SH, Gruben D, Koncz T, Wallenstein GV, Krishnaswami S, Bradley JD, Wilkinson B; ORAL Standard Investigators (2012). Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 367: 508–519.
- Wang IM, Lin H, Goldman SJ, Kobayashi M (2004). STAT-1 is activated by IL-4 and IL-13 in multiple cell types. Mol Immunol. 41: 873–884.
- Wang W, Hansbro PM, Foster PS, Yang M (2011). An alternate STAT6-independent pathway promotes eosinophil influx into blood during allergic airway inflammation. PLoS One. 6: e17766.
- Wang Y, Li Y, Shan J, Fixman E, McCusker C (2011). Effective treatment of experimental ragweed-induced asthma with STAT-6-IP, a topically delivered cell-penetrating peptide. Clin Exp Allergy. 41: 1622–1630.
- Wohlmann A, Sebastian K, Borowski A, Krause S, Friedrich K (2010). Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol Chem. 391: 181–186.
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 126: 375–387.
- Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J, Lin A (2013). Inactivation of BAD by IKK Inhibits TNFα-Induced Apoptosis Independently of NF-κB Activation. Cell. 152: 304–315.
- Yang IV, Tomfohr J, Singh J, Foss CM, Marshall HE, Que LG, McElvania-Tekippe E, Florence S, Sundy JS, Schwartz DA (2012). The clinical and environmental determinants of airway transcriptional profiles in allergic asthma. Am J Respir Crit Care Med. 185: 620–627.
- Yusuf D, Butland SL, Swanson MI, Bolotin E, Ticoll A, Cheung WA, Zhang XY, Dickman CT, Fulton DL, Lim JS, Schnabl JM, Ramos OH, Vasseur-Cognet M, de Leeuw CN, Simpson EM, Ryffel GU, Lam EW, Kist R, Wilson MS, Marco-Ferreres R, Brosens JJ, Beccari LL, Bovolenta P, Benayoun BA, Monteiro LJ, Schwenen HD, Grontved L, Wederell E, Mandrup S, Veitia RA, Chakravarthy H, Hoodless PA, Mancarelli MM, Torbett BE, Banham AH, Reddy SP, Cullum RL, Liedtke M, Tschan MP, Vaz M, Rizzino A, Zannini M, Frietze S, Farnham PJ, Eijkelenboom A, Brown PJ, Laperrière D, Leprince D, de Cristofaro T, Prince KL, Putker M, del Peso L, Camenisch G, Wenger RH, Mikula M, Rozendaal M, Mader S, Ostrowski J, Rhodes SJ, Van Rechem C, Boulay G, Olechnowicz SW, Breslin MB, Lan MS, Nanan KK, Wegner M, Hou J, Mullen RD, Colvin SC, Noy PJ, Webb CF, Witek ME, Ferrell S, Daniel JM, Park J, Waldman SA, Peet DJ, Taggart M, Jayaraman PS, Karrich JJ, Blom B, Vesuna F, O’Geen H, Sun Y, Gronostajski RM, Woodcroft MW, Hough MR, Chen E, Europe-Finner GN, Karolczak-Bayatti M, Bailey J, Hankinson O, Raman V, LeBrun DP, Biswal S, Harvey CJ, DeBruyne JP, Hogenesch JB, Hevner RF, Héligon C, Luo XM, Blank MC, Millen KJ, Sharlin DS, Forrest D, Dahlman-Wright K, Zhao C, Mishima Y, Sinha S, Chakrabarti R, Portales-Casamar E, Sladek FM, Bradley PH, Wasserman WW (2012). The transcription factor encyclopedia. Genome Biol. 13: R24.
- Zandvoort A, Postma DS, Jonker MR, Noordhoek JA, Vos JT, van der Geld YM, Timens W (2006). Altered expression of the Smad signalling pathway: Implications for COPD pathogenesis. Eur Respir J. 28: 533–541.
- Zandvoort A, Postma DS, Jonker MR, Noordhoek JA, Vos JT, Timens W (2008). Smad gene expression in pulmonary fibroblasts: indications for defective ECM repair in COPD. Respir Res. 9: 83.
- Zhang N, Truong-Tran QA, Tancowny B, Harris KE, Schleimer RP (2007). Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol. 179: 578–589.
- Zhang S, Lukacs NW, Lawless VA, Kunkel SL, Kaplan MH (2000). Cutting edge: Differential expression of chemokines in Th1 and Th2 cells is dependent on STAT6 but not STAT4. J Immunol. 165: 10–14.
- Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, Thiessen N, Pettersson S, Jones SJ, Knapp S, Yang H, Chin KC (2012). Bromodomain-containing Protein 4 (BRD4) Regulates RNA Polymerase II Serine 2 Phosphorylation in Human CD4 + T Cells. J Biol Chem. 287: 43137–43155.
- Zheng W, Flavell RA (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 89: 587–596.
- Zhong H, May MJ, Jimi E, Ghosh S (2002). The phosphorylation status of nuclear NF-kappaB determines its association with CBP/p300 or HDAC-1. Mol Cell. 9: 625–636.
- Zhou L, Kawate T, Liu X, Kim YB, Zhao Y, Feng G, Banerji J, Nash H, Whitehurst C, Jindal S, Siddiqui A, Seed B, Wolfe JL (2012). STAT6 phosphorylation inhibitors block eotaxin-3 secretion in bronchial epithelial cells. Bioorg Med Chem. 20: 750–758.
- Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE (2006). GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 16: 3–10.
- Zhu Y, Chen L, Huang Z, Alkan S, Bunting KD, Wen R,Wang D, Huang H (2004). Cutting edge: IL-5 primes Th2 cytokine-producing capacity in eosinophils through a STAT5-dependent mechanism. J Immunol. 173: 2918–2922.