
Abstract
Desmosomes are intercellular junctions that provide strong adhesion or hyper-adhesion in tissues. Here, we discuss the molecular and structural basis of this with particular reference to the desmosomal cadherins (DCs), their isoforms and evolution. We also assess the role of DCs as regulators of epithelial differentiation. New data on the role of desmosomes in development and human disease, especially wound healing and pemphigus, are briefly discussed, and the importance of regulation of the adhesiveness of desmosomes in tissue dynamics is considered.
INTRODUCTION
Animal body tissues are amazingly tough. This is evident in human epidermis, which ranges from 0.07 mm to 0.12 mm in thickness over the general body surface, increasing to 0.8 mm on the palms and 1.4 mm on the soles. The epidermis is subjected to continuous rubbing and pressure. The living layers of the epidermis are composed of cells, keratinocytes (˜20 μm in diameter) surrounded by plasma membranes (˜70-nm thick). How can such a tissue resist physical stress? There are three contributory structures: (1) the underlying dermis, composed of resilient proteins, collagen, elastin and fibrillin, to which the epidermis is anchored by hemidesmosomes; (2) the cytoskeletal proteins, mainly keratins, which give the cells internal strength; and (3) the adhesive links, which both cement the cells together and provide surface attachments for the keratin filaments, creating a tissue-wide scaffolding support.
The principal adhesive links are intercellular junctions called desmosomes, and the purpose of this article is to discuss their structure and function in tissues. Desmosomes are abundant in stress-bearing tissues including cardiac muscle and stratified epithelia. They are less abundant in simple epithelia, meninges and follicular dendritic cells, but are absent in nervous tissue, skeletal and smooth muscle and endothelia, although they are present in at least some endothelia of fish (CitationFawcett, 1961).
Tissues are dynamic structures, and there is therefore a conflict between toughness, which is vital for survival of the organism, and the necessity for malleability in development, turnover and damage repair. On the one hand, it seems crucial for the cells to be locked together to provide strength and resilience; on the other, relative cellular motility seems desirable for modelling, replacement and remodelling. The desmosomal lock, which binds cells together, must be capable of unlocking.
Since this article deals only with specific aspects, we refer the reader to other recent reviews for broader coverage of the topic. These include desmosome assembly and dynamics (CitationNekrasova & Green, 2013), mechanistic basis of heart diseases involving desmosomes (CitationAl-Jassar et al., 2013) pemphigus (CitationKitajima, 2013; CitationWaschke, 2008), desmosomes and cancer (CitationChidgey & Dawson, 2007), as well as more general reviews (CitationDubash & Green, 2011; CitationKowalczyk & Green, 2013; CitationThomason et al., 2010).
DESMOSOMAL ADHESION
Adhesion molecules and some evolutionary considerations
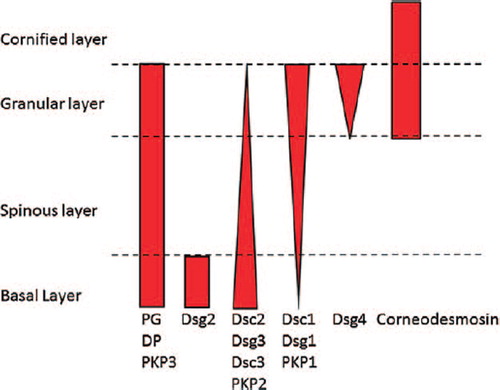
Adhesion in desmosomes is mediated by the desmosomal cadherins (DCs), desmocollin (Dsc) and desmoglein (Dsg). In mammals, there are multiple DC isoforms; for example, in humans, there are three Dscs (1–3) and four Dsgs (1–4) (). Dsc2 and Dsg2 are ubiquitous in desmosome-bearing tissues, although there is now some disagreement as to whether Dsg2 occurs in the basal layer of interfollicular epidermis or is confined to hair follicles (CitationHartlieb et al., 2013). The other isoforms are primarily expressed in stratified epithelia; Dsc1, Dsg1 and Dsg4 predominate in the upper layers, and Dsc3 and Dsg3 in the lower layers. In the zebrafish, however, which also has stratified epithelia, there is only one Dsc (zDsc) and two closely related zDsgs (zDsgα and zDsgβ) (CitationGoonesinghe et al., 2012). Curiously, zDsc is the orthologue of mammalian Dsc1 and the both zDsgs are orthologues of mammalian Dsg2.
Figure 1. Diagram showing the distributions of the major desmosomal components and their isoforms in human epidermis. In the cornified layer, desmosomal adhesion is mediated by corneodesmosin, which is not discussed in this review.
Dscs and Dsgs show ˜30% amino acid identity to each other and to Type1 cadherins, which they resemble more closely in their extracellular (EC) domains. Cytoplasmically, Dscs show alternative splicing of a stop codon-containing mini exon that, when spliced out, produces a longer “a” form, containing binding sites for other plaque components, mainly plakoglobin (PG), plakophilin (PKP) and desmoplakin (DP), and, when spliced in, a shorter “b” form that supports PKP but not PG and DP binding ( and ). The “a” form supports desmosomal plaque formation, but the “b” form does not (CitationTroyanovsky et al., 1993), leaving the function of the “b” form unresolved. Mice lacking Dsc1 have adhesion defects in the upper epidermis, but mice lacking the “a” and “b” cytoplasmic domains of Dsc1 do not show such defects, suggesting that the EC and transmembrane domains are sufficient to maintain adhesion (CitationCheng et al., 2004; CitationChidgey et al., 2001). Human Dsc genes contain 17 exons, exon 16 encoding the mini exon, but the zebrafish gene contains only 16 exons, lacking the mini exon and therefore a Dsc “b” form (CitationGoonesinghe et al., 2012). Yet, zebrafish form functional desmosomes. ENSEMBL data suggest that Xenopus, the chicken and the zebra finch, also lack a mini exon and thus a “b” form, whereas a mini exon and two spliced forms are present in the duck-billed platypus (D. Garrod; unpublished). Thus, the “b” form may be strictly mammalian, making it even more intriguing to speculate about its function.
Figure 2. Stick diagrams of the DCs, Dsc2 and Dsg2, showing features referred to in text. EC, extracellular; EA, extracellular anchor; TM, transmembrane; ICS, intracellular cadherin segment; IPL, intracellular proline-rich linker; RUD, repeat unit domain; TD, terminal domain.
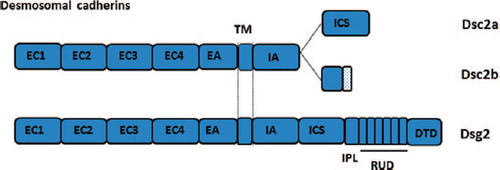
Figure 3. Graph showing reciprocal exponential distributions of Dsc1 and Dsc3 in bovine nasal epidermis as determined by quantitative analysis of immuno-gold labelling (CitationNorth et al., 1996).
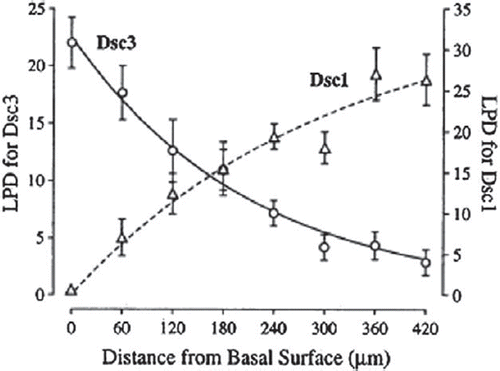
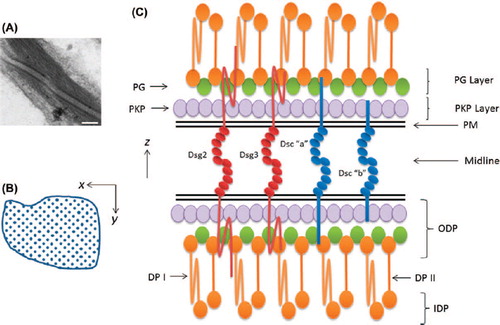
Figure 4. Desmosome structure. Electron micrograph of two desmosomes from human chronic wound epidermis showing the midline between the plasma membranes, the outer dense plaques and the inner dense plaques with attached keratin filaments (bar = 100 nm). Diagram showing the quadratic array of DCs the intercellular region. This had a periodicity of ˜70Å. Diagram (not to scale) showing transverse lamellar arrangement of desmosome. The locations of the molecules are based on immuno-gold labelling and electron tomography (See text).
The cytoplasmic domains of Dsgs consist of a series of subdomains, an intracellular anchor, an intracellular cadherin segment, an intracellular proline-rich linker, a repeat unit domain (RUD) and a terminal domain (TD) (). This unique organisation is present in zebrafish and therefore appears fundamental to desmosome structure and function; true desmosomes do not appear to be present in invertebrates. Apparently normal desmosomes can be formed by cells lacking Dsc and therefore having Dsg as the only DC (CitationKoeser et al., 2003). This suggests that the cytoplasmic domain of Dsg, like Dsc “a”, is sufficient to support desmosome plaque formation.
Possible functions of the DC isoforms
It is not clear why mammals require different isoforms of DCs. Studies of human skin diseases and gene knock-out in mice indicate that the different isoforms have functions that, broadly speaking, correlate with their expression patterns. Thus, in the blistering disease pemphigus foliaceus autoantibodies that target Dsg1 cause loss of cell–cell adhesion (acantholysis) in the granular layer (CitationMahoney et al., 1999). Bacterial exfoliative toxins, serine proteases, show exquisite specificity for Dsg1 and cause blistering only in the superficial epidermis (CitationHanakawa et al., 2004). Autosomal dominant DSG1 mutations cause striate palmoplantar keratoderma, the formation of hyperkeratotic plaques on the fingers, palms and soles (CitationRickman et al., 1999), and homozygous mutations in DSG1 cause severe dermatitis, multiple allergies and metabolic wasting syndrome (SAM), which involves acantholysis in upper epidermis (CitationSamuelov et al., 2013). Knockout of Dsc1 from mice causes acantholysis in the granular layer and epidermal fragility (CitationChidgey et al., 2001).
By contrast, autoantibodies against Dsg3 cause acantholysis between the basal and first suprabasal layers of epidermis and oral mucosa in the blistering disease pemphigus vulgaris (PV) (autoantibodies to Dsg1are generally also required to produce epidermal blistering) (CitationMahoney et al., 1999; CitationShirakata et al., 1998). Knockout of Dsg3 from mice also causes suprabasal acantholysis in oral mucosa and epidermis (CitationKoch et al., 1997). Global knockout of Dsc3 from mice causes death at E2.5, before the first desmosomes form, but conditional knockout from the epidermis gives rise to PV-like suprabasal acantholysis (CitationChen et al., 2008; CitationDen et al., 2006). Pathogenic antibodies to Dsc3 have also been demonstrated in PV (CitationMao et al., 2010), and it is likely that such antibodies also cause pemphigus vegetans (CitationSaruta et al., 2013).
The cleavage planes due to loss of cell adhesion are quite specifically localised, much more so than expression of the various targeted isoforms of DCs. In fact, the territories in which the different isoforms are expressed overlap extensively and in at least one case, the bovine nasal epidermis, Dsc3 and Dsc1 show exquisite exponential reciprocally graded expression () (CitationNorth et al., 1996). The reason for this is unknown. Does it mean that the adhesive properties of desmosomes change gradually with level in the epidermis? For example, is Dsc1 more strongly adhesive than Dsc3, giving stronger adhesion in the upper epidermis than in the lower regions, and if so, why should this be? Surely the epidermis needs to be strong throughout. In the human keratinocyte cell line HaCaT, which expresses Dsg2 and Dsg3, roughly Dsg3 appears equally more important for adhesion than Dsg2, except at high shear rates when Dsg2 is also important (CitationHartlieb et al., 2013). This may suggest differential adhesive properties for the different isoforms, thus supporting a previous suggestion based on differential sorting of mammary epithelial cells (CitationRunswick et al., 2001). More work is needed on the differential adhesive properties of DC isoforms.
Reciprocally graded protein distributions have a widely recognised role in developmental pattern formation (CitationHironaka & Morishita, 2012; CitationKicheva et al., 2012; CitationSt Johnston & Nusslein-Volhard, 1992). Is it possible that the reciprocally graded distributions of DCs play a signalling role in regulating differentiation of stratified epithelia (CitationNorth et al., 1996)? A series of studies in which the DCs Dsg2, Dsg3 and Dsc3 were mis-expressed in mouse epidermis under the involucrin, keratin 1 or keratin 14 promoters showed perturbation of differentiation and proliferation, thus tending to support a role for these proteins in regulating differentiation (CitationAllen et al., 1996; CitationBrennan et al., 2007; CitationElias et al., 2001; CitationHardman et al., 2005; CitationMerritt et al., 2002). However, mis-expression of Dsc1a under the K14 promoter produced no phenotype (CitationHenkler et al., 2001). Inv-Dsg2 keratinocytes showed enhanced survival and anchorage-independent growth dependent on epidermal growth factor receptor (EGFR) activation and NF-κB activity and activation of multiple cell survival pathways, in the mice which were more susceptible to tumour promotion (CitationBrennan et al., 2007). K14-Dsc3 keratinocytes showed enhanced beta-catenin signalling (CitationHardman et al., 2005).
A powerful case has been made from culture experiments for a role of Dsg1 in regulating epidermal differentiation. Thus, Dsg1 promotes differentiation by suppressing EGFR/extracellular signaling-regulating kinases (ERK) signalling, an effect mediated though the cytoplasmic domain of the DC (CitationGetsios et al., 2009). Further, Dsg1 interacts with Erbin, another differentiation-related protein, to disrupt Ras-SHOC2 complexes, thus diminishing ERK signalling and driving differentiation (CitationHarmon et al., 2013). In turn, Dsg1 promotion of differentiation is regulated by the GEF breakpoint cluster region (Bcr) which regulates Rho/MAL control of Dgs1 expression (CitationDubash et al., 2013). In this context, it is interesting that striate palmoplantar keratoderma patients deficient in Dsg1 show increased Ras-SHOC2 activity and decreased SHOC2-Erbin co-localisation (CitationHarmon et al., 2013).
Thus, there is considerable evidence that DCs can participate in the regulation of differentiation signals. Do they regulate epidermal differentiation in vivo? Knockout of mouse Dsg3 or Dsc3 produced adhesive defects in interfollicular epidermis, but no defects in epidermal differentiation (CitationChen et al., 2008; CitationKoch et al., 1997). Knockout of Dsc1 also produced adhesion defects and slight differentiation defects, probably secondary consequences of trauma caused by the adhesion defects (CitationChidgey et al., 2001). There has not yet been a knockout of Dsg1 but failure of Dsg1 membrane expression in SAM syndrome results in psoriasiform dermatitis with alternating para- and orthokeratosis and hypo- and hypergranulosis (CitationSamuelov et al., 2013). On the basis of knockout evidence so far available, it would seem then that the DCs are not major regulators of epidermal differentiation in mice. On the other hand, the association of mutations in human DSG1 and DSC2 with palmoplantar keratoderma (PPK), and the latter with woolly hair, suggests that they contribute to fine regulation of differentiation (CitationRickman et al., 1999; CitationSimpson et al., 2009).
Little is known about how DC gene expression is regulated. Dsg1, Dsc3 and Dsc2 genes possess epithelial-specific promoters containing binding sites for transcription factors, the AP-1, AP-2 and Sp1 families, that regulate keratinocyte-specific gene expression (CitationAdams et al., 1998; CitationMarsden et al., 1997; CitationSilos et al., 1996). CitationSmith et al. (2004) showed that Dsc1 and Dsc3 are differentially activated by epidermally expressed members of the CCAAT/enhancer-binding protein (C/EBP) family; both are activated by C/EBPδ, but Dsc1 is specifically activated by C/EBPα and Dsc3 by C/EBPβ (CitationSmith et al., 2004). This may contribute to the differential expression of these genes in epidermis. Furthermore, Dsc2 and Dsc3 expression are differentially regulated by PG; PG activates both genes but suppresses Dsc3 expression in the presence of Lef-1 (CitationTokonzaba et al., 2013). Additionally, c-rel a component of the ectodysplasin-A (EDA)/ectodysplasin-A receptor (EDAR)/NF-κB signalling cascade activates Dsc2 expression in conjunction with PG/Lef-1. This is seen as a mechanism for differential regulation of Dsc2/Dsc3 particularly in hair follicle development.
The mechanism of adhesion
Adhesive binding by the DCs is directly mediated by their EC domains, which resemble those of Type1 cadherins. As shown by sequence analysis, homology modelling and determination of the NMR structure of hDsg2, the EC domains consist of five subdomains (EC1–5) of which at least the first four have a Type1 cadherin-like β-barrel structure. Furthermore, the inter-domain regions possess low-affinity calcium-binding sites. These are occupied at physiological EC calcium concentrations of > 1 mM and are functional because they maintain the molecules in an extended, adhesion-competent configuration (CitationKoch et al., 1997; CitationNagar et al., 1996; CitationPokutta et al., 1994). When the sites are unoccupied, the EC domains adopt a flexible, non-adhesive conformation.
Because of this, cadherin and DC adhesion is conventionally referred to as “calcium-dependent.” Calcium dependence provides a useful, if artificial, tool for the cell biologist, enabling manipulation of cell adhesion and junction formation in culture (CitationHennings & Holbrook, 1983). Cells generally adhere to surfaces and proliferate when the calcium concentration of the medium ≤ 0.1 mM but neither desmosomes nor adherens junctions form. Restoring a physiological calcium concentration then promotes adhesion and initiates rapid junction formation. Similarly, chelation of EC calcium can be used to dissociate cells. However, calcium dependence is unlikely to provide an adhesion-regulatory mechanism in vivo because EC calcium is maintained relatively constant, always above that necessary for cadherin-mediated adhesion. The normal range of calcium concentration in human serum is 2.12–2.62 mM, and remedial treatment is given if it falls below about 1.9 mM.
Trans interaction (i.e. cell–cell) by classical cadherins involves strand-swapping by the EC1 domains of where the side chains of conserved tryptophan residues (Trp2) are inserted into hydrophobic pockets of apposed molecules. The Trp residue and hydrophobic pocket are conserved in DCs, and we have shown that mutations affecting either abolish adhesive binding (CitationNie et al., 2011). Thus, similar strand-swapping is likely to be involved in desmosomal adhesion.
Type 1 cadherins participate in homophilic adhesion, which appears to be energetically favoured by the strand-swapping mechanism (CitationChen et al., 2005). Adhesion between DCs has long been regarded as both homo- and heterophilic, but the evidence for this was based on studies of the interactions of complete or incomplete recombinant EC domains by biophysical methods or of incomplete molecules in cells that do not form desmosomes (CitationChitaev & Troyanovsky, 1997; CitationSyed et al., 2002; CitationWaschke et al., 2007). It seems that all desmosomes in vivo contain at least one Dsc and one Dsg, and their ratio appears to be critical for adhesion (CitationGetsios et al., 2004). Furthermore, where different isoforms of the DCs are co-expressed in tissues, they are mixed in the same desmosomes (CitationNorth et al., 1996). Thus, multiple types of homo- and heterophilic binding are potentially possible. For example, in HaCaT cells, which express similar amounts of Dsc2, Dsc3, Dsg2 and Dsg3, all co-localised in desmosomes, nonspecific binding would allow 10 possible interactions. Could desmosomes really be that disorganised?
To determine the nature of DC binding in desmosome-forming cells, we treated HaCaT cells with a membrane-impermeable, homobifunctional cross-linker (CitationNie et al., 2011). Since the desmosomes were in their calcium-dependent phase, we could show, by chelating calcium, that the desmosomes were extracellularly cross-linked. The cells were then solubilised, and immunoprecipitation was carried out with antibodies to each of the DCs. Blotting of the immunoprecipitates showed that each was recognised only by the antibody as used for IP. In other words, each DC bound exclusively to itself. Further experiments showed that the binding was in trans. We concluded that adhesive binding by the DCs was homophilic and isoform-specific; of the 10 possible combinations only four were found, Dsc2-Dsc2, Dsc3-Dsc3, Dsg2-Dsg2 and Dsg3-Dsg3. We do not understand the significance of this adhesive specificity but speculate that it is fundamental to the structural organisation and strength of desmosomal adhesion.
DESMOSOME STRUCTURE
Desmosomes were first identified as densities between adhering cells by CitationBizzozero (1864) but no further understanding was made until the advent of electron microscopy. Desmosomes have an extremely regular structure (). In the transverse or “z” direction, desmosomes have a symmetrical, layered structure (CitationOdland, 1958). The space between the plasma membranes of the two cells contributing to the desmosome is sometimes called the desmosomal core or desmoglea. Halfway between the membranes lies a density, the midline, where adhesive binding occurs because the N-termini of the DCs are located there (CitationAl-Amoudi et al., 2007; CitationShimizu et al., 2005). Lanthanum infiltration and electron tomography of vitreous sections have demonstrated that the DC EC domains are arranged in the en face or “x-y” plane in a quadratic array with a repeat of ˜70 Å, which is very close to that found in the C-cadherin crystal structure (CitationAl-Amoudi et al., 2007; CitationBoggon et al., 2002; CitationGarrod et al., 2005; CitationRayns et al., 1969). The inter-membrane distance is ˜35 nm, also very close to the 38.5 nm determined from the C-cadherin crystal structure (CitationAl-Amoudi et al., 2007).
The desmosomal plaques also have a layered structure, resolved into an outer dense plaque(ODP), with its inner face about 20 nm from the membrane, and an inner dense plaque (IDP), which joins the IFs about 50 nm from the membrane (CitationAl-Amoudi et al., 2011; CitationNorth et al., 1999). Immuno-gold labelling showed the ODP as a region of multiple protein–protein interactions (CitationNorth et al., 1999). The key observations were as follows: (i) PKP lies close to the plasma membrane; (ii) PG and the N-terminus of DP are further from the membrane and overlap with the C-terminus of Dsc “a”, as well as with the entire cytoplasmic domain of Dsg3, which lies in the ODP; (iii) the C-terminus of Dsc “b” is closer to the membrane and spatially separated from PG and DP; (iv) the C-terminus of DP lies in the IDP, roughly 40 nm from the membrane. This is consistent with the predicted length of the shorter sliced form, DPII, and suggests that DPI is coiled or folded. Broadly speaking, the locations of these molecules are consistent with their interactions determined in vitro. We have since determined that the RUD domain of Dsg1/2 (which have larger cytoplasmic domains than Dsg3) is located in the ODP near its inner face and the TD internal to the ODP but not extending to the IDP (Scothern and Garrod; unpublished).
Electron tomography of the ODP produced a molecular map with a resolution of 3.2 nm (CitationAl-Amoudi et al., 2011). This showed a 2D interconnected quasiperiodic lattice with a similar spatial organisation to the EC side. On the basis of the above-mentioned immuno-gold labelling, the transverse organisation was resolved into an outer 4 nm-thick PKP layer and an inner, denser, 8 nm-thick PG layer, which also contains the N-termini of DP molecules.
HYPER-ADHESION
We have coined the term hyper-adhesion to distinguish the strong adhesion mediated by mature desmosomes from what we believe to be the weaker adhesion mediated by Type1 cadherins at adherens junctions (CitationGarrod et al., 2005; CitationKimura et al., 2007). Hyper-adhesion is defined experimentally by the complete resistance of desmosomes to disruption by chelation of EC calcium with EGTA (CitationGarrod, 2013). This property is characteristic of most, possibly all, desmosomes in adult mammalian tissues (CitationGarrod et al., 2005; CitationKimura et al., 2007; CitationThomason et al., 2012; CitationWallis et al., 2000). Newly formed desmosomes in culture are calcium dependent. At this time chelation of EC calcium causes desmosomes to split into halves through the plane of adhesion. However, when epithelial cells are maintained in confluent culture, desmosomes acquire hyper-adhesion without changing their molecular composition (CitationKimura et al., 2007). HaCaT cells with hyper-adhesive desmosomes were found to be more strongly adhesive than those with calcium-dependent desmosomes (CitationKimura et al., 2007).
The molecular basis of hyper-adhesion is not understood. However, studies of epidermal wound healing and mouse embryonic development suggest that it is somehow linked to the presence of a midline; hyper- adhesive desmosomes appear to have a prominent midline whereas calcium dependent desmosomes do not (CitationGarrod et al., 2005; CitationKimura et al., 2012; CitationThomason et al., 2012) and see above and below). This implies that the arrangement of the DC EC domains in the intercellular space can be dynamically regulated, in turn regulating the adhesive strength of the desmosomes. It seems probable that transmembrane signals emanating from the desmosomal plaque are involved in this regulation. Thus, desmosomal adhesiveness is regulated by protein kinase C alpha (PKCα), activation-promoting calcium dependence and inhibition-promoting hyper-adhesion (CitationGarrod et al., 2005; CitationKimura et al., 2007; CitationThomason et al., 2012; CitationWallis et al., 2000). Multiple signals may be involved since inhibition of tyrosine phosphatases (i.e. promotion of tyrosine phosphorylation) also promotes hyper-adhesion (CitationGarrod et al., 2008).We suggest that phosphorylation of one or more plaque components causes rearrangement within the plaque and transmits a signal to the EC domains. Desmosomes lacking PKP1 or PG do not seem to acquire hyper-adhesion (CitationSouth et al., 2003) (McHarg, Mueller and Garrod: unpublished) so these plaque components may be important in its regulation. On the other hand, desmosomes lacking DP or carrying DP mutations that prevent IF attachment seem to acquire a midline (CitationJonkman et al., 2005; CitationSumigray & Lechler, 2012). Phosphorylation does, however, regulate DP-keratin binding and keratins have been shown to regulate PKCα activity and DP phosphorylation (CitationHobbs & Green, 2012; CitationKroger et al., 2013). Enhanced association of keratin with DP through blocking DP phosphorylation provides another mechanism for strengthening desmosomal adhesion (CitationHobbs & Green, 2012).
The importance of hyper-adhesion in vivo: desmosomes in wound healing
Hyper-adhesion, which locks epidermal cells together, seems incompatible with cell migration and therefore with wound re-epithelialisation. There is also some evidence for desmosome down-regulation in wound healing (CitationAllen & Potten, 1975; CitationCroft & Tarin, 1970; CitationGarrod et al., 2005). A series of studies have now shown that desmosomal adhesion changes from hyper-adhesive to calcium dependent in wound edge epithelium.
First, wounding a hyper-adhesive monolayer of MDCK cells caused desmosomes at the wound edge to become rapidly calcium dependent and this change was propagated to cells hundreds of micrometres from the edge (CitationWallis et al., 2000). Furthermore, the change appeared to be signalled by PKCα. Second, on wounding mouse epidermis, desmosomes in the wound-edge epithelium lost hyper-adhesiveness, becoming calcium dependent (CitationGarrod et al., 2005). In doing so, they lost the midline and the intercellular space narrowed by 10% (CitationGarrod et al., 2005). Transition to calcium dependence was accompanied by translocation of PKCα from cytosol to desmosomal plaques suggesting its activation. Third, CitationThomason et al. (2012) provided direct evidence of the role of PKCα in wound re-epithelialisation. Thus, PKCα −/− mice showed delayed re-epithelialisation, whereas bitransgenic mice over-expressing constitutively active PKCα showed accelerated re-epithelialisation. Furthermore, these effects were associated with delayed loss of hyper-adhesion in PKCα−/− mice and accelerated loss of hyper-adhesion in CA-PKCα mice. Moreover, in acute human epidermal wounds, PKCα localised to desmosomes at the wound edge. This was associated with switch of desmosomes to calcium dependence. However, in chronic wounds, desmosomes failed to switch from the hyper-adhesive state and PKCα remained cytoplasmic. These results suggest that manipulation of PKC signalling could provide a novel therapeutic approach for chronic wounds.
The normal presence and distribution of desmosomal structural proteins is important to desmosomes functions. CitationBeaudry et al. (2010) used mice lacking the desmosomal structural component Perp in order to demonstrate the contribution of desmosomal adhesion to wound healing (CitationBeaudry et al., 2010). Perp conditional knockout mice showed delayed wound healing relative to controls. Furthermore, loss of Perp did not impair keratinocyte proliferation and actually enhanced keratinocyte migration in vitro. Thus, Perp's role in cell adhesion is essential for wound closure.
The importance of hyper-adhesion in vivo: PV
In PV, a potentially fatal blistering disease of skin and mucous membranes, autoantibodies to Dsg1 and Dsg3 cause acantholysis (CitationAmagai et al., 1991; CitationDing et al., 1999). Suggested alternative, but not necessarily mutually exclusive mechanisms, for acantholysis include direct disruption of desmosomal adhesion due to steric hindrance by the autoantibodies and activation of outside–in signalling by autoantibody binding (CitationMuller et al., 2008; CitationWaschke, 2008).
Ultrastructural analysis of human PV appears to show that direct disruption of desmosomal adhesion is not the primary event (CitationDiercks et al., 2009). Rather, there is extensive loss of cell–cell adhesion in inter-desmosomal regions and possible intracellular cleavage behind the desmosomal plaque that might indicate a weakening of the cytoskeleton, perhaps through a signalling mechanism involving plakoglobin (CitationDiercks et al., 2009; CitationMuller et al., 2008). By contrast, abundant split desmosomes with inserted keratin filaments were found in a mouse model of pemphigus (CitationShimizu et al., 2004).
Work in which keratinocytes were treated PV autoantibodies in culture showed that desmosomes were down-regulated and Dsg 3 was depleted and internalised (CitationCalkins et al., 2006; CitationCirillo et al., 2008; CitationCirillo et al., 2006; CitationDelva et al., 2008; CitationYamamoto et al., 2007). However, all of this work was presumably carried out with keratinocytes possessing calcium-dependent rather than hyper-adhesive desmosomes. A study in which keratinocytes with calcium dependent desmosomes and those with hyper-adhesive desmosomes were compared showed that hyper-adhesion inhibited PV autoantibody-induced acantholysis and internalisation of adhesion molecules including Dsg 3 and E-cadherin (CitationCirillo et al., 2010). Furthermore, over-expression of PKP1 in keratinocytes caused the desmosomes to become hyper-adhesive and protected them from dissociation by PV antibodies (CitationTucker et al., 2013). This result is intriguing in relation to previous observations that loss of PKP1 destabilises desmosomes (CitationSouth et al., 2003). Moreover, PKP1 () is predominantly expressed in the upper layers of epidermis where acantholysis does not occur in PV. However, Kimura et al. found that keratinocytes became hyper-adhesive in confluent culture without any change in the level of PKP1 expression (CitationKimura et al., 2007).
While the primary effect of PV autoantibodies does not appear to be direct disruption of existing desmosomal adhesion, they could inhibit de novo desmosome assembly, which must be a continuous process in stratified epithelia. During the progress of disease, this would be expected to result in gradual down-regulation of desmosomes and loss of cell–cell adhesion. It would therefore be interesting to know whether the above-mentioned results obtained with calcium-dependent keratinocytes in culture have any counterpart in vivo. It could also be the case that the primary loss of inter-desmosomal adhesion found in PV might resemble epidermal wounding and thus cause activation of PKC and consequent weakening of desmosomal adhesion as we have described (CitationGarrod et al., 2005). In this case, inhibition of PKC could provide a novel therapy for pemphigus (CitationCirillo et al., 2010; CitationKitajima, 2013).
DESMOSOMES IN DEVELOPMENT
Desmosomes are essential for embryonic development. Deletion of the genes for PG, PKP2 or DP in mice results in embryonic death principally because of failure of intercellular adhesion (CitationBierkamp et al., 1996; CitationGallicano et al., 1998; CitationGrossmann et al., 2004; CitationRuiz et al., 1996). On the other hand, the early embryonic lethal effects caused by deletion of Dgs2 or Dsc3 are more probably due to signalling defects. Deletion of Dsg2 causes death at around the time of implantation because of a defect in embryonic stem cell proliferation, while that of Dsc3 causes death prior to E2.5, which precedes first desmosome assembly in the trophectoderm (CitationDen et al., 2006; CitationEshkind et al., 2002). Of further developmental significance may be the demonstration that desmosomal adhesion regulates cell positioning during mammary gland morphogenesis (CitationRunswick et al., 2001).
Since desmosomes appear early in mouse tissue development and since developing tissues must remain malleable to participate in morphogenetic movements, we hypothesised that initial weak adhesion would be followed by acquisition of hyper-adhesion (CitationKimura et al., 2012). We showed that epidermal desmosomes were calcium dependent until embryonic day 12 (E12) and became hyper-adhesive by E14. Similarly, blastocyst trophectodermal desmosomes were calcium-dependent on E3 but became hyper-adhesive by E4.5. In both, development of hyper-adhesion was accompanied by the appearance of a midline supporting previous evidence that hyper-adhesiveness depends on the organised arrangement of DCs. By contrast, adherens junctions remained calcium dependent throughout development, but tight junctions became calcium independent as desmosomes mature. Using protein kinase C (PKC) activation and PKCα−/− mice, we provide evidence suggesting that conventional PKC isoforms are involved in developmental progression to hyper-adhesiveness. Furthermore, regulation of desmosomal adhesion by PKC may be important in trophoblast migration during implantation. It appears that tissue stabilisation is one of several roles played by desmosomes in animal development.
Because desmosomes are essential in mammalian development and they appear at the mid gastrula stage of Fundulus development, it seemed likely that they may also be important in fish early development. We therefore examined the role of DCs in zebrafish early development (CitationGoonesinghe et al., 2012). Both zfDsc and zfDsgα were present as maternal and zygotic transcripts, whereas zfDsgβ was first expressed from 8 hours post-fertilisation (hpf). All three transcripts were present throughout subsequent stages. Morpholino knockdown of both zfDsc and zfDsgα expression produced similar defects in epiboly, axis elongation and somite formation, associated with abnormal desmosomes or reduced desmosome numbers. These results demonstrate an important role for DCs and desmosomes in the early morphogenesis of the zebrafish embryo and provide a basis for more detailed analysis of this role. Knockdown of PG in zebrafish gave rise to heart defects at 48 hpf (CitationMartin et al., 2009).
CONCLUSION
The year 2014 is the 150th anniversary of the first brief description of these intercellular connections with the light microscope (CitationBizzozero, 1864), and we still know rather little about their role in tissues. They are quite difficult to study in vivo because they are extremely insoluble and can only really be resolved with the electron microscope. We still need to understand many things. In particular, how are desmosomal dynamics regulated in vivo and how does that relate to the dynamics of tissues during normal homeostasis, during development and during disease? What precisely is the role of desmosomes in cellular signalling and the regulation of differentiation? Why do mammals have multiple isoforms of DCs and what impact do they have on the two previous questions?
Declaration of interest: The authors report no declaration of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adams MJ, Reichel MB, King IA, Marsden MD, Greenwood MD, Thirlwell H, Arnemann J, Buxton RS, Ali RR (1998). Characterization of the regulatory regions in the human desmoglein genes encoding the pemphigus foliaceous and pemphigus vulgaris antigens. Biochem J. 329: 165–174.
- Al-Amoudi A, Castano-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS (2011). The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci USA. 108:6480–6485.
- Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS (2007). The molecular architecture of cadherins in native epidermal desmosomes. Nature. 450: 832–837.
- Al-Jassar C, Bikker H, Overduin M, Chidgey M (2013). Mechanistic basis of desmosome-targeted diseases. J Mol Biol. 425: 4006–4022.
- Allen E, Yu QC, Fuchs E (1996). Mice expressing a mutant desmosomal cadherin exhibit abnormalities in desmosomes, proliferation, and epidermal differentiation. J Cell Biol. 133: 1367–1382.
- Allen TD, Potten CS (1975). Desmosomal form, fate, and function in mammalian epidermis. J Ultrastruct Res. 51: 94–105.
- Amagai M, Klaus-Kovtun V, Stanley JR (1991). Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 67: 869–877.
- Beaudry VG, Ihrie RA, Jacobs SB, Nguyen B, Pathak N, Park E, Attardi LD (2010). Loss of the desmosomal component perp impairs wound healing in vivo. Dermatol Res Pract. 2010: 759731.
- Bierkamp C, McLaughlin KJ, Schwarz H, Huber O, Kemler R (1996). Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 180: 780–785.
- Bizzozero G (1864). Delle celluie cigliate del reticolo malpigphiano dell'epidermide, delle mucose e dei cancroidi. Ann Univ Meal. 190: 110–118.
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L (2002). C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 296: 1308–1313.
- Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, O’Brien T, Uitto J, Rodeck U, Mahoney MG (2007). Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci. 120: 758–771.
- Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP (2006). Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 281: 7623–7634.
- Chen CP, Posy S, Ben-Shaul A, Shapiro L, Honig BH (2005). Specificity of cell-cell adhesion by classical cadherins: Critical role for low-affinity dimerization through beta-strand swapping. Proc Natl Acad Sci USA. 102:8531–8536.
- Chen J, Den Z, Koch PJ (2008). Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 121: 2844–2849.
- Cheng X, Mihindukulasuriya K, Den Z, Kowalczyk AP, Calkins CC, Ishiko A, Shimizu A, Koch PJ (2004). Assessment of splice variant-specific functions of desmocollin 1 in the skin. Mol Cell Biol. 24: 154–163.
- Chidgey M, Brakebusch C, Gustafsson E, Cruchley A, Hail C, Kirk S, Merritt A, North A, Tselepis C, Hewitt J, Byrne C, Fassler R, Garrod D (2001). Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J Cell Biol. 155: 821–832.
- Chidgey M, Dawson C (2007). Desmosomes: a role in cancer?Br J Cancer. 96: 1783–1787.
- Chitaev NA, Troyanovsky SM (1997). Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J Cell Biol. 138: 193–201.
- Cirillo N, Campisi G, Gombos F, Perillo L, Femiano F, Lanza A (2008). Cleavage of desmoglein 3 can explain its depletion from keratinocytes in pemphigus vulgaris. Exp Dermatol. 17: 858–863.
- Cirillo N, Femiano F, Gombos F, Lanza A (2006). Serum from pemphigus vulgaris reduces desmoglein 3 half-life and perturbs its de novo assembly to desmosomal sites in cultured keratinocytes. FEBS Lett. 580: 3276–3281.
- Cirillo N, Lanza A, Prime SS (2010). Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res. 316: 580–592.
- Croft CB, Tarin D (1970). Ultrastructural studies of wound healing in mouse skin. I. Epithelial behaviour. J Anat. 106: 63–77.
- Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP (2008). Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J Biol Chem. 283: 18303–18313.
- Den Z, Cheng X, Merched-Sauvage M, Koch PJ (2006). Desmocollin 3 is required for pre-implantation development of the mouse embryo. J Cell Sci. 119: 482–489.
- Diercks GF, Pas HH, Jonkman MF (2009). The ultrastructure of acantholysis in pemphigus vulgaris. Br J Dermatol. 160: 460–461.
- Ding X, Diaz LA, Fairley JA, Giudice GJ, Liu Z (1999). The anti-desmoglein 1 autoantibodies in pemphigus vulgaris sera are pathogenic. J Invest Dermatol. 112: 739–743.
- Dubash AD, Green KJ (2011). Desmosomes. Curr Biol. 21: R529–531.
- Dubash AD, Koetsier JL, Amargo EV, Najor NA, Harmon RM, Green KJ (2013). The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J Cell Biol. 202: 653–666.
- Elias PM, Matsuyoshi N, Wu H, Lin C, Wang ZH, Brown BE, Stanley JR (2001). Desmoglein isoform distribution affects stratum corneum structure and function. J Cell Biol. 153: 243–249.
- Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE (2002). Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol. 81: 592–598.
- Fawcett DW (1961). Intercellular bridges. Exp Cell Res. Suppl 8: 174–187.
- Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998). Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 143: 2009–2022.
- Garrod DR (2013). The assay that defines desmosome hyper-adhesion. J Invest Dermatol. 133: 576–577.
- Garrod DR, Berika MY, Bardsley WF, Holmes D, Tabernero L (2005). Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci. 118: 5743–5754.
- Garrod DR, Fisher C, Smith A, Nie Z (2008). Pervanadate stabilizes desmosomes. Cell Adh Migr. 2: 161–166.
- Getsios S, Amargo EV, Dusek RL, Ishii K, Sheu L, Godsel LM, Green KJ (2004). Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation. 72: 419–433.
- Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, Cornwell M, Green KJ (2009). Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. 185: 1243–1258.
- Goonesinghe A, Luan XM, Hurlstone A, Garrod D (2012). Desmosomal cadherins in zebrafish epiboly and gastrulation. BMC Dev Bioly. 12: 1.
- Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004). Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 167: 149–160.
- Hanakawa Y, Schechter NM, Lin C, Nishifuji K, Amagai M, Stanley JR (2004). Enzymatic and molecular characteristics of the efficiency and specificity of exfoliative toxin cleavage of desmoglein 1. J Biol Chem. 279: 5268–5277.
- Hardman MJ, Liu K, Avilion AA, Merritt A, Brennan K, Garrod DR, Byrne C (2005). Desmosomal cadherin misexpression alters beta-catenin stability and epidermal differentiation. Mol Cell Biol. 25: 969–978.
- Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, Sarig O, Sprecher E, Green KJ (2013). Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest. 123: 1556–1570.
- Hartlieb E, Kempf B, Partilla M, Vigh B, Spindler V, Waschke J (2013). Desmoglein 2 is less important than desmoglein 3 for keratinocyte cohesion. PloS One. 8: e53739.
- Henkler F, Strom M, Mathers K, Cordingley H, Sullivan K, King I (2001). Trangenic misexpression of the differentiation-specific desmocollin isoform 1 in basal keratinocytes. J Invest Dermatol. 116: 144–149.
- Hennings H, Holbrook KA (1983). Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp Cell Res. 143: 127–142.
- Hironaka K, Morishita Y (2012). Encoding and decoding of positional information in morphogen-dependent patterning. Curr Opin Genet Dev. 22: 553–561.
- Hobbs RP, Green KJ (2012). Desmoplakin regulates desmosome hyperadhesion. J Invest Dermatol. 132: 482–485.
- Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, Pas HH (2005). Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 77: 653–660.
- Kicheva A, Bollenbach T, Wartlick O, Julicher F, Gonzalez-Gaitan M (2012). Investigating the principles of morphogen gradient formation: from tissues to cells. Curr Opin Genet Dev. 22: 527–532.
- Kimura TE, Merritt AJ, Garrod DR (2007). Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol. 127: 775–781.
- Kimura TE, Merritt AJ, Lock FR, Eckert JJ, Fleming TP, Garrod DR (2012). Desmosomal adhesiveness is developmentally regulated in the mouse embryo and modulated during trophectoderm migration. Dev Biol. 369: 286–297.
- Kitajima Y (2013). New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci. 29: 1–13.
- Koch AW, Pokutta S, Lustig A, Engel J (1997). Calcium binding and homoassociation of E-cadherin domains. Biochemistry. 36: 7697–7705.
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR (1997). Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 137: 1091–1102.
- Koeser J, Troyanovsky SM, Grund C, Franke WW (2003). De novo formation of desmosomes in cultured cells upon transfection of genes encoding specific desmosomal components. Exp Cell Res. 285: 114–130.
- Kowalczyk AP, Green KJ (2013). Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 116: 95–118.
- Kroger C, Loschke F, Schwarz N, Windoffer R, Leube RE, Magin TM (2013). Keratins control intercellular adhesion involving PKC-alpha-mediated desmoplakin phosphorylation. J Cell Biol. 201: 681–692.
- Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR (1999). Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 103: 461–468.
- Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, Ishii N, Hashimoto T, Amagai M, Zone JJ, Payne AS (2010). Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. 177: 2724–2730.
- Marsden MD, Collins JE, Greenwood MD, Adams MJ, Fleming TP, Magee AI, Buxton RS (1997). Cloning and transcriptional analysis of the promoter of the human type 2 desmocollin gene (DSC2). Gene. 186: 237–247.
- Martin ED, Moriarty MA, Byrnes L, Grealy M (2009). Plakoglobin has both structural and signalling roles in zebrafish development. Dev Biol. 327: 83–96.
- Merritt AJ, Berika MY, Zhai W, Kirk SE, Ji B, Hardman MJ, Garrod DR (2002). Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Mol Cell Biol. 22: 5846–5858.
- Muller EJ, Williamson L, Kolly C, Suter MM (2008). Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation?J Invest Dermatol. 128: 501–516.
- Nagar B, Overduin M, Ikura M, Rini JM (1996). Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 380: 360–364.
- Nekrasova O, Green KJ (2013). Desmosome assembly and dynamics. Trends Cell Biol. 23: 537–546.
- Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D (2011). Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem. 286: 2143–2154.
- North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, Hatzfeld M, Green KJ, Magee AI, Garrod DR (1999). Molecular map of the desmosomal plaque. J Cell Sci. 112: 432–4336.
- North AJ, Chidgey MA, Clarke JP, Bardsley WG, Garrod DR (1996). Distinct desmocollin isoforms occur in the same desmosomes and show reciprocally graded distributions in bovine nasal epidermis. Proc Natl Acad Sci USA. 93: 7701–7705.
- Odland GF (1958). The fine structure of the interrelationship of cells in the human epidermis. J Biophys Biochem Cytol. 4: 529–538.
- Pokutta S, Herrenknecht K, Kemler R, Engel J (1994). Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur J Biochemistry. 223: 1019–1026.
- Rayns DG, Simpson FO, Ledingham JM (1969). Ultrastructure of desmosomes in mammalian intercalated disc; appearances after lanthanum treatment. J Cell Biol. 42: 322–326.
- Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RA, Leigh IM, Arnemann J, Magee AI, Kelsell DP, Buxton RS (1999). N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet. 8: 971–976.
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W (1996). Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 135: 215–225.
- Runswick SK, O’Hare MJ, Jones L, Streuli CH, Garrod DR (2001). Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol. 3: 823–830.
- Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, Koetsier JL, Gat A, Goldberg I, Bergman R, Spiegel R, Eytan O, Geller S, Peleg S, Shomron N, Goh CS, Wilson NJ, Smith FJ, Pohler E, Simpson MA, McLean WH, Irvine AD, Horowitz M, McGrath JA, Green KJ, Sprecher E (2013). Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 45: 1244–1248.
- Saruta H, Ishii N, Teye K, Ono F, Ohyama B, Koga H, Ohata C, Furumura M, Tsuruta D, Hashimoto T (2013). Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 149: 1209–1213.
- Shimizu A, Ishiko A, Ota T, Saito H, Oka H, Tsunoda K, Amagai M, Nishikawa T (2005). In vivo ultrastructural localization of the desmoglein 3 adhesive interface to the desmosome mid-line. J Invest Dermatol. 124: 984–989.
- Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T (2004). IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol. 122: 1145–1153.
- Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K (1998). Lack of mucosal involvement in pemphigus foliaceus may be due to low expression of desmoglein 1. J Invest Dermatol. 110: 76–78.
- Silos SA, Tamai K, Li K, Kivirikko S, Kouba D, Christiano AM, Uitto J (1996). Cloning of the gene for human pemphigus vulgaris antigen (desmoglein 3), a desmosomal cadherin. Characterization of the promoter region and identification of a keratinocyte-specific cis-element. J Biol Chem. 271: 17504–17511.
- Simpson MA, Mansour S, Ahnood D, Kalidas K, Patton MA, McKenna WJ, Behr ER, Crosby AH (2009). Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology. 113: 28–34.
- Smith C, Zhu K, Merritt A, Picton R, Youngs D, Garrod D, Chidgey M (2004). Regulation of desmocollin gene expression in the epidermis: CCAAT/enhancer-binding proteins modulate early and late events in keratinocyte differentiation. Biochem J. 380: 757–765.
- South AP, Wan H, Stone MG, Dopping-Hepenstal PJ, Purkis PE, Marshall JF, Leigh IM, Eady RA, Hart IR, McGrath JA (2003). Lack of plakophilin 1 increases keratinocyte migration and reduces desmosome stability. J Cell Sci. 116: 3303–3314.
- St Johnston D, Nusslein-Volhard C (1992). The origin of pattern and polarity in the Drosophila embryo. Cell. 68: 201–219.
- Sumigray KD, Lechler T (2012). Desmoplakin controls microvilli length but not cell adhesion or keratin organization in the intestinal epithelium. Mol Biol Cell. 23: 792–799.
- Syed SE, Trinnaman B, Martin S, Major S, Hutchinson J, Magee AI (2002). Molecular interactions between desmosomal cadherins. The Biochemical journal. 362: 317–327.
- Thomason HA, Cooper NH, Ansell DM, Chiu M, Merrit AJ, Hardman MJ, Garrod DR (2012). Direct evidence that PKCalpha positively regulates wound re-epithelialization: correlation with changes in desmosomal adhesiveness. J Pathol. 227: 346–356.
- Thomason HA, Scothern A, McHarg S, Garrod DR (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem J. 429: 419–433.
- Tokonzaba E, Chen J, Cheng X, Den Z, Ganeshan R, Muller EJ, Koch PJ (2013). Plakoglobin as a regulator of desmocollin gene expression. J Invest Dermatol. 133: 2732–2740.
- Troyanovsky SM, Eshkind LG, Troyanovsky RB, Leube RE, Franke WW (1993). Contributions of cytoplasmic domains of desmosomal cadherins to desmosome assembly and intermediate filament anchorage. Cell. 72: 561–574.
- Tucker DK, Stahley SN, Kowalczyk AP (2013). Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J Invest Dermatol.
- Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D (2000). The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell. 11: 1077–1092.
- Waschke J (2008). The desmosome and pemphigus. Histochem Cell Biol. 130: 21–54.
- Waschke J, Menendez-Castro C, Bruggeman P, Koob R, Amagai M, Gruber HJ, Drenckhahn D, Baumgartner W (2007). Imaging and force spectroscopy on desmoglein 1 using atomic force microscopy reveal multivalent Ca(2+)-dependent, low-affinity trans-interaction. J Membr Biol. 216: 83–92.
- Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y (2007). Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chemi. 282: 17866–17876.