
Abstract
Arrhythmogenic cardiomyopathy (AC) is a primary myocardial disorder characterized by a high incidence of ventricular arrhythmias often preceding the onset of ventricular remodeling and dysfunction. Approximately 50% of patients diagnosed with AC have one or more mutations in genes encoding desmosomal proteins, although non-desmosomal genes have also been associated with the disease. Increasing evidence implicates remodeling of intercalated disk proteins reflecting abnormal responses to mechanical load and aberrant cell signaling pathways in the pathogenesis of AC. This review summarizes recent advances in understanding disease mechanisms in AC that have come from studies of human myocardium and experimental models.
INTRODUCTION
Arrhythmogenic cardiomyopathy is a primary myocardial disease characterized by ventricular arrhythmias and sudden cardiac death (CitationCorrado et al., 2000). Originally described as a right ventricular disease (ARVC), it is now recognized to include left ventricular and biventricular forms which are often misdiagnosed as dilated cardiomyopathy or myocarditis. In light of this broader phenotypic spectrum, the term arrhythmogenic cardiomyopathy (AC) has been adopted (CitationSen-Chowdhry et al., 2010).
AC has a prevalence of 1:1000 to 1:5000 in the general population, but it accounts for 11–22% of sudden cardiac deaths among young athletes (CitationRomero et al., 2013). It is the major cause of sudden death among athletes in Northern Italy (CitationThiene et al., 1988) and accounts for 17% of sudden cardiac deaths in young people (≤ 35 years) in the United States (CitationShen et al., 1994). AC is a familial disease in at least 50% of cases and is usually inherited as an autosomal dominant trait (CitationSen-Chowdhry et al., 2007). The overall prevalence may be underestimated because wide phenotypic variation, age-related progression, and low genetic penetrance may obscure diagnosis (CitationSen-Chowdhry et al., 2007). Currently, the diagnosis of AC rests upon fulfilling a complex set of criteria established by an International Task Force, which although relatively specific are not highly sensitive (CitationMarcus et al., 2010).
AC is a highly arrhythmogenic disease. Arrhythmias usually arise as the first manifestation of disease, and typically precede structural remodeling of the myocardium (CitationSaffitz et al., 2010). This so-called “concealed” phase is unique among the non-ischemic cardiomyopathies. In hypertrophic cardiomyopathy, for example, arrhythmic risk appears to be related at least in part to the underlying substrate of myocyte disarray, hypertrophy, fibrosis, and small vessel disease. In dilated cardiomyopathy, arrhythmias occur in the context of significant left ventricular remodeling and contractile dysfunction. By contrast, there is something fundamentally arrhythmogenic about early AC, in which frequent arrhythmias occur in otherwise apparently normal hearts (CitationSen-Chowdhry et al., 2010). As the disease progresses, degenerative changes in cardiac myocytes associated with inflammation and accumulation of fibrofatty scar tissue become more prominent. Thus, AC exhibits features of both the inherited arrhythmia syndromes such as long QT and the non-ischemic cardiomyopathies characterized by complex myocardial pathology (CitationSen-Chowdhry et al., 2010).
GENETICS OF AC
Autosomal dominant inheritance in AC was first described in 1987 in a report on eight Italian families (CitationNava et al., 1987). The first genetic locus linked to AC was identified at 14q23-q24 in 1994 after evaluation of a large Venetian family (CitationRampazzo et al., 1994). It was not until 1998, however, that analysis of patients from the Greek island of Naxos led to identification of the first causative gene mutation in AC (CitationCoonar et al., 1998). The so-called Naxos disease is a highly penetrant recessive syndrome characterized by the clinical triad of ARVC, woolly hair, and keratoderma involving pressure areas of the palms and soles. The cutaneous phenotype is expressed from infancy, thereby unequivocally identifying affected individuals and ensuring accurate linkage analysis. The cardiac symptoms characteristically develop from adolescence to early adulthood although arrhythmias have been documented in young children (CitationCoonar et al., 1998). The disease allele was mapped to 17q21 and shown to involve a homozygous two-base-pair deletion in the gene encoding the desmosomal protein plakoglobin (γ-catenin) (CitationMcKoy et al., 2000). This first association of a desmosomal gene mutation with AC paved the way for identification of disease-causing mutations in other desmosomal genes.
A mutation in the desmoplakin gene, DSP, resulting in truncation of the C-terminal domain was subsequently implicated in another recessive cardio-cutaneous syndrome described in families from Ecuador (CitationPurkis et al., 2000). The so-called Carvajal syndrome consists of palmoplantar keratoderma, woolly hair, and a biventricular cardiomyopathy that exhibits clinical features of dilated cardiomyopathy (CitationCarvajal-Huerta, 1998). Clinical and pathological characterization of the Carvajal syndrome is limited, but frequent and complex ventricular arrhythmias have been documented in pre-adolescence (CitationCarvajal-Huerta, 1998). Pathological features include biventricular dilatation with focal aneurysms, and myocyte degeneration and replacement fibrosis (albeit without adipose tissue) preferentially affecting sub-epicardial and mid-myocardial layers while sparing the sub-endocardium (CitationKaplan et al., 2004a). Focal ventricular aneurysms and sub-epicardial/ mid-myocardial prominence are typical features of the myocardial pathology of AC.
The first dominantly inherited mutation in DSP was identified in 2002 in an ARVC family of Italian descent. This mutation (S299R) was predicted to modify a putative phosphorylation site in the N-terminal domain of desmoplakin and interfere with the protein's binding to plakoglobin (CitationRampazzo et al., 2002). Three additional dominant mutations in DSP were reported in 2005 (R1775I, R1255K and c.423–1G> A) in ARVC families characterized by a high occurrence of sudden cardiac death and prominent left ventricular involvement (CitationBauce et al., 2005). Soon after, another mutation was identified (2034insA) in a family showing predominant left ventricular disease, giving rise to the term “arrhythmogenic left ventricular cardiomyopathy” (CitationNorman et al., 2005). Yang et al. reported that four additional variants (V30M, Q90R, W233X, and R2834H) are also linked to biventricular forms of the disease (CitationYang et al., 2006).
Once the focus of the gene hunt was directed to the desmosome, mutations in AC families were discovered in genes encoding plakophilin-2 (PKP2), desmoglein-2 (DSG2), and desmocollin-2 (DSC2) (CitationThiene et al., 2007). PKP2 appears to be the most commonly mutated gene in ARVC. In the Netherlands, for example, PKP2 mutations may account for up to 70% of index cases (Citationvan Tintelen et al., 2006). Prudence, however, must be applied in distinguishing truly pathogenic mutations from background genetic noise, especially in the case of missense mutations. A recent study on a large series of ARVC patients showed that 21% of patients carry missense mutations in desmosomal genes versus a remarkably high 16% of controls (CitationKapplinger et al., 2011). In some cases, a single sequence variant may be insufficient to cause AC by itself, but may contribute to disease expression when combined with another sequence variant in the same gene (compound heterozygosity) or in another gene (digenic heterozygosity) (CitationXu et al., 2010).
Mutations in non-desmosomal genes have also been associated with AC, the most convincing of which include those encoding transmembrane protein 43 (TMEM43) (CitationMerner et al., 2008) and phospholamban (PLN) (Citationvan der Zwaag et al., 2012). The S358L mutation in TMEM43, first identified in a large founder population in Newfoundland, causes a fully penetrant non-classic form of the disease associated with a high incidence of premature death and heart failure in survivors (CitationMerner et al., 2008). The PLN mutation R14del, first described in Greek families with dilated cardiomyopathy and heart failure(CitationHaghighi et al., 2006), was recently identified in a substantial number of Danish patients clinically diagnosed with ARVC or dilated cardiomyopathy phenotypes, further supporting a broader phenotypic spectrum for AC (Citationvan der Zwaag et al., 2012).
Other extra-desmosomal genes have been associated with AC including those encoding desmin (CitationOtten et al., 2010), transforming growth factor beta-3 (CitationBeffagna et al., 2005) and the cardiac ryanodine receptor (CitationTiso et al., 2001), although the latter appears to be more closely linked to catecholaminergic polymorphic ventricular tachycardia (CitationMarks et al., 2002). It is currently possible to identify mutations in one or more of the five major desmosomal genes in ˜ 60% of probands who fulfill International Task Force criteria for AC. Presumably, other disease alleles yet to be described may be responsible for at least some of the remaining patients with AC.
MORE THAN A DISEASE OF ABNORMAL CELL–CELL ADHESION
Since being recognized as a non-ischemic cardiomyopathy by the World Health Organization in 1994 (CitationMcKenna et al., 1994), AC has become the focus of active investigations of its molecular pathophysiology and disease mechanisms. While there has been significant progress in identifying AC-causing mutations, much less is known about how the mutant proteins actually cause the disease. One leading hypothesis is that abnormal cell–cell adhesion injures the cardiac myocytes and promotes cell death with subsequent replacement by fibrofatty tissue (CitationSaffitz et al., 2009). Such a mechanism almost certainly plays a role. However, desmosomal proteins may fulfill dual roles as structural proteins in adhesion junctions and as signaling molecules, which can alter signaling pathways and thereby modulate pathological gene expression, promote cardiac myocyte apoptosis, and perhaps mediate expression of a fibrogenic and/or an adipogenic phenotype (CitationSaffitz, 2011). Moreover, mononuclear (predominantly T cell) inflammatory infiltrates may be especially prominent in the hearts of AC patients, occurring in nearly 80% in some postmortem series (CitationTabib et al., 2003). The sub-endocardium is commonly affected, suggesting that the inflammatory response may reflect extension of the disease to previously unaffected regions of the myocardium. Active disease progression associated with intense inflammation could play a role in the development of “hot phases”, episodic exacerbations following an interval of clinically quiescent disease (CitationSen-Chowdhry et al., 2010). However, this remains entirely speculative as no definitive experimental evidence exists to link inflammation in AC to disease pathogenesis and/or progression.
Although desmosomes occur in virtually all solid organs, clinical phenotypes related to desmosomal gene mutations appear to be limited to the heart and skin, tissues that experience the greatest mechanical load (CitationSaffitz, 2011). Young competitive athletes with AC have a 5.4-fold relative risk of sudden death compared to non-athletes (CitationBorjesson & Pelliccia, 2009). Strenuous physical activity has been associated with an increased penetrance and acceleration of the disease phenotype (CitationCorrado et al., 2006). The right ventricular apex, an area preferentially affected in ARVC, exhibits heterogeneous fiber orientation and elevated incremental strains, potentially creating a mechanically weak spot in the heart (CitationHariharan et al., 2012). Finally, electron microscopy studies of patient myocardial samples have reported decreased numbers of desmosomes and apparent clefts or widening of various components within the intercalated disks (CitationBasso et al., 2006). All these observations suggest that mechanical stress plays an important role in disease progression and sudden death. However, it should be emphasized that, currently, there is no direct evidence of weakened cell–cell adhesion in the hearts of patients with AC. Thus, despite the interesting potential associations between inflammation and exercise and AC disease pathogenesis, we really have a very limited understanding of the molecular mechanisms responsible for arrhythmias and myocyte injury in AC. Advances in understanding such mechanisms might not only aid in development of preventive measures in AC, but perhaps reveal fundamental new paradigms in sudden death occurring in other, more common forms of heart disease.
DISEASE MECHANISMS IN AC: WHAT HAVE WE LEARNED FROM HUMAN STUDIES?
To gain insights into mechanisms of AC pathogenesis, we have analyzed patient myocardial samples in an effort to discover features of the molecular pathology of this disease spectrum. The great majority of the world-wide human myocardial tissue archive for AC consists of formalin-fixed, paraffin-embedded samples obtained by endomyocardial biopsy or at autopsy. While this limits the repertoire of analytical approaches that may be applied, we have learned a great deal about AC by studying the distribution of intercalated disk proteins using immunohistochemistry.
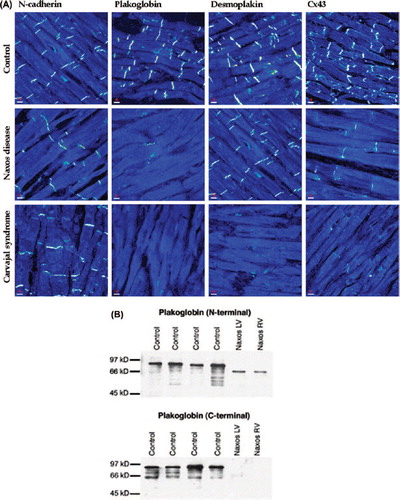
We first determined whether mutant proteins are expressed in AC and if so whether they are localized at cell–cell junctions or are re-distributed to other (intracellular) compartments. We also asked whether a mutation in a single desmosomal protein could affect the localization of other (non-mutant) desmosomal proteins (CitationSaffitz et al., 2009). Because defects in intercellular mechanical coupling are generally associated with abnormal cell–cell communication via gap junctions (CitationSaffitz, 2003), we also determined whether mutations in desmosomal proteins were associated with changes in the distribution of connexins, proteins that form electrical junctions in the myocardium. Initial studies in myocardial samples from patients with the Naxos disease showed that the truncated mutant form of plakoglobin was abundantly expressed in both ventricles, but it failed to localize properly at intercalated disks () (CitationKaplan et al., 2004b). A similar analysis of the heart in the Carvajal syndrome showed that not only the mutant protein, desmoplakin, failed to localize at cell–cell junctions, but so did its non-mutant binding partner plakoglobin () (CitationKaplan et al., 2004a). Thus, a mutation in a single desmosomal protein can affect the localization of other proteins that are not genetically altered (CitationSaffitz et al., 2009). We also observed a marked reduction in the amount of immunoreactive signal for Cx43, the major ventricular gap junction protein, in both the Naxos and Carvajal myocardium () (CitationKaplan et al., 2004a; CitationSaffitz, 2003).
Figure 1. (A) Representative confocal immunofluorescence images of control myocardium and myocardium from a patient with Naxos disease and a patient with Carvajal syndrome. Specific immunoreactive signal for plakoglobin was significantly depressed in both cases compared to controls, as was signal for the major gap junction protein Cx43. Immunoreactive signal for desmoplakin was depressed at intercalated disks in Carvajal syndrome but not in Naxos disease. Signal for the non-desmosomal adhesion protein N-cadherin was present and control-like in both cases. (B) Western Immunoblots using an N-terminal plakoglobin antibody showed that the mutant plakoglobin form (2057del2) is expressed in left and right ventricular myocardium from a patient with Naxos disease. Mutant plakoglobin migrates at a lower molecular weight than the wildtype protein. 2057del2 plakoglobin cannot be detected when a C-terminal plakoglobin antibody is used (reproduced from Kaplan et al. Heart Rhythm 2004; 1) (CitationKaplan et al., 2004b).
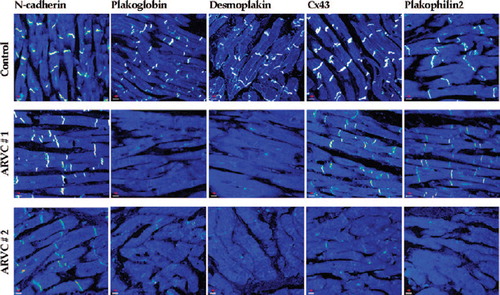
We have subsequently analyzed myocardium from a large number of patients with clinically and/or pathologically documented ARVC related to mutations in one or more of the five desmosomal protein genes and involving autosomal dominant inheritance in most cases (CitationAsimaki et al., 2009). We observed that immunoreactive signal for plakoglobin was reduced at cell–cell junctions in the great majority of cases, independent of the underlying pathogenic mutation. It also occurred in cases that fulfilled diagnostic criteria but in which no desmosomal gene mutation could be identified by genetic screening. This consistent finding was associated with decreased amounts of immunoreactive signal for desmoplakin or plakophilin-2 in some cases while in others the signal appeared normal (). Reduced junctional plakoglobin signal occurred diffusely in both ventricles including areas that appeared histologically normal (CitationAsimaki et al., 2009). A similar reduction was not seen in myocardial samples from patients with end-stage heart failure due to hypertrophic, dilated or ischemic cardiomyopathies(CitationAsimaki et al., 2009), although subsequent studies have shown that junctional plakoglobin signal is diminished at cell–cell junctions in myocardium from patients with sarcoidosis or giant cell myocarditis, both highly arrhythmogenic forms of granulomatous myocarditis (CitationAsimaki et al., 2011). Taken together, these observations suggest that redistribution of plakoglobin is a consistent feature in AC and raise the possibility of common arrhythmogenic mechanisms in AC and some forms of granulomatous myocarditis (CitationAsimaki et al., 2009; CitationAsimaki et al., 2011). Furthermore, as discussed below, redistribution of plakoglobin from junctional to intracellular and/or intranuclear sights may be responsible for modulating Wnt signaling pathways, which experimental studies have implicated in AC disease pathogenesis (CitationGarcia-Gras et al., 2006).
Figure 2. Representative confocal immunofluorescence images of control myocardium and myocardium from two patients with autosomal dominant ARVC. Specific immunoreactive signal for plakoglobin was depressed at cell–cell junctions in the great majority of cases regardless of the underlying pathogenic mutation. Signal for desmoplakin and plakophilin2 varied, while signal for N-cadherin was always present and indistinguishable from controls. The majority of cases examined showed gap junction remodeling as evidenced by decreased junctional signal for Cx43 (reproduced from Asimaki et al. NEJM 2009; 360:1078) (CitationAsimaki et al., 2009).
An extensive literature exists on the dependence of cell–cell electrical coupling at gap junctions on normal mechanical coupling by intercellular adhesion junctions (CitationSaffitz, 2003). Such dependency is probably of particular importance in the myocardium in which extremely large gap junctions, presumably required for safe impulse propagation, are surrounded by extensive points of cell–cell adhesion within intercalated disks (CitationSaffitz, 2003). Initial observations of diminished Cx43 immunoreactive signal at cell–cell junctions in the Naxos disease (CitationSaffitz, 2003) and Carvajal syndrome (CitationKaplan et al., 2004a) () have now been confirmed in additional cases of autosomal dominant ARVC associated with mutations in various desmosomal proteins () (CitationSaffitz, 2009). Gap junction remodeling in AC occurs diffusely in the myocardium and has been shown in at least one case of the Naxos disease to precede the development of myocardial degeneration and remodeling (CitationPolychronopoulou et al., 2002). These observations suggest that gap junction remodeling is a consistent feature of AC and raise the possibility that it may contribute to abnormal tissue electrophysiology during the concealed phase of the disease in which arrhythmias occur in the absence of structural changes (CitationAsimaki & Saffitz, 2012). It should be emphasized, however, that thus far no studies have been reported on functional consequences of the apparent gap junction remodeling seen by immunohistochemistry. Thus, the contributions, if any, of gap junction remodeling to arrhythmogenesis in AC remain a matter of speculation.
Very little is known about the molecular pathology of AC caused by mutations in non-desmosomal genes. The TMEM43 protein contains four transmembrane segments and a large hydrophilic domain that is remarkably conserved among species. It is widely expressed, particularly in the placenta, and in most tissues localizes at the inner nuclear membrane and endoplasmic reticulum where it has been shown to interact with emerin and lamins (CitationBengtsson & Otoo, 2008). Immunoreactive signal for plakoglobin was found to be depressed at intercalated in myocardial samples from three ARVC patients with TMEM43 mutations, whereas PKP2, Cx43, and emerin appeared to be normally distributed (CitationChristensen et al., 2011). The implications of these observations are uncertain but they add weight to the notion that redistribution of plakoglobin from junctional to intracellular and/or intranuclear sites may be an important component of the underlying disease pathway in AC.
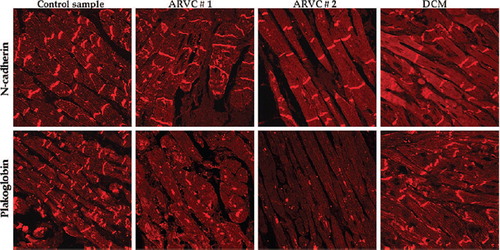
PLN regulates the sarcoplasmic reticulum Ca+2 pump (SERCA2a) in cardiac muscle and is thus important for maintaining Ca+2 homeostasis (CitationMacLennan & Kranias, 2003). Three different PLN mutations have been linked to a dilated cardiomyopathy phenotype associated with malignant ventricular arrhythmias and interstitial fibrosis (CitationSchmitt et al., 2003; CitationHaghighi et al., 2003; CitationPosch et al., 2009). Given the phenotypic overlap between PLN mutation carriers diagnosed with dilated cardiomyopathy and classical ARVC, a large population of patients diagnosed with both disease entities was screened for mutations in this gene (Citationvan der Zwaag et al., 2012). A 3-base pair deletion (R14del) was identified in 39 (15%) dilated cardiomyopathy and 12 (12%) ARVC index patients, none of whom had mutations in desmosomal genes. Interestingly, samples from PLN R14del carriers diagnosed with ARVC showed reduced immunoreactive signal for plakoglobin at cardiac IDs in the majority of cases (5 of 7). By contrast, only 1 of 9 R14del carriers diagnosed with dilated cardiomyopathy showed evidence of plakoglobin re-distribution () (Citationvan der Zwaag et al., 2012). These observations suggest that diminished plakoglobin signal at intercalated disks may track with the ARVC phenotype rather than genotype.
Figure 3. Immunofluorescence images of endomyocardial biopsy samples from two patients diagnosed with ARVC and one with idiopathic dilated cardiomyopathy, all carrying the PLN R14del mutation, compared to those of a control sample. Immunoreactive signal for plakoglobin at cell–cell junctions was significantly depressed in the ARVC subjects compared to those of controls and the dilated cardiomyopathy patient (reproduced from Van der Zwaag et al. Eur J Heart Fail 2012; 14:1204) (Citationvan der Zwaag et al., 2012).
As detailed in the next section, new information is emerging from experimental models that expression of mutations linked to AC is associated with reduced Na+ current density (CitationRizzo et al., 2012). Although the responsible mechanisms are not known, this observation could have important implications for understanding arrhythmogenesis in AC. Reduced INa has not been documented in patients although reduced signal for Nav1.5 at intercalated disks has been shown by immunohistochemistry in myocardium from patients with AC (CitationNoorman et al., 2013). Taken together with observations of gap junction remodeling, this raises the possibility that decreased INa combined with reduced electrical coupling could affect impulse propagation and thereby contribute to arrhythmias early in the disease process (CitationDelmar, 2012). This is a promising area of investigation.
DISEASE MECHANISMS IN AC: WHAT HAVE WE LEARNED FROM EXPERIMENTAL MODELS?
Perhaps the most important insight to emerge from experimental models of AC is the potential role of altered Wnt signaling in disease pathogenesis (CitationGarcia-Gras et al., 2006). Like its homolog β-catenin, plakoglobin (γ-catenin) functions as both an adhesion junction protein and a nuclear signaling molecule modulating Wnt pathways (CitationRubenstein et al., 1997). The N-terminus plays a key role in determining stability and degradation of armadillo proteins. This region appears to be a target for N- and O-glycosylation (CitationHatsell et al., 2003) and is also involved in the ubiquitin–proteasomal degradation pathway (CitationSadot et al., 2000). Wnt pathways are involved in fundamental biological processes including cardiac development and differentiation, hypertrophy, cell–cell adhesion, cytoskeletal re-arrangement, and calcium homeostasis (Citationvan de Schans et al., 2008). Under basal conditions, cytoplasmic plakoglobin undergoes phosphorylation at N-terminal serine residues by glycogen synthase kinase 3β (GSK-3β) which targets it for ubiquitylation and proteasomal degradation. Activation of the canonical Wnt signaling pathway inhibits GSK-3β which allows plakoglobin to accumulate within the cell and enter the nucleus where it may interact with Tcf/Lef transcription factors and alter gene expression. The precise role of plakoglobin once it enters the nucleus is still incompletely understood but the prevailing view is that nuclear localization of plakoglobin suppresses canonical Wnt signaling and its downstream targets through Tcf/Lef1 transcription factors (CitationRubenstein et al., 1997). In this context, Garcia-Gras et al. showed that siRNA-mediated suppression of desmoplakin expression in an atrial cell line promotes nuclear accumulation of plakoglobin associated with a 2-fold reduction in canonical Wnt signaling (CitationGarcia-Gras et al., 2006). The resulting phenotype also included increased expression of adipogenic and fibrogenic genes and intracellular accumulation of fat droplets (CitationGarcia-Gras et al., 2006). This important study was the first to implicate abnormal Wnt signaling in the pathogenesis of AC. In subsequent studies, these authors provided evidence that suppression of the canonical Wnt pathway in progenitor cells in the heart causes a switch to an adipogenic instead of a myogenic fate which could explain the often prominent accumulation of mature adipocytes in areas of myocyte damage in AC (CitationLombardi et al., 2011).
Transgenic mice with cardiac-specific expression of the V30M or Q90R N-terminal desmoplakin mutations are embryonic lethal (CitationYang et al., 2006). On the contrary, transgenic mice with cardiac-specific expression of a C-terminal desmoplakin mutation (R2834H) are viable. These animals show increased cardiac myocyte apoptosis, fibrosis and lipid accumulation as well as biventricular enlargement and dysfunction (CitationYang et al., 2006). Lyon et al. used the ventricular myosin light chain-2-Cre system to ablate desmoplakin specifically from cardiac myocytes, a mouse model designated as DSP-cKO(CitationLyon et al., 2013). Following gene deletion, animals developed a dramatic phenotype characterized by biventricular dilatation, extensive myocyte loss with fibrosis and ultrastructural abnormalities at intercalated disks. DSP-cKO mice also exhibited ventricular arrhythmias that were exacerbated in response to exercise and catecholamine stimulation. Finally, the mice showed RV conduction defects and abnormal electrical wavefront propagation, presumably associated with reduction in expression of the gap junction proteins Cx40 and Cx43 (CitationLyon et al., 2013). While this mouse model exhibits some major features reminiscent of biventricular AC in patients with severe desmoplakin mutations, it should be remembered that germline deletion of desmosomal proteins including plakoglobin (CitationBierkamp et al., 1996), desmoplakin (CitationGallicano et al., 1998), and plakophilin-2 (CitationGrossmann et al., 2004) are embryonic lethal in mice. Furthermore, the best available evidence indicates that mutant desmosomal proteins are expressed in various forms of AC in patients suggesting that disease pathogenesis may be mediated by dominant-negative effects rather than by null alleles.
The idea that exercise exacerbates the disease phenotype in AC noted above in the DSP-cKO model has also been investigated in mice with heterozygous deletion of the plakoglobin gene (CitationKirchhof et al., 2006). By 10 months of age, plakoglobin+/− mice show enlarged right ventricles, but endurance training (8 weeks of daily swimming) caused premature right ventricular dilatation and dysfunction at 5–6 months of age (CitationKirchhof et al., 2006).
Transgenic mice with cardiac-specific over- expression of an ARVC-causing mutant form of DSG2 (N271S) recapitulate clinical features of the disease including premature death, spontaneous ventricular arrhythmias, cardiac dysfunction and biventricular dilatation, and aneurysms (CitationPilichou et al., 2009). Hearts of mice heterozygous for a PKP2-null allele show mild ultrastructural changes but no histological or gross anatomical abnormalities (CitationCerrone et al., 2012). However, ventricular myocytes exhibit changes in INa and the Na+ channel blocker flecainide provoked ventricular arrhythmias and death in PKP2-deficient animals, but not in controls (CitationCerrone et al., 2012).
While clinical experience has suggested that exercise increases disease expression in patients with AC and disease phenotypes are intensified in some mouse models subjected to exercise, relatively little is known about the responsible molecular mechanisms. We have shown that HEK293 cells expressing two different mutations in plakoglobin (2057del2 which causes Naxos disease, or S39_K40insS which causes a dominant form of ARVC without associated cutaneous abnormalities) show abnormal responses to short-term cyclical stretch (CitationHuang et al., 2008). Whereas normal cells increase the amount immunoreactive signal for plakoglobin and Cx43 at cell–cell junctions after even brief intervals of stretch, these responses are significantly blunted in cells expressing mutant desmosomal proteins (CitationHuang et al., 2008). It has also been shown that knockdown of PKP2 expression in neonatal rat ventricular myocytes in vitro reduces cell–cell adhesion strength (CitationSato et al., 2011). These studies must be viewed with caution, however. Responses in HEK cells may not be the same as in cardiac myocytes (CitationHuang et al., 2008) and, as emphasized previously, phenotypes observed after genetic ablation strategies may not reflect disease profiles in patients caused by expression of mutant desmosomal proteins. Moreover, the underlying mechanisms remain poorly understood.
Most experimental studies of AC have focused on myocardial injury and molecular pathology with relatively little attention paid to potential changes in cellular electrophysiology that may contribute to the highly arrhythmogenic phenotype in AC. However, recent studies have begun to shed light on this important area.
Using cultured cardiac myocytes, Sato et al. have provided some evidence that PKP2 interacts directly with Nav1.5 and that knockdown of PKP2 expression promotes changes in the INa and the velocity of impulse propagation (CitationDeo et al., 2011, CitationSato et al., 2009). Finally, Kim et al. generated iPS cell lines from a patient with clinical ARVC and a homozygous (c.2484C> T) mutation in PKP2 that causes cryptic splicing with a 7-nucleotide deletion in exon 12 leading to frame-shift of the carboxy-terminal amino acids (CitationKim et al., 2013). The derived cardiac myocytes abnormal plakoglobin nuclear translocation and decreased Wnt signaling. This important study was the first to characterize cardiac myocytes derived from a patient with AC. Many technical limitations apply to such studies but it can be anticipated that additional insights into disease mechanisms in patients will come from further studies using this approach.
CONCLUSIONS
AC has an unusually dramatic arrhythmogenic phenotype, which is manifest early in the natural history of the disease, often preceding the development of significant ventricular remodeling or contractile dysfunction. The identification of desmosomal gene mutations in more than 50% of index patients has focused attention on the possibility that abnormal cell–cell adhesion may play a critical role in myocardial injury and arrhythmogenesis. At the same time, increasing evidence has implicated abnormal Wnt signaling, perhaps related to redistribution of junctional plakoglobin to intracellular/intranuclear sites. Moreover, AC may be caused by mutations in non-desmosomal genes, and the genetic basis of the disease is undefined in a significant number of patients. It remains to be determined to what extent changes in electrical coupling contribute to the highly arrhythmogenic phenotype in AC and how gap junction remodeling interacts with other potential arrhythmogenic mechanisms in this deadly disease. One promising avenue for future investigation is more detailed elucidation of the manifold mechanisms implicated in dysregulated Wnt signaling pathways. How a mutation in a Ca+ 2 homeostasis protein such as PLN or a nuclear membrane protein such as TMEM43 promotes displacement of plakoglobin from cell–cell junctions must be elucidated. The exact role of plakoglobin in the nucleus and the panel of genes whose expression it may control must be determined. The relationship between genetic, epigenetic, and environmental factors, such as exercise, and the role they play in modifying disease manifestation also requires further investigation. Advances in these areas could provide new insights into arrhythmogenesis not only in AC but also in other, more common forms of heart disease, and identify much needed mechanism-based therapies to treat or prevent sudden death.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Asimaki A, Saffitz JE (2012). Gap junctions and arrhythmogenic cardiomyopathy. Heart Rhythm. 9: 992–995.
- Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey-Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, Stevenson WG, McKenna WJ, Gautam S, Remick DG, Calkins H, Saffitz JE (2011). Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 4: 743–752.
- Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE (2009). A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 360: 1075–1084.
- Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon A, Wozniek O, Corrado D, Datiento L, Danieli GA, Valente M, Nava A, Thiene G, Rampazzo A (2006). Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 27: 1847–1854.
- Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A (2005). Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 26: 1666–1675.
- Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A (2005). Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 65: 366–373.
- Bengtsson L, Otoo H (2008). LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci. 121: 536–548.
- Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R (1996). Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 180: 780–785.
- Borjesson M, Pelliccia A (2009). Incidence and aetiology of sudden cardiac death in young athletes: an international perspective. Br J Sports Med. 43: 644–648.
- Carvajal-Huerta L (1998). Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 39: 418–421
- Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M (2012). Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 95: 460–468.
- Christensen AH, Andersen CB, Tybjaerg-Hansen A, Haunso S, Svendsen JH (2011). Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet. 80: 256–264.
- Coonar AS, Protonotarios N, Tsatsopoulou A, Needham EW, Houlston RS, Cliff S, Otter MI, Murday VA, Mattu RK, McKenna WJ (1998). Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 97: 2049–2058.
- Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G (2006). Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. Jama-J Am Med Assoc. 296: 1593–1601.
- Corrado D, Basso C, Thiene G (2000). Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart. 83: 588–595.
- Delmar M (2012). Desmosome- ion channel interactions and their possible role in arrhythmogenic cardiomyopathy. Pediatr Cardiol. 33: 975–979.
- Deo M, Sato PY, Musa H, Lin X, Pandit SV, Delmar M, Berenfeld O (2011). Relative contribution of changes in sodium current versus intercellular coupling on reentry initiation in 2-dimensional preparations of plakophilin- 2-deficient cardiac cells. Heart Rhythm. 8: 1740–1748.
- Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998). Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 143: 2009–2022.
- Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ (2006). Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 116: 2012–2021.
- Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004). Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 167: 149–160.
- Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW II, MacLennan DH, Kremastinos DT, Kranias EG (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 103: 1388–1393.
- Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias E (2003). Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 111: 869–876.
- Hariharan V, Provost J, Shah S, Konofagou E, Huang H (2012). Elevated strain and structural disarray occur at the right ventricular apex. Cardiovas Eng Technol. 3: 52–61.
- Hatsell S, Medina L, Merola J, Haltiwanger R, Cowin P (2003). Plakoglobin is O-glycosylated close to the N-terminal destruction box. J Biol Chem. 278: 37745–37752.
- Huang H, Asimaki A, Lo D, McKenna W, Saffitz J (2008). Disparate effects of different mutations in plakoglobin on cell mechanical behavior. Cell Motil Cytoskeleton. 65: 964–978.
- Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. (2004a).Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 13: 26–32.
- Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. (2004b). Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 1: 3–11.
- Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MG, Bhuiyan Z, Bikker H, Wiesfeld AC, Hauer RN, van Tintelen JP, Jongbloed JD, Calkins H, Judge DP, Wilde AA, Ackerman MJ (2011). Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 57: 2317–2327.
- Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen HS (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 494: 105–110.
- Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, Breithardt G, Ruiz P, Wichter T, Levkau B (2006). Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 114: 1799–1806.
- Lombardi R, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ (2011). Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 109: 1342–1353.
- Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, Gu Y, Evans SM, Omens JH, Peterson KL, McCulloch AD, Sheikh F (2013). Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet. [Epub ahead of print]
- MacLennan DH, Kranias EG (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 4: 566–577.
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W (2010). Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation. 121: 1533–1541.
- Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ (2002). Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 190: 1–6.
- McKenna WJ, Thiene G, Nava A, Fontaliron F, Blomstrom-Lundquist G, Fontaine G, Camerini F; on behalf of the Task Force of the working group myocardial and pericardial disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology (1994). Diagnosis of arrhythmogenic right ventricular dysplasia cardiomyopathy. Br Heart J. 71: 215–218.
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000). Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355: 2119–2124.
- Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL (2008). Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 82: 809–821.
- Nava A, Scognamiglio R, Thiene G, Canciani B, Daliento L, Buja G, Stritoni P, Fasoli G, Dalla Volta S (1987). A polymorphic form of familial arrhythmogenic right ventricular dysplasia. Am J Cardiol. 59: 1405–1409.
- Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA (2013). Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 10: 412–419.
- Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, Sen-Chowdhry S, Rowland E, Crosby A, McKenna WJ (2005). Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 112: 636–642.
- Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP (2010). Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 7: 1058–1064.
- Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJ, de Bakker JM, Tan HL, Valente M, Nava A, Wilde AA, Moorman AF, Thiene G, Bezzina CR (2009). Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med. 206: 1787–1802.
- Polychronopoulou S, Tsatsopoulou A, Papadhimitriou SI, Panagiotou JP, Anastasakis A, Paterakis G, Anagnostou D, Protonotarious N, Haidas SA (2002). Myelodysplasia and Naxos disease: a novel pathogenetic association?Leukemia. 16: 2335–2337.
- Posch MG, Perrot A, Geier C, Boldt LH, Schmidt G, Lehmkuhl HB, Hetzer R, Dietz R, Gutberlet M, Haverkamp W, Ozcelik C (2009). Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm. 6: 480–486.
- Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000). Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 9: 2761–2766.
- Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G (1994). The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 3: 959–962.
- Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA (2002). Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 71: 1200–1206.
- Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 95: 409–418.
- Romero J, Mejia-Lopez E, Manrique C, Lucariello R (2013). Arrhythmogenic right ventricular cardiomyopathy (ARVC/D): a systematic literature review. Clin Med Ins Cardiol l7: 97–114.
- Rubenstein A, Merriam J, Klymkowsky MW (1997). Localizing the adhesive and signaling functions of plakoglobin. Dev Genet. 20: 91–102.
- Sadot E, Simcha I, Iwai K, Ciechanover A, Geiger B, Ben-Ze'ev A (2000). Differential interaction of plakoglobin and β-catenin with the ubiquitin-proteasome system. Oncogene. 19: 1992–2000.
- Saffitz JE (2003). Dependence of electrical coupling on mechanical coupling in cardiac myocytes. In: Thiene G, Dessina AC (eds). Advances in Cardiovascular Medicine. Padua, Italy: Universitá degli Studi di Padova . pp. 15–28.
- Saffitz JE, Asimaki A, Huang H (2010). Arrhythmogenic right ventricular cardiomyopathy: new insights into mechanisms of disease. Cardiovasc Pathol. 19: 166–170.
- Saffitz JE, Asimaki A, Huang H (2009). Arrhythmogenic right ventricular cardiomyopathy: new insights into disease mechanisms and diagnosis. J Investig Med. 57: 861–864.
- Saffitz JE (2009). Arrhythmogenic cardiomyopathy and abnormalities of cell-to-cell coupling. Heart Rhythm. 8: S62–S65.
- Saffitz JE (2011). The pathobiology of arrhythmogenic cardiomyopathy. Annu Rev Pathol. 6: 299–321.
- Sato PY, Coombs W, Lin XM, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M (2011). Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 109: 193–U160.
- Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, Isom LL, Delmar M (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 105: 523–526.
- Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 299: 1410–1413.
- Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ (2010). Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 61: 233–253.
- Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ (2007). Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 115: 1710–1720.
- Shen WK, Edwards WD, Hammill SC, Gersh BJ (1994). Right ventricular dysplasia: a need for precise pathological definition for interpretation of sudden death. Am Coll Cardiol. 23: 34A.
- Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P (2003). Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/ or dysplasia. Circulation. 108: 3000–3005.
- Thiene G, Corrado D, Basso C (2007). Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J Rare Dis. 2: 45–61.
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988). Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 318: 129–133.
- Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A (2001). Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 10: 189–194.
- van de Schans VA, Smits JF, Blankesteijn WM (2008). The Wnt/frizzled pathway in cardiovascular development and disease: friend or foe?Eur J Pharmacol. 585: 338–345.
- van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck-Zwarts KY, Lekanne dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP (2012). Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 14: 1199–1207.
- van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN (2006). Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 113: 1650–1658.
- Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, Danieli GA, Lorenzon A, Nava A, Bauce B, Thiene G, Basso C, Calkins H, Gear K, Marcus F, Towbin JA; Multidisciplinary Study of Right Ventricular Dysplasia Investigators (2010). Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 55: 587–597.
- Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA (2006). Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 99: 646–655.