Abstract
In the skin epidermis, adhesion to the underlying basement membrane is mediated through trans-membrane integrin receptors. In addition to a structural role, integrins can signal in a bi-directional manner though the membrane and thus play a crucial role in cell adhesion, migration, proliferation, and differentiation. In this review we will discuss the role of integrins and their network of partner proteins in normal skin development, and how dysregulation influences disease states such as skin blistering disorders and cancers. We also discuss major integrin-specific therapeutic advances that have been made over the past few years in treating these skin disorders as well as targeting angiogenesis, neo-vasculature, and tumorigenesis.
INTRODUCTION
The skin
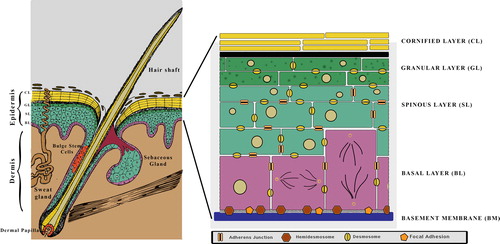
The mammalian skin comprises of the epidermis, dermis, and its associated appendages (). The epidermis and dermis is separated by a basement membrane (BM) that is rich in Collagen IV and Laminin 332. In development, the epidermis arises from the embryonic ectoderm whereas the dermis is mesodermal in origin (CitationFuchs & Raghavan, 2002). The primary function of the skin is to protect the organism from harmful microorganisms and chemicals from the outside and prevent water loss from the inside. This remarkable feat is achieved by the epidermis, which is a stratified squamous epithelia and provides a water-tight barrier from the external environment. The basal cells of the epidermis are proliferative and once these withdraw from the cell cycle and detach from the BM, they trigger a process of terminal differentiation. This gives rise to the spinous, granular layers, and eventually the cornified layer which is comprised of enucleated cells that are sloughed off. The epidermis is constantly renewing every 2–3 weeks and the inter- follicular epidermal stem cells are critical for this process. The other class of stem cells that are found in the skin are the slow-cycling bulge stem cells, which primarily contribute to the hair follicular cycle (CitationBlanpain & Fuchs, 2009). In addition to forming a barrier, the skin also has to withstand mechanical stress, and achieves this by maintaining robust cell–cell and cell–substratum adhesions. In the skin the cell–substratum adhesions are mediated by integrins.
Figure 1. The skin and epidermis: The skin is made up of the epidermis, underlying dermis and its associated appendages (hair follicles and sebaceous glands). The epidermis is a stratified squamous epithelia comprising of a proliferative basal layer (BL) and post-mitotic cells that make up the Spinous, Granular and Cornified layers.
Integrins and epithelia
Integrins are a large family of heterodimeric trans- membrane receptors, comprising of αβ subunits that link the extracellular matrix (ECM) to the cellular cytoskeleton. Integrins signal through the cell membrane in a bi-directional manner. These signaling events regulate cellular processes such as cell adhesion, migration, proliferation, differentiation, and apoptosis. In mammals, 18 α and 8 β integrin subunits have been identified that combine to form 24 different heterodimers that bind to ECM ligands (CitationHynes, 2002). The epithelial BM is composed of a characteristic set of large extracellular glycoproteins; Laminin, Type IV collagen, entactin/nidogen, and perlecan, which lie in direct contact with the cell surfaces (CitationTimpl & Brown, 1996). These molecules are typically arranged in an ordered fashion. A number of BM proteins interact with one another and some isoforms of Type IV collagen and laminin can self-assemble into lattice-like polymeric networks in physiological buffers (CitationTimpl & Brown, 1996). Of these 24 integrin pairs a small subset are expressed in the skin epidermis and can be broadly classified as β1, β4 (which partners with α6) or αv-containing integrins. The predominant epithelial integrins are α3β1 and α6β4 both of which are receptors for Laminin 332, a major component of the epithelial BM (CitationWatt, 2002). The β4 and β1 containing integrins are associated with two distinct cell–substratum adhesive structures. Integrin α6β4 forms the central core of the hemidesmosome, which connects the ECM to the keratin cytoskeleton network within the keratinocytes. The αβ1 containing integrins on the other hand form the core of the focal adhesions (FAs), which connect the ECM to the actin cytoskeleton network with the basal keratinocytes. While most epithelial integrins are constitutively expressed in the skin, the expression of integrins such as α5β1 and αvβ6 is induced during wound healing and other pathological conditions.
HOMEOSTASIS AND DISEASE STATE
Hemidesmosomes and associated disorders
Hemidesmosomes (HDs) appear as electron-dense structures in electron micrographs and are found at the basal side of the basal keratinocytes in close physical proximity to the BM. HDs are nucleated by integrin α6β4, which binds to Laminin 332. The transmembrane protein BP180 (also known as collagen XVII) associates with β4 integrin and also binds to Laminin 332 (). Intracellularly, there are two plakin proteins, plectin, and BP230/BPAG1 that provide connections to the keratin intermediate filament cytoskeletal network (CitationMargadant et al., 2010; CitationWickstrom et al., 2011). Tetraspanin CD151 is a transmembrane protein that is associated with the α6 subunit. Additionally anchoring fibrils which are comprised of Collagen VII serve to stabilize the adhesion of integrins and BP230 to Laminin 332. The function of the hemidesmosome is primarily structural, that is, to provide strong and stable adhesion between the basal cells and the BM. This is borne out by the fact that mutations or loss of several components of the HD results in severe skin blistering Epidermolysis Bullosa (EB). EB is a serious genetic skin-blistering disease that affects 1 in 50,000 new borns around the globe. The differing underlying mechanism for this dysfunction leads to its classification into three distinct subcategories—EB simplex (EBS), junctional EB (JEB), and dystrophic EB (DEB).
Figure 2. Schematic Representation of the Hemidesmosome: It is composed of a network of structural proteins which provides a firm connection between the basal keratinocytes and the dermis; the basal keratinocytes are connected to the BM via the hemidesmosome-anchoring filament complex, which in turn is connected to the dermis via anchoring fibrils. Various forms of EB (skin blistering diseases) are a result of defect at different levels of this network. BP230, Dystonin/BPAG1; BP180, Collagen XVII; DEB, Dystrophic EB; JEB, Junctional EB; EBS, EB simplex.
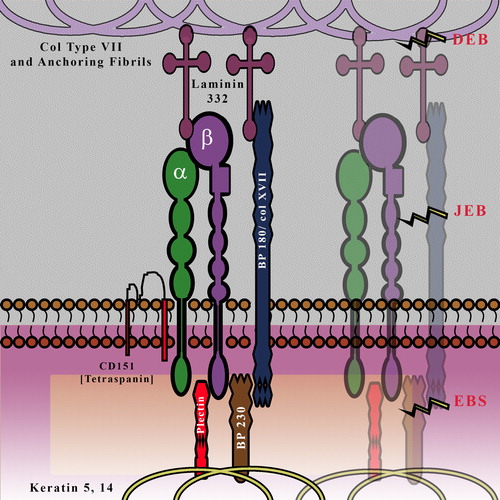
In its most prevalent form, EBS manifests as blistering due to fragility of the basal layer of epidermal keratinocytes, caused by mutations in keratin 5 (KRT5) or 14 (KRT14). EBS is characterized by fragility of the skin (and mucosal epithelia) that results in non-scarring blisters caused by little or no trauma. Patients suffering from EB due to Plectin (PLEC1) mutation develop muscular dystrophy later on in adulthood (CitationCharlesworth et al., 2003; CitationSmith et al., 1996). In JEB, the split occurs within the basal lamina, often in the lumina lucida, and can be caused by multiple genetic mutations; one of the three genes contributing to Laminin-332 (LAMA3, LAMB3, and LAMC2) is the culprit in the lethal Herlitz form of JEB (H-JEB), whereas mutational silencing of α6β4 integrin (ITGB4, ITGA6) leads to JEB with pyloric atresia (JEB-PA) and collagen XVII (COL17A1) mutations are known to cause non-Herlitz form of JEB (nH-JEB) (CitationBruckner-Tuderman & Has, 2013). The main clinical signs encompass skin and mucosal blistering and/or imperfect amelogenesis, along with some rare cases of systemic dysfunctioning (CitationLaimer et al., 2010; CitationYancey & Hintner, 2010). In DEB, recessive (RDEB) or dominant (DDEB), the tissue disruption occurs at the anchoring fibrils just below the BM and the molecular cause is now known to be defective collagen VII (COL7A1). Symptoms include severe skin and mucosal blistering and scarring, chronic wounds, nail dystrophy, and scarring alopecia (CitationFine et al., 2008; CitationShinkuma et al., 2011). In addition to these EB dermatoses (), there are rare multi-organ disorders such as EB-congenital nephrotic syndrome-interstitial lung disease, postulated to be caused by homozygous mutation in the ITGA3 gene (CitationHas et al., 2012).
Table 1. EB types and genes responsible (CitationFine et al., 2008).
Research focused on treatment for EB (of various types) is also aimed at understanding its link to carcinomas. Most of the proteins implicated in the disease also have major cellular signaling functions; integrins being arguably the most significant among these. Thus, disruption of such crucial structural proteins also delinks cellular machinery and leads to tumorigenesis more often than not. Disrupted wound healing pattern observed in EB cases is another reason for their propensity toward Squamous Cell carcinomas (SCCs). Chronic wound-state inflammation, as seen in severe cases of DEB, can lead to tumor-formation. Keratinocyte migration during wound healing is governed by downstream effectors of β1 under feedback from growth factor-cues. Even in integrin β1-null mice, wound re-population (although delayed) has been found to be effected by hypertrophy involving β1-retaining cells, which indicates the indispensability of β1 in the reconstructive process (CitationPiwko-Czuchra et al., 2009). This makes it imperative for researchers to target the wound-healing process as well; a better understanding of the regenerative process will help abate tumorigenesis. This also includes the structural integrity of EB skin as loss-of-barrier can enhance infection as well as exposure to carcinogens; frail BM structure can aid spread of cancerous cells easily via underlying/newly formed blood stream.
At present there is no definite cure for EB, and conventional treatment is limited to chronic-wound care. The inherent fragility and turnover of skin makes the disease very hard to tackle permanently, and its inclination for developing SCCs makes it potentially deadly. Hence, treatments being developed have to be multi-faceted, while mitigating adverse immune-response, so as to maintain efficacy.
FAs and signaling diseases
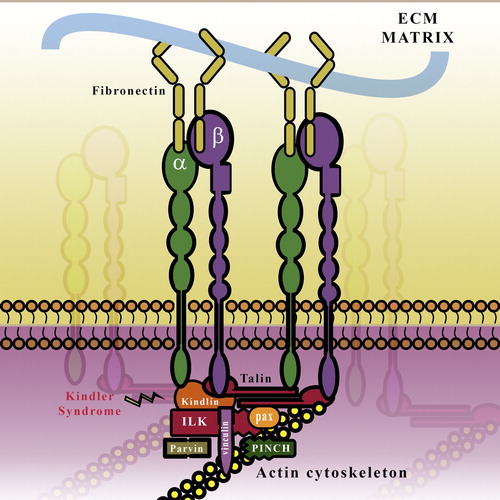
FAs are cell–substratum adhesions that are not readily visualized in vivo. It is, however, the primary way by which cells in culture attach to the underlying ECM and serves both structural as well as signaling functions. Like the HD, the FA complex is also nucleated by integrins (αβ1, αvβ5, and αvβ6 in keratinocytes). However a major difference is, whereas HD integrin α6β4 binds to a single ECM ligand, Laminin 332, the FA integrins associate with a myriad of ECM ligands. In keratinocytes, integrin α5β1 is the major fibronectin receptor whereas α3β1 is the Laminin 332 receptor and α2β1 is the receptor for Collagen IV (CitationMargadant et al., 2010). Additionally, FAs have much more complex structures (). At last count, there were over 180 proteins that were associated with the integrin “adhesome” (CitationWolfenson et al., 2013). These can be broadly classified as scaffolding proteins, such as paxillin which mediates its connection to the actin cytoskeletal network, and signaling proteins such as focal adhesion kinase (FAK) which modulate cell–substratum adhesion. A small subset of the proteins found in the integrin adhesome are physically associated with the integrin tails, and these can be either activators (such as talin and kindlin) or inactivators (such as SHARPIN and Filamin).
Figure 3. Schematic Representation of the FA complex: Integrins bind to components of the ECM such as collagen and Fibronectin (FN). The integrins are linked to the actin cytoskeleton and are stabilized by the binding of proteins such as Talin, Kindlin, and the IPP (ILK-PINCH-parvin) complex. ILK, Integrin-linked Kinase; PINCH, Particularly interesting new cysteine–histidine rich protein; Pax, Paxillin.
Studies from several labs have revealed that integrins can exist in low- (inactive) and high-affinity (active) states (CitationCalderwood et al., 2013) (). In its low-affinity state, the integrin extracellular domain is in a bent conformation and thereby unable to bind to ECM proteins. Endogenous integrin inactivators such as Filamins, ICAP, and DOK1 are known to inhibit by binding to the β-cytoplasmic tail of the heterodimer and directly preventing talin association (CitationKiema et al., 2006; CitationWegener et al., 2007). SHARPIN binds to conserved sequences in the α1-, α2-, and α5- subunit of β1-integrins inhibiting activation of the integrins by preventing association with talin and kindlin (CitationBouvard et al., 2013; CitationRantala et al., 2011).
Figure 4. Schematic of integrin inactivation and activation states: In the low-affinity “inactive” state, integrin cytoplasmic tails are blocked from binding to talin and kindlin by cytoplasmic proteins such as Filamins and SHARPIN; Inside-out signaling is characterized by cytoplasmic docking of talin and kindlin on the β tail of integrins to activate extracellular domain into open conformation; Outside–in signaling is characterized by ECM ligand binding to integrin which brings about formation of cytoplasmic signaling complex. Subsequently, integrin clustering precipitates the formation of a high-affinity “active” state.
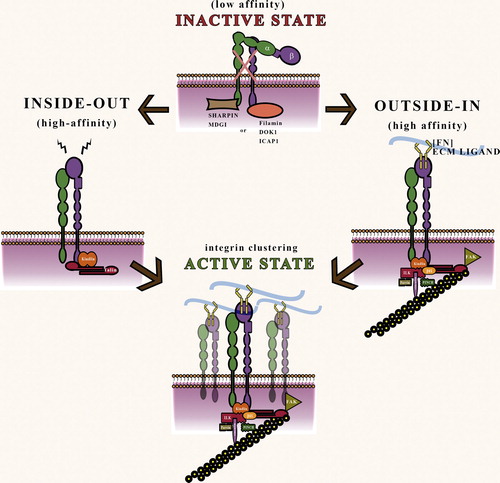
The high-affinity state of the integrin is characterized by an extended, open conformation due to the binding of the adaptor protein Talin and Kindlin to specific NPxY binding sites in the β tail (). The activated integrins bind ECM ligands, which in turn triggers a signaling cascade referred to as outside–in signaling, the primary function of which is to establish a stable integrin–actin connection. The outside–in signaling cascade which results in the recruitment of adaptor proteins and cytoskeletal rearrangement is also mediated through talin and kindlin.
Talin is a FERM-domain containing protein; loss of talin expression results in cells unable to undergo sustained cell spreading, indicating defects in integrin-mediated cell adhesion. Studies have shown that Talin is a key adaptor linking the actin cytoskeleton with the ECM by virtue of being able to directly bind both the β1 tail and F-actin.
Kindlins are another group of FERM-domain containing proteins. In mammals, there are three kindlin family members: kindlin-1 (URP1), kindlin-2 (MIG2), and kindlin-3 (URP2). Kindlin-1 is expressed in epithelial tissues such as skin, kidney, and intestinal epithelia and plays an important role in keratinocyte migration, proliferation, and adhesion (CitationHerz et al., 2006; CitationKloeker et al., 2004; CitationLai-Cheong et al., 2010). Kindlin-2 is widely expressed, with highest amount in skeletal and smooth muscles, while Kindlin-3 is associated with podosomes, which are integrin-rich adhesion sites in hematopoietic cells (CitationLai-Cheong et al., 2008; CitationMoser et al., 2008). Despite sharing 62% homology, kindlin-1 and -2 are found to have non-overlapping functionality in keratinocytes due to inherent differential association with integrin β6 as opposed to β1 (CitationBandyopadhyay et al., 2012).
Kindler syndrome (KS) is a rare skin disorder caused by loss-of-function mutation in KIND1 and results in extensive blistering along with symptoms such as erythema, atrophy, alopecia, and abnormal pigmentation. It is a curious disease for epidermal experts, as it is the first case of a disease caused by mutations in a (signaling-based) FA-associated protein that manifests as a structural disease akin to EB. KS has been reported to also affect gastrointestinal and urinary epithelia while causing webbing of fingers and toes (CitationSiegel et al., 2003; CitationUssar et al., 2008; CitationWhite & McLean, 2005).
The ILK-PINCH-parvin (IPP) complex is critical for the association of integrins to the actin-cytoskeletal network. The molecular interaction between integrin-linked kinase (ILK) and integrins has been a subject of intense study, with opposing views on its purported linkage via the IPP complex leading to integrin activation (CitationLegate et al., 2006; CitationStanchi et al., 2009; CitationWickstrom et al., 2010; CitationWu, 2004). It is known that ablation of ILK in epidermal compartment has a similar effect to that of integrin β1 deletion (CitationBrakebusch et al., 2000; CitationRaghavan et al., 2000); consistent with BM disruption as well as impairment of progenitors during downward migration in hair follicle (CitationLorenz et al., 2007; CitationNakrieko et al., 2008), however a causal link is yet to be satisfactorily established. As the molecular axis connecting ILK and other integrin partners to the ECM-integrin tether gets resolved, we will be in a better position to develop targeted therapeutics for treating integrin-related signaling disorders. For example, PINCH is one such molecule of interest, having been implicated in a diverse array of diseases including renal failure, cardiomyopathy, and tumorigenesis through Ras activation as well as inhibition of apoptosis (CitationChen et al., 2008; CitationDougherty et al., 2008).
Tetraspanin (CD151), an important allied membrane protein, can associate both with α3β1 and α6β4 in keratinocytes, and is thought to thus regulate integrin-mediated cellular migration (via α3β1) or matrix adhesion (via α6β4) (CitationZevian et al., 2011). The function of CD151 is most likely linked to α3β1 functioning; as targeted deletion of CD151 in mice did not cause any overt HD phenotype but resulted in severe kidney dysfunction; similar to α3β1 ablations (CitationSachs et al., 2006; CitationWright et al., 2004). A mutation in the CD151 gene in humans has been described to cause localized skin blistering, in addition to deafness and renal failure (CitationKaramatic Crew et al., 2004). The dissociation of CD151–integrin complex seems to permit remodeling of epithelial cell interactions with the ECM and play a major role in cell migration (CitationChometon et al., 2006). It is unsurprising, then, that it has been implicated in multiple metastatic carcinomas (CitationDeng et al., 2012; CitationDevbhandari et al., 2011; CitationKwon et al., 2012; CitationTakeda et al., 2011).
Integrins, associated proteins, and cancer
As many solid tumors originate from epithelial cells, the epithelial integrins (including α6β4, α6β1, αvβ5, α2β1, and α3β1) are generally retained in the cancer state, though expression levels are invariably altered. These integrins not only typically mediate epithelial cell adhesion to the BM, but also affect migration, proliferation, and survival in tumor cells. While many integrins can enhance cell survival; some can, conversely, initiate apoptotic cascades (CitationDesgrosellier & Cheresh, 2010). Studies have shown that although some integrins, such as αvβ3 and α6β4, enhance tumor progression, paradoxically, others such as α5β1 inhibit oncogene-induced transformation (CitationGiancotti & Ruoslahti, 1990; CitationPetitclerc et al., 1999; CitationVarner et al., 1995). This paradox can be partly explained by the role played by ligation state in integrin activity. The phenomenon of integrin-mediated death or IMD (CitationStupack et al., 2001; CitationZhao et al., 2005) can recruit caspases to initiate apoptosis based on adherence to ECM proteins. Indeed, inhibitors targeting unligated integrins can prevent cell–matrix adhesion and induce IMD in susceptible cancer models.
In SCCs of skin as well as oral cavity, altered integrin expression can provide a growth advantage by protecting against apoptosis, while promoting proliferation and invasion. Abnormal integrin expression on differentiated cells can regulate the clonal expansion of cancer cell population from neighboring progenitor cells (CitationJanes & Watt, 2006). Also, inflammation can be triggered by aberrant integrin expression, contributing to tumorigenesis. Desmoplastic exacerbation found in multiple metastases is directly linked to upregulation of integrin α11 and resultant collagen deposition (CitationZhu et al., 2007). New therapeutics targeting the tumor stroma could prove a useful strategy in retarding tumor progression.
The role played by integrins in maintaining normal stem cells, and hence refractory cancer stem cells has made them important targets for chemotherapeutics. Their role is varied, ranging from regulation of stem cell-surface markers such as CD44 (CitationSamanna et al., 2006) to stabilizing cross-talk with oncogene products or growth factors (CitationDesgrosellier & Cheresh, 2010). ECM ligand cross-talk providing resistance against apoptosis is also a major area of therapeutics study (CitationAoudjit & Vuori, 2012).
Integrins are also instrumental in the activation of TGFβ signaling. TGFβ ligands are secreted as inactive complexes with a latency-associated peptide (LAP). Owing to its RGD motif, LAP was found to be a ligand for multiple integrins. In basal cell carcinomas (BCCs), upregulation of αvβ6 expression correlates with increased TGFβ1 activation, fibrosis, and a dense stroma, resulting in aggressive prognosis. Further studies have shown the important role of integrin αvβ6 in activating TGFβ1 in vivo, contributing to tumor growth and maintenance (CitationBates et al., 2005; CitationMcCarty et al., 2008; CitationVan Aarsen et al., 2008). Also, other integrins are involved at the effector end of TGFβ1 signaling, as suggested by studies showing that epithelial-mesenchymal transition (EMT) of TGFβ1- stimulated HSC-4 (hOSCC) cells was regulated by integrin α3β1 (CitationSaito et al., 2013). It is very likely that aberrant integrin activity, at either end of the signaling spectrum, is an important step in many other pathological states.
Although not directly oncogenic themselves, integrins can cooperate with certain oncogenes during tumorigenesis. For example, integrin α6β4 has been known to interact with ErbB2 (HER2) via TGFβ-induced clustering to enhance invasiveness in Breast Cancers by activating STAT3 and JUN, leading to cell-polarity loss and hyperplasia (CitationGuo et al., 2006; CitationWang et al., 2009). Likewise, integrin β1 is implicated in Polyoma T oncoprotein- mediated breast cancer (CitationWhite et al., 2004), while integrin α1 is observed in KRAS-G12D-induced Lung tumors (CitationMacias-Perez et al., 2008). The pathway and cross-talk of oncogenes with integrins differs for different cancers depending on host cell type. Nevertheless, this provides yet another way of limiting oncogene activation and hence is a viable drugging strategy.
Kindlin
The evidence that KIND1 gene plays an important role in cancer progression is the observation of increased KIND1 mRNA expression in up to 60% and 70% of lung and colon cancers, respectively (CitationWeinstein et al., 2003). Subsequent gene expression microarray studies of RNA profiles of TGFβ1-treated HMEC cells showed that KIND1 is a TGFβ1 inducible gene. This TGFβ1-induced enhancement of Kindlin-1 expression can help explain propensity for EMT (CitationKloeker et al., 2004). The cutaneous fibrosis and secretions observed in KS has been attributed to increased ECM-deposition due to immunogenically upregulated paracrine signaling by null-keratinocytes to dermal myofibroblasts (CitationHeinemann et al., 2011). Also, the fragility disorder is observed to increase the predisposition for epithelial carcinomas in adulthood (CitationFine et al., 2009; CitationLai-Cheong et al., 2009).
In breast cancer cell line TMX2-28, knockdown of KIND2 by RNAi leads to a significant reduction of cancer cell invasiveness (CitationGozgit et al., 2006). However, gene expression profiling showed reduced KIND2 mRNA expression in several colonic carcinoma cell lines such as RKO, HT-29, DLD-1, LoVo, and HCT-116 as well as in the HT-1080 cell line, a highly metastatic fibrosarcoma cell line (CitationShi & Wu, 2008). Studies have also shown that kindlin-2 may suppress mesenchymal cancer cell invasion by reducing the level of secreted urokinase-type plasminogen activator, which plays an important role in proteolytic degradation of the ECM and tumor cell invasion (CitationDuffy, 2002; CitationShi & Wu, 2008). Kindlin-2 was found to mechanistically interact with β-catenin and TCF4 to enhance Wnt signaling (CitationYu et al., 2012) in tumors. Thus, kindlin-2 shows variable functioning in different cancer models.
ILK
The central role played by ILK makes it an ideal candidate in coordinating the signals from growth factors and integrins that control both matrix assembly and E-cadherin expression. ILK has been found to have proto-oncogenic functions, as multiple siRNA studies targeting ILK have resulted in abrogation of multiple cancers (CitationLi et al., 2013; CitationLiu et al., 2012; CitationZhu et al., 2012). The newly discovered association of ILK in microtubule dynamics (CitationLim et al., 2013) can account for its hand in cell-cycle derailment observed in multiple carcinomas, and small molecule inhibitors such as QLT-0267 could demonstrate new avenues of therapy. It is also known to influence the β-catenin/TCF transcriptional activity (CitationOloumi et al., 2004). Overexpression in tumors can thus result in EMT, and more aggressive invasiveness. ILK has been implicated in BSC progression (CitationPapanikolaou et al., 2010) and gliomal invasion (CitationLiang et al., 2013) with particular influence on EMT by downregulating E-cadherin.
Growth factor receptors (GFRs)
Interaction of integrins with growth factor receptors (GFRs) regulate myriad cellular functions such as cell adhesion, migration, invasion, and survival. This interaction can be cooperative, like in the case of complexes formed that activate MAPK cascade (CitationMiyamoto et al., 1996), but also involves mutual activation or surface expression. Several integrins such as the αβ1 family and α6β4 have been found to be important for angiogenesis and hence tumorigenesis, causing increased VEGF/FGF binding due to their cooperation with GFRs. The role of these integrins has been discussed more comprehensively elsewhere (CitationAvraamides et al., 2008).
Crosstalk with Wnt pathway
Crosstalk of integrin–FAK activation with the canonical Wnt pathway through adaptor Grb2 is known (CitationCrampton et al., 2009), and can account for certain heterogeneous cancers. Other FA molecules, significantly kindlin-2 (CitationYu et al., 2012), are known to interact with β-catenin to enhance Wnt signaling. Also, depletion experiments of integrin α3β1 showed a causal link with β-catenin-dependent tumorigenesis, proving that activation downstream from α3β1 involve Rac1/PAK1, MAPK and JNK and promote pro-proliferative signals (CitationCagnet et al., 2013), thus providing new insights into the molecular mechanisms underlying breast cancer progression.
In addition to the role of cell–substratum adhesion in cancer progression, the cross talk with epithelial cell–cell adhesion is a critical facet of tumorigenesis. Crosstalk between CCA and CMA is known to occur via various molecules such as Src, FAK, ILK, and small Rho GTPases (such as CDC42, Rac1, and RhoA which have conventional cytoskeletal roles), as well as calpains and MMPs and actin-modulators Ena/VASP and WASP. While we will not cover this in detail, we would like to point the readers to a comprehensive review that does (CitationCagnet et al., 2013).
Immunity-mediated diseases
Aberrant circulation of various auto-antibodies precipitates the phemphigoid group of diseases; the targeting of HD proteins by these antibodies results in Bullous phemphigoid (BP). Chief molecules, which elicit such an inflammatory response and activation of the complement system include antigens BP180/Col XVII and BP230/Dystonin/BPAG1. BP230's absence incidentally has been shown to enhance hair loss in mice models, pointing to a disruption of normal hair cycling (CitationGuo et al., 1995) while causing hyperproliferation. They are postulated to buttress HD stability and their absence could thus slough off cells internal to the ORS due to integrin-ECM delinkage (CitationOlasz & Yancey, 2008).
Another “auto-immune” disorder plaguing the general population is psoriasis. Although it is likely that the epidermal cell hyperplasia that characterizes the disorder is triggered by the influx of cytokine-releasing inflammatory cells, psoriatic keratinocytes are known to secrete T-cell activating cytokines in vitro. Also, ectopic overexpression of β1 subunit has been shown to mimic psoriatic hyperproliferation (CitationCarroll et al., 1995). It is posited that whereas psoriasis is most likely triggered by infiltrative inflammatory cells, subsequent progression may be augmented by keratinocyte hyperproliferation and their cytokine release (CitationMargadant et al., 2010). Although the importance of integrin α4β7 in leukocyte-based intestinal inflammation has been established, its role in cutaneous inflammation has recently been studied in terms of T-cell migration to the skin (CitationOhmatsu et al., 2010). Such a role would help us better understand the immune response observed in a lymphocyte-mediated skin disease such as atopic dermatitis and eczema.
THERAPEUTICS
In the past decade, tremendous strides have been made in finding new therapeutics to tackle genodermatoses such as EB, as well as heterogeneous carcinomas. The principal mechano-sensory role of integrins at the surface of the cell makes them an important part of the targeting/treatment scheme. Here, we aim to provide a brief overview of such studies that have enhanced the arsenal of treatment options available for various skin disorders.
Gene therapy
This line of therapy relies on individualized genomics-based correction of the mutant gene by introducing copies of a corrected version of the gene in the patient's own skin cells. For DDEB, supressing or silencing the faulty copy of the gene is a challenging and expensive strategy. Supplemental therapy, which tries to supplement the missing protein with a relative so as to abrogate the harmful effects, seems to be helpful in such EB cases. Transfection experiments using supplemental desmin to treat K5-K14 mutant EBS have been successful (CitationD’Alessandro et al., 2004). Transplantations of genetically modified epidermal stem cells have resulted in ex vivo genetic correction of nH-JEB (CitationMavilio et al., 2006) and also LAMB3 correction in H-JEB (CitationRobbins et al., 2001).
Recent study trials have also included treatment with Granulocyte Colony Stimulating Factor (GCSF) to stimulate the bone marrow to produce more granulocytes and stem cells and thus effectively treat DEB [Clinical Trial Identifier NCT01538862]. Also, ex vivo Gene transfer studies are ongoing at Stanford University [NCT01263379] targeting DEB. Specifically, the study aims to successfully graft LZRSE-Col7A1 Engineered Autologous Epidermal Sheets (LEAES) expressing the patient's missing Col VII.
The identification of appropriate drug candidates is more straightforward using siRNA than traditional pharmaceuticals, so development of siRNA-based therapeutics has progressed rapidly. Studies to test efficacy of combining irradiation treatments involving siRNA toward αvβ3 integrin in breast cancers (CitationCao et al., 2006) yielded positive results with enhanced apoptotic response as well as cell cycle arrest. Also, inhibition of ITGB3 by antisense was found to drastically limit metastases in B16 melanoma cells (CitationNasulewicz-Goldeman et al., 2012). Preclinical studies have shown that antisense therapy (to either ITGAV or ITGB3) suppressed the growth of sub-cutaneously injected human hepatocellular carcinoma cells (CitationLi et al., 2007). Much of the research in this field has been toward developing better delivery platforms dependent on integrin binding, rather than target integrins themselves. This has been particularly important in leukocyte-directed siRNA systems which depend on liposomal delivery having hyaluronan modified with anti-integrin mAb for receptivity (CitationPeer et al., 2008). Self-assembling RGD-ligand nanoparticles have shown good specificity in targeting tumor neovasculature expressing specific integrins (CitationSchiffelers et al., 2004) and can be used to deliver siRNA inhibiting VEGFR2 expression (and thereby angiogenesis) selectively. This is a vast and uncharted field, both from an integrin-targeting as well as an integrin-assisted targeting point of view.
Protein therapy
Another strategy being used involves treating these fragility disorders by providing enough of the correctly functioning protein. For example, RDEB treatment using intradermal injection of recombinant Collagen VII has ameliorative effects (CitationRemington et al., 2009) in murine models. The junctional fragility of EB makes engineered substitutes a highly worthwhile treatment option. Recently, a Phase III study for efficacy of ABH001 (a bio-resorbable scaffold derived from neonatal dermal fibroblasts) in patients with non-healing EB wounds [NCT01749306] was initiated. HP802-247 is another investigational allogeneic living human cell bioformulation that consists of a fibrinogen solution and a cell preparation containing a mixture of growth-arrested allogeneic epidermal keratinocytes and dermal fibroblasts. Another living cell-based product called Apligraf® has been commercialized for ulcers which consists of a bi-layered cell-based technology, composed of layers of differentiated keratinocytes and fibroblasts seeded in a collagen matrix, which is purported to close wounds faster [NCT00587223].
Cell therapy
This is an allied field of therapy; it involves providing donor cells which carry a correct copy of the gene from a healthy donor, or alternatively transplanting stem cells to re-form the skin strata. Reprograming of skin cells into induced pluripotent stem cells (iPSCs) for correcting deficiencies is another avenue at this forefront of therapeutics.
Local cell therapy are common where injection of donor fibroblast skin cells directly into the skin can improve wound healing, reduce blistering, and strengthen the skin. The benefits are limited to the areas injected and effects are temporary. Phase II trials of a fibroblast therapy for treating DEB as a collaboration between Kings College London, Guys and St Thomas Hospital and Intercytex have found promising results with increase in erosion-healing after treatment (CitationPetrof et al., 2013). Allogeneic mesenchymal stromal cells have been intradermally injected into two RDEB patients to effect re-epithelialization of ulcerated skin by replenishing Col VII (CitationConget et al., 2010).
Stem cell therapy involves transfer of pluripotent adult or induced stem cells into a patient to express missing factors. Umbilical cord blood-derived unrestricted somatic stem cells have been used to re-express missing Col VII and enhance wound healing in RDEB model mice (CitationLiao et al., 2013). Clinical trials (Phase II) of bone marrow transplantation (BMT) from healthy donors into children with RDEB at the University of Minnesota [NCT00478244] and Columbia University [NCT00881556] yielded preliminary positive results (CitationKiuru et al., 2010). Allogeneic BMT in six RDEB patients showed early clinical benefits regarding healing of lesions, with production of Col VII (CitationWagner et al., 2010). Phase II gene therapy trials with Allogeneic Stem Cell Transplantation are underway for patients of DEB to correct for mutational deficiencies and reduce skin fragility [NCT01033552]. Also, therapeutics technology to induce somatic cells into functional keratinocytes through patient-specific iPSCs to treat EB has been successfully implemented (CitationItoh et al., 2011; CitationTolar et al., 2011; CitationTolar et al., 2013). Another angle of therapeutics seeks to correct RDEB mutations via iPSCs by a “gene-correction” approach, whereby the gene is edited back to the correct normal sequence without disrupting surrounding DNA. This is achieved using transcription activator-like effector nucleases (TALENs), engineered proteins which function as molecular scissors and introduce normal sequences in-frame. Preliminary results using RDEB patient-derived cells show that it is possible to use TALENs to mediate a permanent gene repair in iPSCs in the laboratory which can then be introduced into a skin model (CitationOsborn et al., 2013). This, along with utilizing revertant mosaicism (CitationPasmooij et al., 2007) could pave the way for safer and more effective genetic correction of dermatoses.
Overall, SC therapy shows promise in obtaining sustainable treatment options, but the high-risk involved in pre-implantation procedures (such as radio/chemo-therapy) and post-transplant infection or rejection cannot be ignored.
Molecular or drug therapy
A very promising area of research, molecular drug therapy aims to synthesize or mimic molecules/drugs which can increase or decrease the activity of EB-associated genes, so that the skin microenvironment can revert to a normal state.
The extracellular ligand-binding domains are the only regions accessible using antibody-based therapeutics, which constitute a large portion of the integrin antagonists in clinical use and under development. A similar method of antagonism involves the binding of molecules or antibodies in close proximity to the natural ligand binding site. This strategy provides steric hindrance of the ligand binding site such that endogenous binding site is obscured and ligand binding is prevented. Targeting the intracellular “tails” of integrins and their post-translational modifications represents a major, yet underdeveloped, area of therapeutic approach to antagonism, owing to the large signaling machinery that revolves around and is influenced by it. Hence downstream effects on cytoskeletal dynamics or turn-over of FAs, as well as nuclear signaling can be manipulated pharmacologically in inflammatory and cell-cycle disorders. Also, targeting the metal-ion binding region can influence the activation state of the integrin heterodimer and hence is a significant area of future study.
Therapeutic integrin-related monoclonal antibodies (mAbs) are being tested for various diseases such as cancer, arthritis, Crohn's disease, multiple sclerosis (MS), coronary syndromes as well as psoriasis (). They are very pragmatic due to their specificity and affinity, while also having longer lifetimes as compared with small peptides.
Table 2. Members of integrin family and potential therapeutic use of antagonists (CitationPerdih & Dolenc, 2010).
The majority of integrin-antagonists target the angiogenic process essential to metastases. During tumor-enhancing processes such as angiogenesis, integrins are responsible for endothelial cell migration, proliferation, differentiation through FAK/Src/p130Cas, and Raf/MEK/Erk signaling pathways (CitationAvraamides et al., 2008). In addition, ligated state of integrin molecules promotes the increased expression of pro-survival molecules such as Bcl-2 and FLIP, increases NFkB signaling and decreases p53 activation. Thus, αv integrin antagonists are a hotbed of anti-angiogenesis research. A mAb molecule undergoing Phase I trials is MEDI-522 (Abergin) which targets αvβ3 integrin in patients with malignant melanomas [NCT00111696]. There have been multiple integrin antagonists in use which target αIIb-β3 integrins on platelets and megakaryocytes; including Abciximab, Eptifibatide, and Tirofiban. Numerous α5β1 integrin mAb drugs are currently under development (some are in clinical use) for the treatment of angiogenesis in cancers. Principle among them is the antibody drug Volociximab. Through its inhibition of α5β1, it induces apoptosis in vitro and in animal tumor models of neovasculature. Phase II trials involving volociximab in combination with regimens of Dacarbazine have been completed for the treatment of metastatic melanoma [NCT00099970]. Also Phase I trials have been completed for volociximab in combination with a standard treatment of carboplatin, paclitaxel, and bevacizumab in non-small cell lung cancer [NCT00666692]. Phase II trials in metastatic melanomas and renal cell carcinomas and platinum resistant ovarian cancer have been terminated. Phase I trials of volociximab for the treatment of macular degeneration have been completed and study results are expected [NCT00782093]. Another molecule under trials is the integrin α5β1-antagonist JSM6427 for treating neovascularization due to Age-related macular degeneration (AMD) [NCT00536016]. An mAb against integrin αvβ6, 6.3G9, was found to arrest the growth of human pharyngeal carcinoma cells both in vitro and in vivo (CitationVan Aarsen et al., 2008).
Peptide-based drugs may have high target affinity but may lack specificity, as the same ligand sequences are shared among many integrin heterodimers. Hence, steric constraints and flanking sequences are currently under study to enhance specificity of these integrin-targeted peptides. Another problem with peptides is their lowered stability in vivo. This circulation-lifetime can be enhanced in peptidomimetics by incorporation of D configuration-amino acids that are not recognized by host peptidases and hydrolases. RGD peptides such as Cilengitide Merck (EMD 121974), are being increasingly used as potent αvβ3 and αvβ5 inhibitors, due to their effect on cell–matrix adhesion and cytoskeletal dynamics through the FAK-Src-Akt pathway (CitationYamada et al., 2006) and by disrupting VE-cadherin (CitationAlghisi et al., 2009). There are multiple Phase II underway recruiting cilengitide, both as a single agent and along with standard therapies, in treatment of melanomas and glioblastomas (CitationNabors et al., 2007; CitationReardon et al., 2008; CitationStupp et al., 2010). Unfortunately, the Phase III trial of cilengitide along with temozolomide did not meet primary endpoint in patients with newly diagnosed glioblastoma patients [NCT00689221]. This underscores the complexity of such a heterogeneous disease as cancer, and emphasizes the challenges faced in drug development.
Small molecule inhibitors are low-MW synthetic drugs having specific affinity for suitable bio-chemical pathway components, and can provide targeted response inside a cell. They can be mass-produced by pharmacophore modeling, bio-isosteric substitution, and high-throughput library screening (CitationMillard et al., 2011). However, these molecules tend to be zwitterionic in nature, and can thus encounter limitations in bioavailability and integrin selectivity in vivo. There are multiple statins, including endostatin, tumstatin, and angiostatin that target integrins in angiogenic processes, and simvastatin that initiates integrin-targeted apoptosis in carcinomas (CitationEliceiri & Cheresh, 2001; CitationRehn et al., 2001).
Various non-RGD peptides targeting α5β1, ATN-161 being principal among them, are also being scrutinized due to their purported property to block metastases in breast cancers (CitationCianfrocca et al., 2006; CitationKhalili et al., 2006). Another such peptide undergoing pre-clinical studies is integrin αvβ3 antagonist PSK1404 (CitationZhao et al., 2007).
Endogenous integrin-inhibitors such as β tail-centric Filamins, DOK1 and α tail-centric SHARPIN, MDGI can be developed as viable therapeutic agents. A small cytosolic protein: Mammary-derived growth Inhibitor was found to interact with α-subunits and thus supress β1 Integrin activity and associated invasiveness by a different approach in Breast cancer patients (CitationNevo et al., 2010). This discovery was followed by studies on SHANK-associated RH domain-interacting protein (SHARPIN) which found it to be a general endogenous inhibitor of β1 integrins in vivo. Increased β1-integrin activity was observed in keratinocytes of Sharpin- compromised mice and suppression of SHARPIN in human peripheral blood leukocytes increased the migration on FN-coated surfaces (CitationRantala et al., 2011). Thus, such antagonists have tremendous potential to be utilized in internalization-based inhibition of aberrant integrins in various pathological states. Downstream signaling molecules can also impair integrin-mediated cell–matrix adhesion and motility. RSK2 (AGC Kinase) is one such substrate of ERK; it has been found to inhibit integrin β1 activation, by binding filamin in vitro (CitationGawecka et al., 2012), which results in actin cytoskeletal and FA disruption. This is another layer of modulation that underscores the vertical and horizontal network linking the ECM to cellular rearrangement and motility.
The cooperative involvement of multiple integrins in various diseases has encouraged the use of dual antagonists—single drugs having multiple target action. This can prove more effective and avoid the pharmacokinetic issues involved in taking multiple drugs simultaneously. The targets suitable for dual targeting are either different proteins in the same biochemical pathway with similar active sites, or the same protein involved in different pathways playing significant roles in the pathological state (CitationPerdih & Dolenc, 2010). Drug design should ensure promiscuous binding is prevented to avoid off-target activity. Many such multiple antagonists have been obtained, such as the triple integrin αvβ3/αvβ5/α5β1 antagonist which was found to regulate angiogenesis by MMP-independent apoptosis much better than selective αvβ3 antagonist in Type I collagen RGD-independent ECM (CitationMeerovitch et al., 2003).
Apart from the use of traditional monoclonal antibodies to inhibit integrins in various disorders, there has been a shift toward developing certain single-chain variable fragments (scFv) antagonists. ScFv are fusion proteins of the variable regions of the heavy and light chains of certain immunoglobulins, linked by a peptide chain of ˜12–25 amino acids.) Pre-clinical research is underway to study efficacy of scFv antagonists and their fragment peptides against αvβ6 in SCC (CitationKogelberg et al., 2008). Dimerized scFv (i.e. Diabodies) are known to have dissociation constants up to 40-fold lower than their corresponding scFv, meaning that they have a much higher affinity to their target. Hence these are attractive therapeutic as well as bio-labeling options, holding enormous potential due to their high specificity. Recently, a novel recombinant diabody has been developed in the UK that specifically targets αvβ6 and blocks its function (CitationKogelberg et al., 2013). Such molecules have a range of potential applications in bio-imaging and targeted delivery/inhibition.
Radiotracer RGD-peptide studies for use in studying cardiovascular angiogenesis and fibrosis as well as tumorigenesis has led to considerable excitement. Initial success has been achieved with 18F-Galacto-RGD and 18F-AH111585 that are radiopharmaceutical agents with avidity for αvβ3 and αvβ5 integrin [NCT01813045]. This has been followed by 68Ga-BNOTA-PRGD2 being tested for use as a non-invasive biosensor for PET imaging of αvβ3-rich tumors [NCT01527058]. Bio-imaging and contrast agents are a valuable tool in diagnostics, and integrin-based radio-peptides are crucial to our understanding of disease progression.
CONCLUSIONS AND FUTURE PERSPECTIVES
In the last few decades, tremendous advances have been made in the discovery and development of integrin targeted therapeutics, as well as in our understanding of the cellular partners of these multidimensional molecules. Years of intense research into integrin–ligand interaction (structural and functional) has provided potential applications in the treatment of mutational defects in inflammation, angiogenesis, neo-vascularization, and tumor growth. Some of the challenges in the next decade are to understand how integrins collaborate with other integrins and receptors within a network to deliver cues responsible for cell processes that range from migration to differentiation, and how this is influenced by local microenvironment. In this review, we have attempted to bridge the gap between the normal homeostasis of integrins (and their associated proteins) with the multiple disease states manifested by their dysfunction. Although by no means comprehensive, we have strived to put forward the exciting potential of integrin-based therapeutics while elucidating current study trials.
Although studies of human disorders by transplantation studies and studies using transgenic and/or knockout mice have generated valuable information, the fundamental difference between mouse and human epidermis necessitates more patient-sample studies as well as use of novel iPSC lines to gain better understanding of human phenotypes and underlying dysfunctioning. This will not only increase our understanding of integrin functions in skin homeostasis, stem cell quiescence, and wound healing, but may also lead to new strategies to tackle several human pathologies.
ACKNOWLEDGMENT
We thank members of the Raghavan lab for useful discussions. SH is supported through a Shyam Prasad Mukherjee Fellowship administered by CSIR. SR is supported in part by American Cancer Society grant RSG-12-053-01 and core funds from inStem.
Declaration of Interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Alghisi GC, Ponsonnet L, Ruegg C (2009). The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PLoS One. 4: e4449.
- Aoudjit F, Vuori K (2012). Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract. 2012: 283181.
- Avraamides CJ, Garmy-Susini B, Varner JA (2008). Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 8: 604–617.
- Bandyopadhyay A, Rothschild G, Kim S, Calderwood DA, Raghavan S (2012). Functional differences between kindlin-1 and kindlin-2 in keratinocytes. J Cell Sci. 125: 2172–2184.
- Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM (2005). Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 115: 339–347.
- Blanpain C, Fuchs E (2009). Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 10: 207–217.
- Bouvard D, Pouwels J, De Franceschi N, Ivaska J (2013). Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat Rev Mol Cell Biol. 14: 430–442.
- Brakebusch C, Grose R, Quondamatteo F, Ramirez A, Jorcano JL, Pirro A, Svensson M, Herken R, Sasaki T, Timpl R, Werner S, Fässler R (2000). Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 19: 3990–4003.
- Bruckner-Tuderman L, Has C (2013). Disorders of the cutaneous basement membrane zone-The paradigm of epidermolysis bullosa. Matrix Biol. (Epub ahead of print).
- Cagnet S, Faraldo MM, Kreft M, Sonnenberg A, Raymond K, Glukhova MA (2013). Signaling events mediated by alpha3beta1 integrin are essential for mammary tumorigenesis. Oncogene. (Epub ahead of print).
- Calderwood DA, Campbell ID, Critchley DR (2013). Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. 14: 503–517.
- Cao Q, Cai W, Li T, Yang Y, Chen K, Xing L, Chen X (2006). Combination of integrin siRNA and irradiation for breast cancer therapy. Biochem Biophys Res Commun. 351: 726–732.
- Carroll JM, Romero MR, Watt FM (1995). Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 83: 957–968.
- Charlesworth A, Gagnoux-Palacios L, Bonduelle M, Ortonne JP, De Raeve L, Meneguzzi G (2003). Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J Invest Dermatol. 121: 1344–1348.
- Chen K, Tu Y, Zhang Y, Blair HC, Zhang L, Wu C (2008). PINCH-1 regulates the ERK-Bim pathway and contributes to apoptosis resistance in cancer cells. J Biol Chem. 283: 2508–2517.
- Chometon G, Zhang ZG, Rubinstein E, Boucheix C, Mauch C, Aumailley M (2006). Dissociation of the complex between CD151 and laminin-binding integrins permits migration of epithelial cells. Exp Cell Res. 312: 983–995.
- Cianfrocca ME, Kimmel KA, Gallo J, Cardoso T, Brown MM, Hudes G, Lewis N, Weiner L, Lam GN, Brown SC, Shaw DE, Mazar AP, Cohen RB (2006). Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer. 94: 1621–1626.
- Conget P, Rodriguez F, Kramer S, Allers C, Simon V, Palisson F, Gonzalez S, Yubero MJ (2010). Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 12: 429–431.
- Crampton SP, Wu B, Park EJ, Kim JH, Solomon C, Waterman ML, Hughes CC (2009). Integration of the beta-catenin-dependent Wnt pathway with integrin signaling through the adaptor molecule Grb2. PLoS One. 4: e7841.
- D’Alessandro M, Morley SM, Ogden PH, Liovic M, Porter RM, Lane EB (2004). Functional improvement of mutant keratin cells on addition of desmin: an alternative approach to gene therapy for dominant diseases. Gene Ther. 11: 1290–1295.
- Deng X, Li Q, Hoff J, Novak M, Yang H, Jin H, Erfani SF, Sharma C, Zhou P, Rabinovitz I, Sonnenberg A, Yi Y, Zhou P, Stipp CS, Kaetzel DM, Hemler ME, Yang XH (2012). Integrin-associated CD151 drives ErbB2-evoked mammary tumor onset and metastasis. Neoplasia. 14: 678–689.
- Desgrosellier JS, Cheresh DA (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 10: 9–22.
- Devbhandari RP, Shi GM, Ke AW, Wu FZ, Huang XY, Wang XY, Shi YH, Ding ZB, Xu Y, Dai Z, Fan J, Zhou J (2011). Profiling of the tetraspanin CD151 web and conspiracy of CD151/integrin beta1 complex in the progression of hepatocellular carcinoma. PLoS One. 6: e24901.
- Dougherty GW, Jose C, Gimona M, Cutler ML (2008). The Rsu-1-PINCH1-ILK complex is regulated by Ras activation in tumor cells. Eur J Cell Biol. 87: 721–734.
- Duffy MJ (2002). Urokinase-type plasminogen activator: a potent marker of metastatic potential in human cancers. Biochem Soc Trans. 30: 207–210.
- Eliceiri BP, Cheresh DA (2001). Adhesion events in angiogenesis. Curr Opin Cell Biol. 13: 563–568.
- Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, McGrath JA, Mellerio JE, Murrell DF, Shimizu H, Uitto J, Vahlquist A, Woodley D, Zambruno G (2008). The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 58: 931–950.
- Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C (2009). Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986–2006. J Am Acad Dermatol. 60: 203–211.
- Fuchs E, Raghavan S (2002). Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 3: 199–209.
- Gawecka JE, Young-Robbins SS, Sulzmaier FJ, Caliva MJ, Heikkila MM, Matter ML, Ramos JW (2012). RSK2 protein suppresses integrin activation and fibronectin matrix assembly and promotes cell migration. J Biol Chem. 287: 43424–43437.
- Giancotti FG, Ruoslahti E (1990). Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell. 60: 849–859.
- Gozgit JM, Pentecost BT, Marconi SA, Otis CN, Wu C, Arcaro KF (2006). Use of an aggressive MCF-7 cell line variant, TMX2-28, to study cell invasion in breast cancer. Mol Cancer Res. 4: 905–913.
- Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E (1995). Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 81: 233–243.
- Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, Giancotti FG (2006). Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 126: 489–502.
- Has C, Sparta G, Kiritsi D, Weibel L, Moeller A, Vega-Warner V, Waters A, He Y, Anikster Y, Esser P, Straub BK, Hausser I, Bockenhauer D, Dekel B, Hildebrandt F, Bruckner-Tuderman L, Laube GF (2012). Integrin alpha3 mutations with kidney, lung, and skin disease. N Engl J Med. 366: 1508–1514.
- Heinemann A, He Y, Zimina E, Boerries M, Busch H, Chmel N, Kurz T, Bruckner-Tuderman L, Has C (2011). Induction of phenotype modifying cytokines by FERMT1 mutations. Hum Mutat. 32: 397–406.
- Herz C, Aumailley M, Schulte C, Schlotzer-Schrehardt U, Bruckner-Tuderman L, Has C (2006). Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J Biol Chem. 281: 36082–36090.
- Hynes RO (2002). Integrins: bidirectional, allosteric signaling machines. Cell. 110: 673–687.
- Itoh M, Kiuru M, Cairo MS, Christiano AM (2011). Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A. 108: 8797–8802.
- Janes SM, Watt FM (2006). New roles for integrins in squamous-cell carcinoma. Nat Rev Cancer. 6: 175–183.
- Karamatic Crew V, Burton N, Kagan A, Green CA, Levene C, Flinter F, Brady RL, Daniels G, Anstee DJ (2004). CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood. 104: 2217–2223.
- Khalili P, Arakelian A, Chen G, Plunkett ML, Beck I, Parry GC, Donate F, Shaw DE, Mazar AP, Rabbani SA (2006). A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther. 5: 2271–2280.
- Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, Campbell ID, Ylanne J, Calderwood DA (2006). The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 21: 337–347.
- Kiuru M, Itoh M, Cairo MS, Christiano AM (2010). Bone marrow stem cell therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 28: 371–382, xii–xiii.
- Kloeker S, Major MB, Calderwood DA, Ginsberg MH, Jones DA, Beckerle MC (2004). The Kindler syndrome protein is regulated by transforming growth factor-beta and involved in integrin-mediated adhesion. J Biol Chem. 279: 6824–6833.
- Kogelberg H, Tolner B, Thomas GJ, Di Cara D, Minogue S, Ramesh B, Sodha S, Marsh D, Lowdell MW, Meyer T, Begent RH, Hart I, Marshall JF, Chester K (2008). Engineering a single-chain Fv antibody to alpha v beta 6 integrin using the specificity-determining loop of a foot-and-mouth disease virus. J Mol Biol. 382: 385–401.
- Kogelberg H, Miranda E, Burnet J, Ellison D, Tolner B, Foster J, Picon C, Thomas GJ, Meyer T, Marshall JF, Mather SJ, Chester K (2013). Generation and Characterization of a Diabody Targeting the alphavbeta6 Integrin. PLoS One. 8: e73260.
- Kwon MJ, Park S, Choi JY, Oh E, Kim YJ, Park YH, Cho EY, Nam SJ, Im YH, Shin YK, Choi YL (2012). Clinical significance of CD151 overexpression in subtypes of invasive breast cancer. Br J Cancer. 106: 923–930.
- Lai-Cheong JE, Ussar S, Arita K, Hart IR, McGrath JA (2008). Colocalization of kindlin-1, kindlin-2, and migfilin at keratinocyte focal adhesion and relevance to the pathophysiology of Kindler syndrome. J Invest Dermatol. 128: 2156–2165.
- Lai-Cheong JE, Tanaka A, Hawche G, Emanuel P, Maari C, Taskesen M, Akdeniz S, Liu L, McGrath JA (2009). Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 160: 233–242.
- Lai-Cheong JE, Parsons M, McGrath JA (2010). The role of kindlins in cell biology and relevance to human disease. Int J Biochem Cell Biol. 42: 595–603.
- Laimer M, Lanschuetzer CM, Diem A, Bauer JW (2010). Herlitz junctional epidermolysis bullosa. Dermatol Clin. 28: 55–60.
- Legate KR, Montanez E, Kudlacek O, Fassler R (2006). ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 7: 20–31.
- Li J, Tan H, Dong X, Xu Z, Shi C, Han X, Jiang H, Krissansen GW, Sun X (2007). Antisense integrin alphaV and beta3 gene therapy suppresses subcutaneously implanted hepatocellular carcinomas. Dig Liver Dis. 39: 557–565.
- Li Q, Li C, Zhang YY, Chen W, Lv JL, Sun J, You QS (2013). Silencing of integrin-linked kinase suppresses in vivo tumorigenesis of human ovarian carcinoma cells. Mol Med Rep. 7: 1050–1054.
- Liang F, Zhang S, Wang B, Qiu J, Wang Y (2013). Overexpression of integrin-linked kinase (ILK) promotes glioma cell invasion and migration and down-regulates E-cadherin via the NF-kappaB pathway. J Mol Histol. (Epub ahead of print).
- Liao Y, Itoh M, Yang A, Zhu H, Roberts S, Highet AM, Latshaw S, Mitchell K, van de Ven C, Christiano A, Cairo MS (2013). Human cord blood derived unrestricted somatic stem cells promote wound healing and have therapeutic potential for patients with recessive dystrophic epidermolysis bullosa. Cell Transplant. (Epub ahead of print).
- Lim S, Kawamura E, Fielding AB, Maydan M, Dedhar S (2013). Integrin-linked kinase regulates interphase and mitotic microtubule dynamics. PLoS One. 8: e53702.
- Liu Q, Xiao L, Yuan D, Shi X, Li P (2012). Silencing of the integrin-linked kinase gene induces the apoptosis in ovarian carcinoma. J Recept Signal Transduct Res. 32: 120–127.
- Lorenz K, Grashoff C, Torka R, Sakai T, Langbein L, Bloch W, Aumailley M, Fassler R (2007). Integrin-linked kinase is required for epidermal and hair follicle morphogenesis. J Cell Biol. 177: 501–513.
- Macias-Perez I, Borza C, Chen X, Yan X, Ibanez R, Mernaugh G, Matrisian LM, Zent R, Pozzi A (2008). Loss of integrin alpha1beta1 ameliorates Kras-induced lung cancer. Cancer Res. 68: 6127–6135.
- Margadant C, Charafeddine RA, Sonnenberg A (2010). Unique and redundant functions of integrins in the epidermis. FASEB J. 24: 4133–4152.
- Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, Maruggi G, Ferrari G, Provasi E, Bonini C, Capurro S, Conti A, Magnoni C, Giannetti A, De Luca M (2006). Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 12: 1397–1402.
- McCarty JH, Barry M, Crowley D, Bronson RT, Lacy-Hulbert A, Hynes RO (2008). Genetic ablation of alphav integrins in epithelial cells of the eyelid skin and conjunctiva leads to squamous cell carcinoma. Am J Pathol. 172: 1740–1747.
- Meerovitch K, Bergeron F, Leblond L, Grouix B, Poirier C, Bubenik M, Chan L, Gourdeau H, Bowlin T, Attardo G (2003). A novel RGD antagonist that targets both alphavbeta3 and alpha5beta1 induces apoptosis of angiogenic endothelial cells on type I collagen. Vascul Pharmacol. 40: 77–89.
- Millard M, Odde S, Neamati N (2011). Integrin targeted therapeutics. Theranostics. 1: 154–188.
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM (1996). Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 135: 1633–1642.
- Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R (2008). Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 14: 325–330.
- Nabors LB, Mikkelsen T, Rosenfeld SS, Hochberg F, Akella NS, Fisher JD, Cloud GA, Zhang Y, Carson K, Wittemer SM, Colevas AD, Grossman SA. (2007). Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 25: 1651–1657.
- Nakrieko KA, Welch I, Dupuis H, Bryce D, Pajak A, St Arnaud R, Dedhar S, D’Souza SJ, Dagnino L (2008). Impaired hair follicle morphogenesis and polarized keratinocyte movement upon conditional inactivation of integrin-linked kinase in the epidermis. Mol Biol Cell. 19: 1462–1473.
- Nasulewicz-Goldeman A, Uszczynska B, Szczaurska-Nowak K, Wietrzyk J (2012). siRNA-mediated silencing of integrin beta3 expression inhibits the metastatic potential of B16 melanoma cells. Oncol Rep. 28: 1567–1573.
- Nevo J, Mai A, Tuomi S, Pellinen T, Pentikainen OT, Heikkila P, Lundin J, Joensuu H, Bono P, Ivaska J (2010). Mammary-derived growth inhibitor (MDGI) interacts with integrin alpha-subunits and suppresses integrin activity and invasion. Oncogene. 29: 6452–6463.
- Ohmatsu H, Kadono T, Sugaya M, Tomita M, Kai H, Miyagaki T, Saeki H, Tamaki K, Steeber DA, Tedder TF, Sato S (2010). alpha4beta7 Integrin is essential for contact hypersensitivity by regulating migration of T cells to skin. J Allergy Clin Immunol. 126: 1267–1276.
- Olasz EB, Yancey KB (2008). Bullous pemphigoid and related subepidermal autoimmune blistering diseases. Curr Dir Autoimmun. 10: 141–166.
- Oloumi A, McPhee T, Dedhar S (2004). Regulation of E-cadherin expression and beta-catenin/Tcf transcriptional activity by the integrin-linked kinase. Biochim Biophys Acta. 1691: 1–15.
- Osborn MJ, Starker CG, McElroy AN, Webber BR, Riddle MJ, Xia L, DeFeo AP, Gabriel R, Schmidt M, von Kalle C, Carlson DF, Maeder ML, Joung JK, Wagner JE, Voytas DF, Blazar BR, Tolar J (2013). TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 21: 1151–1159.
- Papanikolaou S, Bravou V, Gyftopoulos K, Nakas D, Repanti M, Papadaki H (2010). ILK expression in human basal cell carcinoma correlates with epithelial-mesenchymal transition markers and tumour invasion. Histopathology. 56: 799–809.
- Pasmooij AM, Pas HH, Bolling MC, Jonkman MF (2007). Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J Clin Invest. 117: 1240–1248.
- Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M (2008). Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science. 319: 627–630.
- Perdih A, Dolenc MS (2010). Small molecule antagonists of integrin receptors. Curr Med Chem. 17: 2371–2392.
- Petitclerc E, Stromblad S, von Schalscha TL, Mitjans F, Piulats J, Montgomery AM, Cheresh DA, Brooks PC (1999). Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 59: 2724–2730.
- Petrof G, Martinez-Queipo M, Mellerio JE, Kemp P, McGrath JA (2013). Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomised, vehicle-controlled trial. Br J Dermatol. (Epub ahead of print).
- Piwko-Czuchra A, Koegel H, Meyer H, Bauer M, Werner S, Brakebusch C, Fassler R (2009). Beta1 integrin-mediated adhesion signalling is essential for epidermal progenitor cell expansion. PLoS One. 4: e5488.
- Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E (2000). Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 150: 1149–1160.
- Rantala JK, Pouwels J, Pellinen T, Veltel S, Laasola P, Mattila E, Potter CS, Duffy T, Sundberg JP, Kallioniemi O, Askari JA, Humphries MJ, Parsons M, Salmi M, Ivaska J (2011). SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat Cell Biol. 13: 1315–1324.
- Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, Glantz M, Ravin P, Raizer JJ, Rich KM, Schiff D, Shapiro WR, Burdette-Radoux S, Dropcho EJ, Wittemer SM, Nippgen J, Picard M, Nabors LB (2008). Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 26: 5610–5617.
- Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, Pihlajaniemi T, Alitalo K, Vuori K (2001). Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci U S A. 98: 1024–1029.
- Remington J, Wang X, Hou Y, Zhou H, Burnett J, Muirhead T, Uitto J, Keene DR, Woodley DT, Chen M (2009). Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 17: 26–33.
- Robbins PB, Lin Q, Goodnough JB, Tian H, Chen X, Khavari PA (2001). In vivo restoration of laminin 5 beta 3 expression and function in junctional epidermolysis bullosa. Proc Natl Acad Sci U S A. 98: 5193–5198.
- Sachs N, Kreft M, van den Bergh Weerman MA, Beynon AJ, Peters TA, Weening JJ, Sonnenberg A (2006). Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol. 175: 33–39.
- Saito D, Kyakumoto S, Chosa N, Ibi M, Takahashi N, Okubo N, Sawada S, Ishisaki A, Kamo M (2013). Transforming growth factor-beta1 induces epithelial-mesenchymal transition and integrin alpha3beta1-mediated cell migration of HSC-4 human squamous cell carcinoma cells through Slug. J Biochem. 153: 303–315.
- Samanna V, Wei H, Ego-Osuala D, Chellaiah MA (2006). Alpha-V-dependent outside-in signaling is required for the regulation of CD44 surface expression, MMP-2 secretion, and cell migration by osteopontin in human melanoma cells. Exp Cell Res. 312: 2214–2230.
- Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC (2004). Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 32: e149.
- Shi X, Wu C (2008). A suppressive role of mitogen inducible gene-2 in mesenchymal cancer cell invasion. Mol Cancer Res. 6: 715–724.
- Shinkuma S, McMillan JR, Shimizu H (2011). Ultrastructure and molecular pathogenesis of epidermolysis bullosa. Clin Dermatol. 29: 412–419.
- Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS, Wilhelmsen KC, South AP, Smith FJ, Prescott AR, Wessagowit V, Oyama N, Akiyama M, Al Aboud D, Al Aboud K, Al Githami A, Al Hawsawi K, Al Ismaily A, Al-Suwaid R, Atherton DJ, Caputo R, Fine JD, Frieden IJ, Fuchs E, Haber RM, Harada T, Kitajima Y, Mallory SB, Ogawa H, Sahin S, Shimizu H, Suga Y, Tadini G, Tsuchiya K, Wiebe CB, Wojnarowska F, Zaghloul AB, Hamada T, Mallipeddi R, Eady RA, McLean WH, McGrath JA, Epstein EH (2003). Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet. 73: 174–187.
- Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH, Lane EB (1996). Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 13: 450–457.
- Stanchi F, Grashoff C, Nguemeni Yonga CF, Grall D, Fassler R, Van Obberghen-Schilling E (2009). Molecular dissection of the ILK-PINCH-parvin triad reveals a fundamental role for the ILK kinase domain in the late stages of focal-adhesion maturation. J Cell Sci. 122: 1800–1811.
- Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA (2001). Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol. 155: 459–470.
- Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, Grabenbauer GG, Ochsenbein AF, Simon M, Dietrich PY, Pietsch T, Hicking C, Tonn JC, Diserens AC, Pica A, Hermisson M, Krueger S, Picard M, Weller M (2010). Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 28: 2712–2718.
- Takeda Y, Li Q, Kazarov AR, Epardaud M, Elpek K, Turley SJ, Hemler ME (2011). Diminished metastasis in tetraspanin CD151-knockout mice. Blood. 118: 464–472.
- Timpl R, Brown JC (1996). Supramolecular assembly of basement membranes. Bioessays. 18: 123–132.
- Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, Wagner JE, Blazar BR (2011). Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 131: 848–856.
- Tolar J, Xia L, Lees CJ, Riddle M, McElroy A, Keene DR, Lund TC, Osborn MJ, Marinkovich MP, Blazar BR, Wagner JE (2013). Keratinocytes from induced pluripotent stem cells in junctional epidermolysis bullosa. J Invest Dermatol. 133: 562–565.
- Ussar S, Moser M, Widmaier M, Rognoni E, Harrer C, Genzel-Boroviczeny O, Fassler R (2008). Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 4: e1000289.
- Van Aarsen LA, Leone DR, Ho S, Dolinski BM, McCoon PE, LePage DJ, Kelly R, Heaney G, Rayhorn P, Reid C, Simon KJ, Horan GS, Tao N, Gardner HA, Skelly MM, Gown AM, Thomas GJ, Weinreb PH, Fawell SE, Violette SM (2008). Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res. 68: 561–570.
- Varner JA, Emerson DA, Juliano RL (1995). Integrin alpha 5 beta 1 expression negatively regulates cell growth: reversal by attachment to fibronectin. Mol Biol Cell. 6: 725–740.
- Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, Chen M, Riddle MJ, Osborn MJ, Lund T, Dolan M, Blazar BR, Tolar J (2010). Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 363: 629–639.
- Wang SE, Xiang B, Zent R, Quaranta V, Pozzi A, Arteaga CL (2009). Transforming growth factor beta induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 69: 475–482.
- Watt FM (2002). Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 21: 3919–3926.
- Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID (2007). Structural basis of integrin activation by talin. Cell. 128: 171–182.
- Weinstein EJ, Bourner M, Head R, Zakeri H, Bauer C, Mazzarella R (2003). URP1: a member of a novel family of PH and FERM domain-containing membrane-associated proteins is significantly over-expressed in lung and colon carcinomas. Biochim Biophys Acta. 1637: 207–216.
- White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ (2004). Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 6: 159–170.
- White SJ, McLean WH (2005). Kindler surprise: mutations in a novel actin-associated protein cause Kindler syndrome. J Dermatol Sci. 38: 169–175.
- Wickstrom SA, Lange A, Montanez E, Fassler R (2010). The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase!. EMBO J 29: 281–291.
- Wickstrom SA, Radovanac K, Fassler R (2011). Genetic analyses of integrin signaling. Cold Spring Harb Perspect Biol. 3.
- Wolfenson H, Lavelin I, Geiger B (2013). Dynamic regulation of the structure and functions of integrin adhesions. Dev Cell. 24: 447–458.
- Wright MD, Geary SM, Fitter S, Moseley GW, Lau LM, Sheng KC, Apostolopoulos V, Stanley EG, Jackson DE, Ashman LK (2004). Characterization of mice lacking the tetraspanin superfamily member CD151. Mol Cell Biol. 24: 5978–5988.
- Wu C (2004). The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim Biophys Acta. 1692: 55–62.
- Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (2006). Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery. 59: 1304–1312; discussion 1312.
- Yancey KB, Hintner H (2010). Non-herlitz junctional epidermolysis bullosa. Dermatol Clin. 28: 67–77.
- Yu Y, Wu J, Wang Y, Zhao T, Ma B, Liu Y, Fang W, Zhu WG, Zhang H (2012). Kindlin 2 forms a transcriptional complex with beta-catenin and TCF4 to enhance Wnt signalling. EMBO Rep. 13: 750–758.
- Zevian S, Winterwood NE, Stipp CS (2011). Structure-function analysis of tetraspanin CD151 reveals distinct requirements for tumor cell behaviors mediated by alpha3beta1 versus alpha6beta4 integrin. J Biol Chem. 286: 7496–7506.
- Zhao H, Ross FP, Teitelbaum SL (2005). Unoccupied alpha(v)beta3 integrin regulates osteoclast apoptosis by transmitting a positive death signal. Mol Endocrinol. 19: 771–780.
- Zhao Y, Bachelier R, Treilleux I, Pujuguet P, Peyruchaud O, Baron R, Clement-Lacroix P, Clezardin P (2007). Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 67: 5821–5830.
- Zhu CQ, Popova SN, Brown ER, Barsyte-Lovejoy D, Navab R, Shih W, Li M, Lu M, Jurisica I, Penn LZ, Gullberg D, Tsao MS (2007). Integrin alpha 11 regulates IGF2 expression in fibroblasts to enhance tumorigenicity of human non- small-cell lung cancer cells. Proc Natl Acad Sci U S A. 104: 11754–11759.
- Zhu XY, Liu N, Liu W, Song SW, Guo KJ (2012). Silencing of the integrin-linked kinase gene suppresses the proliferation, migration and invasion of pancreatic cancer cells (Panc-1). Genet Mol Biol. 35: 538–544.