
Abstract
Desmosomes are intercellular junctions that provide tissues with structural stability. These junctions might also act as signaling centers that transmit environmental clues to the cell, thereby affecting cell differentiation, migration, and proliferation. The importance of desmosomes is underscored by devastating skin and heart diseases caused by mutations in desmosomal genes. Recent observations suggest that abnormal desmosomal protein expression might indirectly contribute to skin disorders previously not linked to these proteins. For example, it has been postulated that reduced desmosomal protein expression occurs in patients affected by Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC), a skin fragility disorder caused by mutations in the transcription factor TP63. Currently, it is not clear how these changes in desmosomal gene expression contribute to AEC. We will discuss new approaches that combine in vitro and in vivo models to elucidate the role of desmosomal gene deregulation in human skin diseases such as AEC.
DESMOSOMES—CONTEXT AND HISTORICAL PERSPECTIVE
Desmosomes are multiprotein complexes (junctions) that are assembled at the plasma membrane (). These junctions serve a dual function: they mediate cell–cell attachment and they provide anchorage points for cytoplasmic intermediate filaments (IF). As such, desmosomes and the IF cytoskeleton form a three-dimensional network of structural proteins that confers resistance to tissues and organs that are exposed to significant mechanical stress forces, such as the skin and the heart (CitationPetrof et al., 2012; CitationSchmidt & Koch, 2007; CitationCheng et al., 2005; CitationGaneshan et al., 2010).
Figure 1. Desmosomal structure and composition. (A) Electron micrograph of a desmosome from mouse epidermis. The arrows demarcate the positions of the plasma membranes of neighboring cells. The brackets indicate the location of the cytoplasmic plaques. (B) Schematic representation of the proteins that assemble into desmosomes. Note that the transmembrane components (DSG and DSC) connect on the cytoplasmic surface of the desmosome with the plaque proteins (JUP, DSP and PKP) which in turn link to the intermediate filament (IF) cytoskeleton.
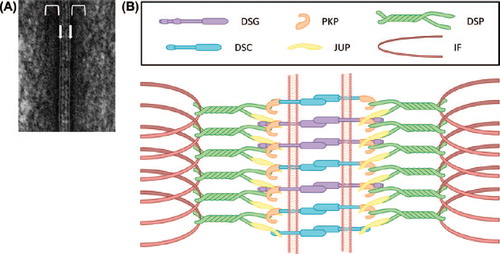
Desmosomes consist of a core of type-1 transmembrane glycoproteins (desmogleins [DSG] and desmocollins [DSC]) that connect neighboring cells by participating in hetero- and/or homophilic interactions in the intercellular space (CitationChitaev et al., 1996; CitationNie et al., 2011). On the cytoplasmic side of the plasma membrane, DSGs and DSCs interact with plaque proteins (desmoplakin [DSP], plakophilins [PKP], and plakoglobin [JUP]) (). These plaque proteins in turn connect DSGs and DSCs to the IF cytoskeleton. Based on their predicted amino acid sequences, DSGs and DSCs are classified as members of the cadherin subfamily, a group of calcium-dependent cell adhesion proteins (CitationKoch et al., 1990; CitationKoch et al., 1991a, Citation1991b; CitationBuxton et al., 1993). Further, DSGs and DSCs each comprise a group of multiple related proteins (DSG1-4 and DSC1-3 in humans [CitationBuxton et al., 1993; CitationMahoney et al., 2006]), each of which exhibit tissue- and cell type-specific expression patterns. For example, all DSG and DSC proteins are expressed in the skin, whereas other tissues and cell types express subsets of desmosomal cadherins [e.g. (CitationKoch et al., 1992; CitationTheis et al., 1993)].
Abnormalities in desmosome structure or function underlie a subset of human disorders, mainly affecting the skin and the heart (CitationPetrof et al., 2012). The first diseases unequivocally linked to impaired desmosome function were the autoimmune disorders pemphigus vulgaris and pemphigus foliaceus (CitationAmagai & Stanley, 2012). In these diseases, auto-antibodies are generated against DSGs, leading to blistering lesions in mucous membranes and the skin. Further, auto-antibodies against DSC3 have been linked to atypical pemphigus (CitationRafei et al., 2011; CitationHatano et al., 2012). In addition to autoimmune responses targeting desmosomal proteins, mutations in seven desmosomal genes have now been linked to disorders affecting the structure and function of skin and skin appendages (CitationPetrof et al., 2012). Further, a severe form of heart disease, arrhythmogenic right ventricular cardiomyopathy (ARVC), has been linked to mutations in desmosomal genes [JUP, DSC2, DSG2, DSP, and PKP2 (CitationPetrof et al., 2012; CitationLi Mura et al., 2013)].
INSIGHTS INTO THE ROLE OF DESMOSOMAL PROTEINS FROM IN VIVO STUDIES
To gain further insight into the role of individual desmosomal proteins, several mouse lines with null mutations in desmosomal genes have been developed since the early 1990s (e.g., CitationGaneshan et al., 2010; CitationCheng et al., 2005; CitationCheng & Koch, 2004). As describing the individual role of each protein analyzed would exceed the scope of this manuscript, we will highlight a few major conclusions derived from these animal studies. Interestingly, although loss of some desmosomal proteins in mice led to viable mice that could be analyzed, loss of others was found to be incompatible with life. Embryonic lethality was noted, for example, in mice with null mutations in Jup, Dsp, and Dsc3 (CitationRuiz et al., 1996; CitationBierkamp et al., 1996; CitationGallicano et al., 2001, Citation1998; CitationDen et al., 2006). To overcome the embryonic lethality, tissue-specific null mutations were introduced into desmosomal genes required for embryogenesis (e.g., CitationChen et al., 2008; CitationVasioukhin et al., 2001; CitationLi et al., 2011). One key finding obtained by analyzing these mice was that many desmosomal proteins are indeed required to maintain tissue adhesion, as demonstrated, for example, by the blistering skin and hair loss phenotypes of Dsg3 and Dsc3 null mice (CitationKoch et al., 1997; CitationChen et al., 2008). In addition, loss of some desmosomal proteins, such as DSP, led to defects in keratinocyte differentiation (CitationVasioukhin et al., 2001). Finally, as expected, severe heart defects were observed when desmosomal proteins expressed in the heart were inactivated in genetically engineered mice (CitationLi et al., 2011).
In many cases, the phenotypes of animals with impaired desmosome function provided the rationale to investigate the role of desmosomal proteins in human skin and heart diseases.
However, we are far from fully understanding the role of desmosomal proteins in human disease; although it is clear that complete loss of desmosomal proteins will lead to severe phenotypes, it is not clear whether more subtle changes in desmosomal gene expression contribute to human diseases. For example, increased as well as decreased expression of desmosomal genes has been observed in human cancers (e.g. CitationChen et al., 2011 and references therein). Developing appropriate animal models will be essential in these cases to establish a causal link between these expression changes and cancer development and progression. In addition, desmosomal abnormalities may be present in skin fragility syndromes with primary defects in upstream regulators of desmosomal genes. Few upstream regulators of desmosomal genes have been identified so far, but their future identification is anticipated to facilitate a more comprehensive understanding of different types of skin fragility disorders (e.g. CitationTokonzaba et al., 2013 and references therein).
In the following section, we will discuss new disease models which will be essential to understand the role of desmosomal proteins in skin fragility. The specific example we will focus on is Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC), an ectodermal dysplasia caused by mutations in the transcription factor-encoding gene TP63 (CitationMcGrath et al., 2001). As outlined below, it has recently been discovered that desmosomal proteins are deregulated in the skin of AEC patients. However, the contribution of these desmosomal defects to the skin fragility observed in affected individuals is currently not known. We will use this disorder as an example to highlight the challenges faced in modeling diseases and the advantages of utilizing a combination of animal and human cell-based models to understand disease mechanisms.
AEC
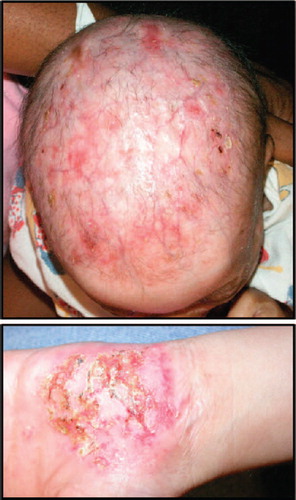
AEC is an ectodermal dysplasia characterized by the presence of severe skin erosions, often located to the scalp (). In addition to skin erosions, clinical features of AEC include abnormalities in appendages such as hair, nail, teeth, sweat glands, and limbs as well as the presence of cleft lip and/or palate. The severe skin erosions place AEC patients at high risk for local and systemic infections, and represent the main cause for the morbidity and mortality associated with AEC (CitationJulapalli et al., 2009; CitationSiegfried et al., 2005; CitationVanderhooft et al., 1993). AEC is caused by dominant mutations in TP63, a gene that encodes multiple transcription factors that are critical for the development and homeostasis of the skin and skin appendages (CitationYang et al., 1998; CitationKoster, 2010). Although all TP63 isoforms can be detected in the epidermis at the transcript level, it is well-established that the ΔNp63α isoform is the predominantly expressed isoform at the protein level, at least under homeostatic conditions (CitationParsa et al., 1999; CitationTruong et al., 2006; CitationYang et al., 1998; CitationTestoni & Mantovani, 2006). Interestingly, AEC-causing TP63 mutations cluster in exons encoding the SAM domain, a domain that is only present in isoforms that contain the α C-terminus, such as ΔNp63α (CitationMcGrath et al., 2001; CitationRinne et al., 2007). Within the epidermis, ΔNp63α expression levels are highest in the basal layer, and its expression declines rapidly during keratinocyte differentiation through phosphorylation- and ubiquitin-dependent protein degradation (CitationBellomaria et al., 2010; CitationBrowne et al., 2011; CitationDi Costanzo et al., 2009; CitationYang et al., 1998). Although the expression pattern is well-described, transcriptional networks controlled by ΔNp63α in normal or diseased skin are still relatively poorly characterized. A review of known pathways controlled by ΔNp63α has been provided elsewhere and is beyond the scope of the current manuscript (CitationKoster, 2010).
Figure 2. Clinical presentation of AEC patients. Scalp and palmar erosions on two patients affected by AEC. Patient images provided by the National Foundation for Ectodermal Dysplasias (NFED).
DEREGULATION OF DESMOSOMAL PROTEINS AND GENES IN AEC
Despite the severity of skin erosions in AEC patients, few studies have addressed the cellular and molecular abnormalities leading to the apparent skin fragility. This is caused, in part, by the lack of availability of skin samples for this rare disorder. We obtained skin biopsies from AEC patients, thus allowing us to interrogate abnormal expression of genes and proteins associated with skin integrity (CitationFete et al., 2009; CitationKoster et al., 2009; CitationBeaudry et al., 2009). Consistent with reports using smaller sample sets, our analysis of AEC patient skin revealed the presence of suprabasal proliferation, impaired terminal differentiation, and abnormal deposition of basement membrane components (CitationKoster et al., 2009; CitationMarinari et al., 2009; CitationClements et al., 2012; CitationBrowne et al., 2011; CitationMcGrath et al., 2001).
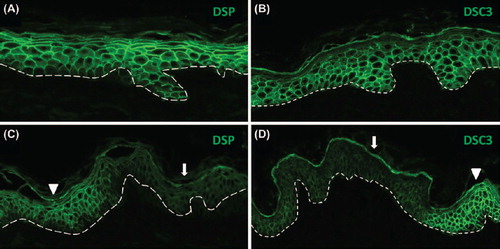
In addition to these abnormalities, the skin fragility exhibited by AEC patients suggested that desmosomal protein expression or localization may be aberrant in AEC patient skin. In support of this hypothesis, several desmosomal genes, including DSP, DSC3, DSG1, and PERP, were found to be under direct transcriptional control of ΔNp63α in keratinocytes (CitationFerone et al., 2013; CitationIhrie et al., 2005). Further, expression of some desmosomal genes was reported to be compromised in AEC patients (CitationBeaudry et al., 2009; CitationFerone et al., 2013), and structural desmosomal abnormalities were observed in one AEC patient (CitationPayne et al., 2005). However, a systematic analysis of desmosomal abnormalities in AEC patient skin has not been reported. Preliminary data from our group demonstrate that expression of DSP and DSC3 is deregulated in a subset of AEC patient skin samples (). These findings are of particular interest since genetic ablation of Dsc3 and Dsp in mouse skin has been associated with loss of cell adhesion (CitationVasioukhin et al., 2001; CitationChen et al., 2008). Further, Dsp null mutations result in keratinocyte differentiation defects in the mouse (CitationVasioukhin et al., 2001), a feature also observed in AEC patient skin (CitationKoster et al., 2009; CitationClements et al., 2012; CitationMarinari et al., 2009).
Figure 3. Focal downregulation of DSP and DSC3 expression in non-lesional skin of an AEC patient. (A,B) Normal human skin and (C, D) skin of an AEC patient. The dashed line demarcates the epidermal–dermal junction. The arrowheads in panels (C) and (D) point to areas of normal DSP and DSC3 staining, respectively, while the arrows indicate area in which expression of either DSC3 or DSP is significantly downregulated.
MODELING AEC IN MICE
Our data, together with published work, clearly demonstrate that desmosomal abnormalities occur in AEC patients. However, these data do not provide insight into the cause or consequence of these abnormalities. To further understand the role of desmosomal protein deregulation in AEC patient skin, it is critical to generate models that replicate the disease. Several mouse models aimed at understanding the role of mutant ΔNp63α proteins expressed in AEC patients (ΔNp63α-AEC) have been generated. The first, generated by our group, is based on the premise that ΔNp63α-AEC proteins function, at least in part, as dominant-negative molecules towards wild-type ΔNp63α (ΔNp63α-wt) protein (CitationKoster et al., 2009; CitationMarinari et al., 2009; CitationLopardo et al., 2008; CitationBrowne et al., 2011). To mimic this dominant-negative effect in a mouse model, we designed genetically engineered mice that allow for the downregulation of ΔNp63 proteins through the inducible expression of a ΔNp63-specific siRNA in the epidermis (CitationKoster et al., 2007). Upon epidermal-specific ΔNp63 downregulation, these mice developed skin erosions within a few days. These erosions mimicked erosions observed in AEC patients on a cellular and molecular level, thus highlighting the value of this model for further understanding AEC (CitationKoster et al., 2009). However, despite the usefulness of this model, it does not take into account that mechanisms other than a dominant-negative effect may contribute to the skin fragility in AEC patients. For example, it has been postulated that ΔNp63α-AEC proteins may harbor gain-of-function effects towards specific genes, although evidence for this hypothesis is currently lacking (CitationChung et al., 2011; CitationBrunner et al., 2002). To overcome limitations of this siRNA-based mouse model, another group generated mice in which an AEC-causing mutation was engineered into the endogenous Trp63 gene locus (CitationFerone et al., 2012). In principle, this model should allow for a more detailed understanding of the role of ΔNp63α-AEC proteins in the epidermis. However, although several features of AEC were replicated in these mice, most notably the presence of a cleft palate, the skin phenotype did not mimic that of AEC patients. In fact, whereas AEC patient skin is hyperplastic and exhibits suprabasal proliferation (CitationKoster et al., 2009; CitationMarinari et al., 2009; CitationClements et al., 2012; CitationBrowne et al., 2011; CitationMcGrath et al., 2001), the epidermis of the mice is hypoplastic and exhibits reduced proliferation (CitationFerone et al., 2012).
NEW APPROACHES OF MODELING COMPLEX DISEASES IN VITRO
As outlined above, mouse models have been extremely informative in modeling aspects of AEC. However, significant discrepancies in the skin phenotype of mice expressing a TP63-AEC mutation and AEC patient skin indicate that the mouse model does not truly mimic all aspects of the disease. One possible explanation is a difference in the biology of mouse and human skin. To overcome this issue, in vitro approaches using human cell-based systems were designed. The Khavari group ectopically expressed ΔNp63α-AEC proteins in 3D skin equivalents generated from discarded human tissue (CitationZarnegar et al., 2012; CitationSen et al., 2012). Interestingly, deregulation of several desmosomal genes was observed in keratinocytes that ectopically expressed the ΔNp63α-AEC protein. Ectopic expression of mutant genes is a potentially powerful approach to assess the function of mutated transcription factors. Disadvantages of this system, however, include the use of viral vectors to express genes which randomly integrate into the keratinocyte genome, mutant gene expression levels that do not reflect the balance of wild-type and mutant transcripts found in patient cells, and the inability to mimic the effects of a possible change in the ratio of different splice variants of TP63 (e.g., ΔNp63α and ΔNp63β) in patient keratinocytes. Lastly, the use of genetically heterogeneous sources of keratinocytes in this type of study is of concern. In fact, genetic background effects are well-known to influence the severity of disease phenotypes as demonstrated by the clinical variability observed in related patients carrying the same TP63 mutation (CitationBertola et al., 2004; CitationClements et al., 2010; CitationDianzani et al., 2003). Further, discarded patient skin is often derived from different body sites. It remains to be determined whether keratinocytes obtained from foreskin and breast tissue, for example, show identical transcriptome changes in response to the expression of mutant TP63. Thus, using patient cells from defined anatomical locations represent an ideal approach to study the disease mechanisms.
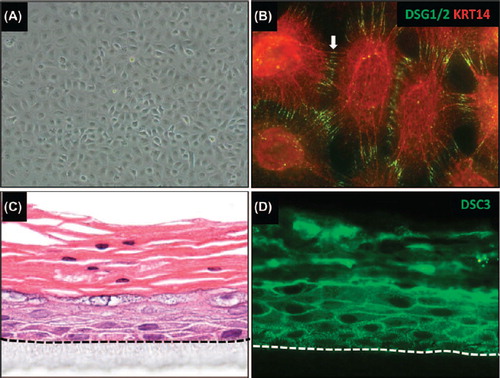
What would be the biological requirements for an ideal human cell-based in vitro model to study the patho-physiology of AEC? The system would (i) harbor disease-causing TP63 mutations and (ii) mimic the AEC skin phenotype at the cellular and the molecular levels. Further, to generate reproducible results in a genetically defined system, the model has to be renewable. Induced pluripotent stem cell (iPSC) technology has provided an ideal tool for the generation of tissue models as it allows for the generation of unlimited supplies of human iPSC that can be differentiated into keratinocytes and 3D epidermal equivalents [(CitationItoh et al., 2011; CitationTolar et al., 2011); ]. To generate iPSC, a set of transcription factors (reprogramming factors) is introduced into somatic cells (e.g., skin fibroblasts isolated from a skin biopsy (CitationPark et al., 2008b; CitationPark et al., 2008a; CitationOkita et al., 2007; CitationTakahashi et al., 2007)). These reprogramming factors dedifferentiate somatic cells to a state similar to that of embryonic stem (ES) cells. These cells can then be differentiated into keratinocytes. Because iPSC can be amplified indefinitely, this technology enables investigators to produce unlimited quantities of patient-derived keratinocytes, thereby circumventing the problem of the limited lifespan and amplification potential of cultured primary human keratinocytes in vitro (CitationGreen et al., 1977; CitationChapman et al., 2010).
Figure 4. Generating in vitro tissue models using iPSC-derived keratinocytes. (A) iPSC-derived keratinocytes were generated following a protocol similar to a previously published procedure (CitationItoh et al., 2011). (B) iPSC-derived keratinocytes express keratin 14 (KRT14) and desmoglein 1/2 (DSG1/2). Note that these cells also express other epithelial markers, such as keratin 5, TP63, and α6β4 integrin (data not shown). (C) in vitro skin equivalent generated from human iPSC-derived keratinocytes on an artificial surface. Note that the epithelium stratified with a notable granular and cornified layer. Parakeratosis (retention of nuclei in the stratum corneum) occurs occasionally in in vitro skin equivalents. (D) Staining of the skin equivalent shown in (C) with an antibody for desmocollin 3 (DSC3). Note that these iPSC-derived skin equivalents also express other epithelial markers such as KRT5/14, KRT1/10, and loricrin (data not shown). The dotted line demarcates the junction between stratified epithelium and the artificial matrix on which the cells are growing.
iPSC-based approaches also allow for the correction of point mutations in the TP63 gene of AEC patients. The development of genome editing tools such as zinc finger nucleases, TALEN and CRISPR/CAS (CitationHockemeyer et al., 2009; CitationHockemeyer et al., 2011; CitationMali et al., 2013; CitationCong et al., 2013) enables us to correct these mutations and generate pairs of conisogenic iPSC-derived keratinocytes that differ only with respect to the presence or absence of a TP63-AEC mutation. The identical genetic background of these pairs of cells facilitates disease pathway analysis using advanced transcriptome and proteome tools. Because this approach eliminates genetic background variation effects, it is conceptually superior to using “normal controls” from donors without TP63 mutations.
A potentially unlimited source of patient iPSC-derived keratinocytes enables us to generate in vitro skin equivalents designed to mimic the histo-pathology of AEC. Further, xenotransplantation approaches (CitationLichti et al., 2008), that is, the generation of human epidermis with iPSC-derived keratinocytes on immunosuppressed mice, will enable researchers to evaluate AEC epidermis in an in vivo environment. Of particular interest with respect to desmosomal proteins is the question of whether these systems will be able to reproduce the aberrant expression patterns of key targets in AEC, such as DSP and DSC3. Next, it will be possible to define the signaling pathways that lead to the deregulation of desmosomal genes in this context using advanced transcriptome and proteome analyses. Finally, using conventional overexpression and knockdown experiments, this in vitro skin equivalent system should be ideally suited to mechanistically define the contribution of individual desmosomal proteins to the skin fragility observed in AEC patient skin.
SUMMARY AND CONCLUSIONS
Although it is undisputed that null mutations in many desmosomal genes can cause severe diseases in humans, it is not clear in which way more subtle changes in desmosomal gene expression or protein synthesis contribute to skin disorders such as AEC. The case of AEC is of particular interest. Several desmosomal genes are targets of the transcription factor TP63. However, only a few desmosomal genes are downregulated in nonlesional skin of AEC patients, in particular DSC3 and DSP. It will be of interest to determine whether reduced expression of these genes contributes to the tissue fragility and keratinocyte differentiation defects observed in the skin of AEC patients. Given that mouse models have limitations in their ability to mimic the AEC skin phenotype, we propose that new human cell-based tissue models of this disease are needed. Using stem cell (iPSC) technology, we now have the tools to generate patient-derived keratinocytes for cell biological, transcriptome, and proteome analysis. This technology also enables us to generate conisogenic pairs of cells from patients that differ only with respect to the presence and absence, respectively, of disease causing mutations such as those causing AEC. These genetically defined cells will be invaluable for identifying disease pathways. Further, the approach described here will be applicable to any monogenetic skin disorder. In the future, this technology will also be essential for generating patient-derived and gene-corrected replacement tissue for transplantation, for example, in cases of severe skin fragility disorders.
ACKNOWLEDGMENTS
We would like to thank the University of Colorado School of Medicine Histology Core (www.medschool.ucdenver. edu/histology) and iPSC Core (www.medschool.ucdenver.edu/iPS) for technical support. We would like to thank the National Foundation for Ectodermal Dysplasias (NFED) for providing patient photographs and assistance in obtaining patient skin biopsies.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
PJK and MIK were supported by a grant from the NFED and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) under Award Numbers R00 AR054696 (MIK), R01 AR061506 (MIK), and R01 AR053892 (PJK). JD is supported by a pre-doctoral fellowship from the Colorado Clinical & Translational Science Institute (TR001081).
References
- Amagai M, Stanley JR (2012). Desmoglein as a target in skin disease and beyond. J Invest Dermatol. 132: 776–784.
- Beaudry VG, Pathak N, Koster MI, Attardi LD (2009). Differential PERP regulation by TP63 mutants provides insight into AEC pathogenesis. Am J Med Genet A. 149A: 1952–1957.
- Bellomaria A, Barbato G, Melino G, Paci M, Melino S (2010). Recognition of p63 by the E3 ligase ITCH: Effect of an ectodermal dysplasia mutant. Cell Cycle. 9: 3754–3763.
- Bertola DR, Kim CA, Albano LM, Scheffer H, Meijer R, van Bokhoven H (2004). Molecular evidence that AEC syndrome and Rapp-Hodgkin syndrome are variable expression of a single genetic disorder. Clin Genet. 66: 79–80.
- Bierkamp C, McLaughlin KJ, Schwarz H, Huber O, Kemler R (1996). Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 180: 780–785.
- Browne G, Cipollone R, Lena AM, Serra V, Zhou H, van Bokhoven H, Dötsch V, Merico D, Mantovani R, Terrinoni A, Knight RA, Candi E, Melino G (2011). Differential altered stability and transcriptional activity of DeltaNp63 mutants in distinct ectodermal dysplasias. J Cell Sci. 124: 2200–2207.
- Brunner HG, Hamel BCJ, van Bokhoven H (2002). The p63 gene in EEC and other syndromes. J Med Genet. 39: 377–381.
- Buxton RS, Cowin P, Franke WW, Garrod DR, Green KJ, King IA, Koch PJ, Magee AI, Rees DA, Stanley JR (1993). Nomenclature of the desmosomal cadherins. J Cell Biol. 121: 481–483.
- Chapman S, Liu X, Meyers C, Schlegel R, McBride AA (2010). Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest. 120: 2619–2626.
- Chen J, Den Z, Koch PJ (2008). Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 121: 2844–2849.
- Chen J, O’Shea C, Fitzpatrick JE, Koster MI, Koch PJ. (2011). Loss of desmocollin 3 in skin tumor development and progression. Mol Carcinog. 51: 535–545.
- Cheng X, Den Z, Koch PJ (2005). Desmosomal cell adhesion in mammalian development. Eur J Cell Biol. 84: 215–223.
- Cheng X, Koch PJ. (2004). In vivo function of desmosomes. J Dermatol. 31: 171–187.
- Chitaev NA, Leube RE, Troyanovsky RB, Eshkind LG, Franke WW, Troyanovsky SM (1996). The binding of plakoglobin to desmosomal cadherins: patterns of binding sites and topogenic potential. J Cell Biol. 133: 359–369.
- Chung J, Grant RI, Kaplan DR, Irwin MS (2011). Special AT-rich Binding Protein-2 (SATB2) Differentially Affects Disease-causing p63 Mutant Proteins. J Biol Chem. 286: 40671–40680.
- Clements SE, Techanukul T, Holden ST, Mellerio JE, Dorkins H, Escande F, McGrath JA (2010). Rapp-Hodgkin and Hay-Wells ectodermal dysplasia syndromes represent a variable spectrum of the same genetic disorder. Br J Dermatol. 163: 624–629.
- Clements SE, Techanukul T, Lai-Cheong JE, Mee JB, South AP, Pourreyron C, Burrows NP, Mellerio JE, McGrath JA (2012). Mutations in AEC syndrome skin reveal a role for p63 in basement membrane adhesion, skin barrier integrity and hair follicle biology. Br J Dermatol. 167: 134–144.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science. 339: 819–823.
- Den Z, Cheng X, Merched-Sauvage M, Koch PJ (2006). Desmocollin 3 is required for pre-implantation development of the mouse embryo. J Cell Sci. 119: 482–489.
- Di Costanzo A, Festa L, Duverger O, Vivo M, Guerrini L, La Mantia G, Morasso MI, Calabrò V (2009). Homeodomain protein Dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle. 8: 1185–1195.
- Dianzani I, Garelli E, Gustavsson P, Carando A, Gustafsson B, Dahl N, Annerén G (2003). Rapp-Hodgkin and AEC syndromes due to a new frameshift mutation in the TP63 gene. J Med Genet. 40: e133.
- Ferone G, Mollo MR, Thomason HA, Antonini D, Zhou H, Ambrosio R, De Rosa L, Salvatore D, Getsios S, van Bokhoven H, Dixon J, Missero C (2013). p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet. 22: 531–543.
- Ferone G, Thomason HA, Antonini D, De Rosa L, Hu B, Gemei M, Zhou H, Ambrosio R, Rice DP, Acampora D, van Bokhoven H, Del Vecchio L, Koster MI, Tadini G, Spencer-Dene B, Dixon M, Dixon J, Missero C (2012). Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signalling in AEC syndrome. EMBO Mol Med. 4: 192–205.
- Fete M, van Bokhoven H, Clements SE, McKeon F, Roop DR, Koster MI, Missero C, Attardi LD, Lombillo VA, Ratovitski E, Julapalli M, Ruths D, Sybert VP, Siegfried EC, Bree AF (2009). International Research Symposium on Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate (AEC) syndrome. Am J Med Genet A. 149A: 1885–1893.
- Gallicano GI, Bauer C, Fuchs E (2001). Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 128: 929–941.
- Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998). Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 143: 2009–2022.
- Ganeshan R, Chen J, Koch PJ (2010). Mouse models for blistering skin disorders. Dermatol Res Pract. 2010: 584353.
- Green H, Rheinwald JG, Sun TT (1977). Properties of an epithelial cell type in culture: the epidermal keratinocyte and its dependence on products of the fibroblast. Prog Clin Biol Res. 17: 493–500.
- Hatano Y, Hashimoto T, Fukuda S, Ishikawa K, Goto M, Kai Y, Takeo N, Okamoto O, Fujiwara S (2012). Atypical pemphigus with exclusively anti-desmocollin 3-specific IgG antibodies. Eur J Dermatol. 22: 560–562.
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, Meng X, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 27: 851–857.
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 29: 731–734.
- Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, Mills AA, Attardi LD. (2005). Perp is a p63-regulated gene essential for epithelial integrity. Cell. 120: 843–856.
- Itoh M, Kiuru M, Cairo MS, Christiano AM (2011). Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A. 108: 8797–8802.
- Julapalli MR, Scher RK, Sybert VP, Siegfried EC, Bree AF (2009). Dermatologic findings of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 149A: 1900–1906.
- Koch PJ, Goldschmidt MD, Walsh MJ, Zimbelmann R, Franke WW (1991a). Complete amino acid sequence of the epidermal desmoglein precursor polypeptide and identification of a second type of desmoglein gene. Eur J Cell Biol. 55: 200–208.
- Koch PJ, Goldschmidt MD, Walsh MJ, Zimbelmann R, Schmelz M, Franke WW (1991b). Amino acid sequence of bovine muzzle epithelial desmocollin derived from cloned cDNA: a novel subtype of desmosomal cadherins. Differentiation. 47: 29–36.
- Koch PJ, Goldschmidt MD, Zimbelmann R, Troyanovsky R, Franke WW (1992). Complexity and expression patterns of the desmosomal cadherins. Proc Natl Acad Sci U S A. 89: 353–357.
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR (1997). Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 137: 1091–1102.
- Koch PJ, Walsh MJ, Schmelz M, Goldschmidt MD, Zimbelmann R, Franke WW (1990). Identification of desmoglein, a constitutive desmosomal glycoprotein, as a member of the cadherin family of cell adhesion molecules. Eur J Cell Biol. 53: 1–12.
- Koster MI (2010). p63 in skin development and ectodermal dysplasias. J Invest Dermatol130: 2352–2358.
- Koster MI, Dai D, Marinari B, Sano Y, Costanzo A, Karin M, Roop DR (2007). p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci U S A. 104: 3255–3260.
- Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR (2009). DeltaNp63 knockdown mice: A mouse model for AEC syndrome. Am J Med Genet A. 149A: 1942–1947.
- Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL (2011). Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol. 31: 1134–1144.
- Li Mura IE, Bauce B, Nava A, Fanciulli M, Vazza G, Mazzotti E, Rigato I, De Bortoli M, Beffagna G, Lorenzon A, Calore M, Dazzo E, Nobile C, Mostacciuolo ML, Corrado D, Basso C, Daliento L, Thiene G, Rampazzo A (2013). Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet. 21: 1226–1231.
- Lichti U, Anders J, Yuspa SH (2008). Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 3: 799–810.
- Lopardo T, Lo Iacono N, Marinari B, Giustizieri ML, Cyr DG, Merlo G, Crosti F, Costanzo A, Guerrini L (2008). Claudin-1 is a p63 target gene with a crucial role in epithelial development. PLoS.ONE. 3: e2715.
- Mahoney MG, Hu Y, Brennan D, Bazzi H, Christiano AM, Wahl JK III (2006). Delineation of diversified desmoglein distribution in stratified squamous epithelia: implications in diseases. Exp Dermatol. 15: 101–109.
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013). RNA-guided human genome engineering via Cas9. Science. 339: 823–826.
- Marinari B, Ballaro C, Koster MI, Giustizieri ML, Moretti F, Crosti F, Papoutsaki M, Karin M, Alema S, Chimenti S, Roop DR, Costanzo A (2009). IKKalpha is a p63 transcriptional target involved in the pathogenesis of ectodermal dysplasias. J Invest Dermatol. 129: 60–69.
- McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H (2001). Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 10: 221–229.
- Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D (2011). Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem. 286: 2143–2154.
- Okita K, Ichisaka T, Yamanaka S (2007). Generation of germline-competent induced pluripotent stem cells. Nature. 448: 313–317.
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008a). Disease-specific induced pluripotent stem cells. Cell. 134: 877–886.
- Park IH, Lerou PH, Zhao R, Huo H, Daley GQ (2008b). Generation of human-induced pluripotent stem cells. Nat.Protoc. 3: 1180–1186.
- Parsa R, Yang A, McKeon F, Green H (1999). Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J. Invest Dermatol. 113: 1099–1105.
- Payne AS, Yan AC, Ilyas E, Li W, Seykora JT, Young TL, Pawel BR, Honig PJ, Camacho J, Imaizumi S, Heymann WR, Schnur RE (2005). Two novel TP63 mutations associated with the ankyloblepharon, ectodermal defects, and cleft lip and palate syndrome: a skin fragility phenotype. Arch.Dermatol. 141: 1567–1573.
- Petrof G, Mellerio JE, McGrath JA (2012). Desmosomal genodermatoses. Br J Dermatol. 166: 36–45.
- Rafei D, Muller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, Eming R (2011). IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol. 178: 718–723.
- Rinne T, Brunner HG, H. van Bokhoven H (2007). p63-associated disorders. Cell Cycle. 6: 262–268.
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Günthert U, Franke WW, Birchmeier W (1996). Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 135: 215–225.
- Schmidt A, Koch PJ (2007). Desmosomes: just cell adhesion or is there more?Cell Adh Migr. 1: 28–32.
- Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA (2012). ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell. 22: 669–677.
- Siegfried E, Bree A, Fete M, Sybert VP (2005). Skin erosions and wound healing in ankyloblepharon-ectodermal defect-cleft lip and/or palate. Arch. Dermatol. 141: 1591–1594.
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131: 861–872.
- Testoni B, Mantovani R (2006). Mechanisms of transcriptional repression of cell-cycle G2/M promoters by p63. Nucleic Acids Res. 34: 928–938.
- Theis DG, Koch PJ, Franke WW (1993). Differential synthesis of type 1 and type 2 desmocollin mRNAs in human stratified epithelia. Int J Dev Biol. 37: 101–110.
- Tokonzaba E, Chen J, Cheng X, Den Z, Ganeshan R, Műller EJ, Koch PJ (2013). Plakoglobin as a Regulator of Desmocollin Gene Expression. J Invest Dermatol. 133: 2732–2740.
- Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, Wagner JE, Blazar BR (2011). Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 131: 848–856.
- Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA (2006). p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes and Development. 20: 3185–3197.
- Vanderhooft SL, Stephan MJ, Sybert VP (1993). Severe skin erosions and scalp infections in AEC syndrome. Pediatr.Dermatol. 10: 334–340.
- Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E (2001). Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 3: 1076–1085.
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, Andrews NC, Caput D, McKeon F (1998). p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 2: 305–316.
- Zarnegar BJ, Webster DE, Lopez-Pajares V, Vander Stoep Hunt B, Qu K, Yan KJ, Berk DR, Sen GL, Khavari PA (2012). Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. Am J Hum Genet. 91: 435–443.