
Abstract
Autoantibodies from patients suffering from the autoimmune blistering skin disease pemphigus can be applied as tools to study desmosomal adhesion. These autoantibodies targeting the desmosomal cadherins desmoglein (Dsg) 1 and Dsg3 cause disruption of desmosomes and loss of intercellular cohesion. Although pemphigus autoantibodies were initially proposed to sterically hinder desmosomes, many groups have shown that they activate signaling pathways which cause disruption of desmosomes and loss of intercellular cohesion by uncoupling the desmosomal plaque from the intermediate filament cytoskeleton and/or by interfering with desmosome turnover. These studies demonstrate that desmogleins serve as receptor molecules to transmit outside-in signaling and demonstrate that desmosomal cadherins have functions in addition to their adhesive properties. Two central molecules regulating cytoskeletal anchorage and desmosome turnover are p38MAPK and PKC. As cytoskeletal uncoupling in turn enhances Dsg3 depletion from desmosomes, both mechanisms reinforce one another in a vicious cycle that compromise the integrity and number of desmosomes.
INTRODUCTION
Pemphigus is a rare but severe disease affecting skin and mucous membranes, and is characterized by pathogenic autoantibodies targeting the desmosomal cadherins desmoglein (Dsg) 1 and Dsg3 (CitationAmagai & Stanley, 2012). Patients with the more common form pemphigus vulgaris (PV) develop antibodies directed against Dsg3 and in some cases also against desmocollin (Dsc) 3 in the mucosal-dominant stage at the onset of the disease (CitationKneisel & Hertl, 2011; CitationMao et al., 2010; CitationRafei et al., 2011). Transition to the mucocutaneous stage also affects the epidermis and usually is associated with appearance of antibodies targeting Dsg1 in addition to anti-Dsg3 antibodies. In pemphigus foliaceus (PF), where flaccid skin blisters are present without mucosal erosions, the autoantibody profile typically comprises Dsg1 as the only antigen from the desmosomal cadherin family. However, over 50 other antigens can react with sera from pemphigus patients of which the pathogenic significance is largely unclear (CitationGrando, 2012).
Using pemphigus as disease model allows studying desmosomal adhesion by application of autoantibody fractions from pemphigus patients. Although siRNA-mediated knockdown of desmosomal components became available to specifically target binding of desmosomes, autoantibodies have several advantages: they can be applied under comparable conditions in different experimental settings both in vivo and in vitro. Moreover, in a reductive approach where antibodies are incubated with cultured keratinocytes, they can be added after cells have formed a stable monolayer and established desmosomes.
ROLE OF SIGNALING INDUCED BY AUTOANTIBODIES IN PEMPHIGUS PATHOGENESIS
Pemphigus antibodies were initially thought to disrupt Dsg trans-interaction but later have been shown to also exert their pathological effects by triggering intracellular signaling pathways (CitationEsaki et al., 1995; CitationSeishima et al., 1995). Direct steric inhibition apparently mimics the so-called tryptophan-swap between interacting Dsg3 molecules which hampers cis- and trans-interaction (CitationDi Zenzo et al., 2012; CitationSpindler et al., 2013; CitationYamagami et al., 2010). As inhibition of Dsg3 binding contributes to loss of cell cohesion in vivo and in vitro (CitationHeupel et al., 2009b, Citation2008; CitationSpindler et al., 2013), it is possible that this mechanism alone is sufficient to cause blister formation in some cases, especially when blistering is mechanically induced (CitationMao et al., 2013; CitationSaito et al., 2012). However, direct inhibition of Dsg1 binding in PF may not be required for loss of cell cohesion (CitationWaschke et al., 2005) and binding of PV autoantibodies to the cell surface alone is not sufficient to cause cell dissociation (CitationCalkins et al., 2006). Interference with Dsg3 trans-interaction is closely followed by activation of signaling (CitationSpindler et al., 2013), indicating that Dsg binding and signaling are connected events. Thus, by now it is widely accepted in the field of pemphigus research that signaling mechanisms are required for and significantly contribute to loss of cell cohesion and blister formation in pemphigus. Many signaling molecules have been associated with pemphigus pathogenesis and shown to contribute to loss of cell adhesion in a variety of model systems (CitationGetsios et al., 2010; CitationWaschke, 2008). These include phospholipase C, Ca2+, and protein kinase C (PKC) (CitationEsaki et al., 1995; CitationOsada et al., 1997; CitationSeishima et al., 1995) as well as plakoglobin regulating the expression of Myc (CitationCaldelari et al., 2001; CitationWilliamson et al., 2006). Rho GTPases act as molecular switches in actin dynamics (CitationGliem et al., 2010; CitationWaschke et al., 2006). The involvement of p38MAPK is well established, which may require its downstream kinase MAPKAP kinase 2 (MK2) (CitationBerkowitz et al., 2005, Citation2006; CitationMao et al., 2013) and epidermal growth factor receptor (EGF-R) and/or Src signaling (CitationBektas et al., 2013; CitationChernyavsky et al., 2007; CitationFrusic-Zlotkin et al., 2006; CitationTsang et al., 2012b). Other molecules implicated in pemphigus are Akt/mTOR and FAK (CitationGil et al., 2012; CitationPretel et al., 2009) as well as JNK (CitationMarchenko et al., 2010), CDK-2 (CitationLanza et al., 2008), PERP (CitationNguyen et al., 2009) and factors associated with apoptotic signaling such as caspases 3, 6, 8, and 9 as well as Bcl/Bax and others (CitationLi et al., 2009; CitationMarchenko et al., 2010). However, for most signaling pathways the precise mechanisms by which they reduce Dsg-mediated binding and desmosomal integrity are not well understood. From the data available, it can be concluded that signaling pathways induce uncoupling of the desmosomal plaque from the intermediate filament cytoskeleton and/or interfere with desmosome turnover. Because apoptotic cell death is not typical in pemphigus skin lesions, the caspase activation observed may not lead to apoptosis but rather participate in the regulation of desmosomal turnover (CitationSchmidt & Waschke, 2009).
Here, we will focus on p38MAPK and PKC, two important players involved in regulation of both desmosomal anchorage and desmosomal turnover.
SIGNALING FUNCTION OF DESMOSOMAL CADHERINS
The role of p38MAPK in pemphigus has been well established by the work of David Rubenstein. It has been demonstrated that p38MAPK is phosphorylated upon autoantibody binding and is activated in pemphigus skin lesions (CitationBerkowitz et al., 2007, Citation2005). Pharmacologic p38MAPK inhibition blocked blister formation in response to both PV–IgG and PF–IgG (CitationBerkowitz et al., 2008, Citation2006). On the other hand, PKC was the first signaling molecule shown to be activated by pemphigus autoantibodies based on the work of Yasuo Kitajima's laboratory (CitationEsaki et al., 1995; CitationSeishima et al., 1995). PKC was found to negatively regulate desmosomal hyper-adhesion, a state in which desmosomes become Ca2+-independent (CitationGarrod et al., 2005; CitationThomason et al., 2010; CitationWallis et al., 2000). Please also see accompanying review by Garrod et al. on this topic. Recently, the significance of PKC signaling in pemphigus pathogenesis was demonstrated by showing that inhibition of PKC abolished loss of keratinocyte adhesion in vitro and blister formation in vivo (CitationCirillo et al., 2010; CitationSpindler et al., 2011).
When investigating the role of signaling mechanisms for regulation of desmosomal adhesion, it is important, but not always possible, to distinguish between signaling mediated by a specific desmosomal cadherin (i.e., primary to loss of cell cohesion) and signaling in response (i.e., secondary) to general loss of cell adhesion. Available data suggest that modulation of extracellular binding of desmosomal cadherins triggers signaling inside the cell (outside in signaling) (CitationMüller et al., 2008). This can be concluded because autoantibodies, which directly interfere with Dsg3 binding, induce rapid alterations of signaling pathways. Furthermore, a small peptide designed to interfere with Dsg interaction (CitationHeupel et al., 2009b) induced activation of p38MAPK (CitationSpindler et al., 2013). It is not entirely clear how this extracellular cue, loss of binding, is transmitted into the cell. In some cases, a specific signaling molecule associates with the Dsg3 complex. This has been shown for Rho GTPases and Src (CitationTsang et al., 2012a, Citation2012b). PKC has been located at the desmosomal plaque via electron microscopy (CitationGarrod et al., 2005), and may directly bind to desmosomal proteins. Also p38MAPK is attached to a complex containing Dsg3 and plakoglobin (CitationSpindler et al., 2013). Importantly, in this complex p38MAPK is activated when challenged with Dsg3-specific antibodies.
Activation of PKC after binding of pemphigus autoantibodies seems to be mediated by a rapid increase of cytosolic Ca2+ which is released from internal stores in a timeframe of seconds (CitationSeishima et al., 1995). This release is stimulated by the phospholipase C/inositol 1,4,5-trisphosphate pathway to finally activate PKC (CitationEsaki et al., 1995). The mechanism of phospholipase C activation is unknown.
MECHANISMS REGULATING THE CYTOSKELETAL ANCHORAGE OF DESMOSOMES
Recent data indicate that p38MAPK and PKC regulate the anchorage of the desmosomal plaque to the keratin filament cytoskeleton. Activation of p38MAPK by PV-IgG or selective stimulation by anisomycin reduced keratin filament insertion at desmosomes leading to enhanced perinuclear localization () (CitationBerkowitz et al., 2005; CitationSpindler et al., 2013). This phenomenon called keratin retraction is closely paralleled by loss of keratinocyte cohesion. RhoA is inhibited in response to pemphigus autoantibodies in p38MAPK-dependent manner and activation of this GTPase blocked blister formation and enhanced insertion of keratin 14 at cell junctions (CitationWaschke, 2008). Recently, RhoA, Rac1, and Cdc42 were shown to directly bind to Dsg3, at least in the non-desmosomal fraction, further indicating that the desmosomal cadherins regulate Rho GTPases (CitationTsang et al., 2012a). Keratin retraction downstream of p38MAPK is dependent on EGF-R activation, () (CitationBektas et al., 2013). This supports the finding that EGF also causes cytokeratin retraction (CitationHeupel et al., 2009a), although in this study EGF-R was not found to be required for pemphigus IgG-induced loss of cell cohesion. Whether MK2 which appears to be important for the p38MAPK-mediated loss of adhesion in pemphigus (CitationMao et al., 2013) is also involved in keratin retraction—for example via inactivation of RhoA—has not been investigated so far. These data suggest that p38MAPK regulates desmosomal adhesion in part at the level of keratin filament dynamics, although this has not been tested directly.
Figure 1. The signaling mechanisms triggered by pemphigus autoantibodies reduce the cytoskeletal anchorage of desmosomes. Under resting conditions, desmogleins and desmocollins in desmosomes are linked to intermediate filaments via desmoplakin and the adaptor proteins plakoglobin and plakophilin. Desmosomal components interact with several signaling molecules: p38MAPK is associated with Dsg3, PKC is sequestered by keratin filaments via RACK1 and active RhoA is bound to extradesmosomal Dsg3. Upon autoantibody binding, p38MAPK is activated within the signaling complex with Dsg3, a process involved in EGF-R activation and RhoA inhibition, which finally causes keratin retraction. In parallel, PKC activation reduces intermediate filament binding to desmoplakin.
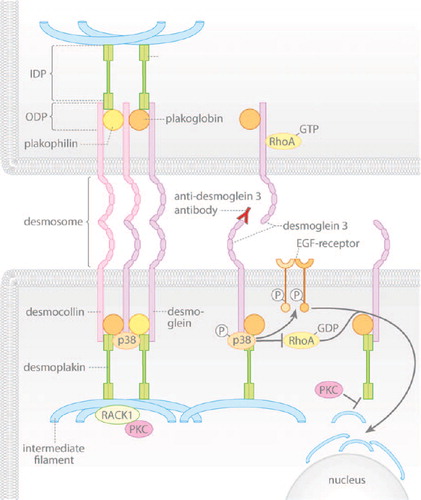
The first insight that desmosomal adhesion can be regulated by keratin filament anchorage came from the finding that a desmoplakin mutant (DP Ser2849Gly), which resides within a PKC consensus sequence close to the keratin-binding domain, drastically enhanced intermediate filament binding and induced hyper-adhesion of desmosomes (CitationHobbs & Green, 2012; CitationMeng et al., 1997). PKC was found to relocate to the desmosomal plaque during acquisition of hyper-adhesion () (CitationGarrod et al., 2005). At the plaque, PKC can be sequestered by keratin filaments via Rack1. Apparently, this is a central mechanism by which keratin filaments stabilize desmosomal adhesion (CitationKröger et al., 2013). Cells expressing this mutant do not lose cell cohesion upon PKC activation and keratinocytes lacking intermediate filaments regain cohesion when PKC is inhibited (CitationHobbs & Green, 2012; CitationKröger et al., 2013). Recent studies from our group indicate that PKC-dependent serine phosphorylation of desmoplakin is increased following PV-IgG incubation and that DP Ser2849Gly reduced loss of cell cohesion and blocked keratin retraction (unpublished observations). Taken together, pemphigus autoantibodies activate p38MAPK, which controls keratin filament dynamics and via PKC-mediated desmoplakin phosphorylation reduces keratin anchorage of Dsg3.
MECHANISMS REGULATING DESMOSOME TURNOVER
After synthesis and posttranslational modification, desmosomal components are transported to the plasma membrane via distinct mechanisms which require differential contributions of the cytoskeleton (CitationNekrasova & Green, 2013). The integral membrane proteins, Dsgs and Dscs, are translocated to the cell surface via a microtubule-dependent mechanism () (CitationNekrasova et al., 2011). Available data suggest that different motor proteins mediate the membrane translocation of these two sets of adhesion molecules. Kinesin 1 binds and transports Dsg2, whereas kinesin 2 is required to target Dsc2 to the membrane. Up to now it is unclear whether these differences in transport are restricted to Dsg2/Dsc2 or are a uniform mechanism for desmogleins vs. desmocollins. Once they arrive at the plasma membrane, the desmosomal cadherins may shift laterally into the desmosome (CitationSato et al., 2000), although the precise mechanisms by which this happens remain unclear. The formation of the cytoplasmic plaque appears to be independent of microtubules. Plakophilin 2 (Pkp2) scaffolds desmoplakin and PKCα in a complex together with intermediate filaments in the cortical region of keratinocytes (CitationBass-Zubek et al., 2008). The relocation of this complex to the membrane requires a properly organized actin cytoskeleton (Figure 2). In this context, RhoA localization at the site of developing desmosomes is necessary, which again is dependent on Pkp2 (CitationGodsel et al., 2010).
Figure 2. Pemphigus autoantibodies interfere with desmosomal cadherin turnover. Gray arrows indicate assembly and disassembly of desmosomal components. Red arrows denote changes in signaling induced by autoantibodies of pemphigus patients. Desmosomal cadherins are transported to the cell membrane in a microtubule-dependent process. Membrane localization of the desmosomal plaque is dependent on plakophilin and on PKC signaling. Pemphigus autoantibodies induce internalization and depletion of extradesmosomal Dsg3 via activation of p38MAPK and EGF-R. Furthermore, activation of PKC may be necessary for shifting Dsg3 out of the desmosome. Additionally, autoantibodies interfere with RhoA signaling, which primarily may impair assembly of desmosomes.
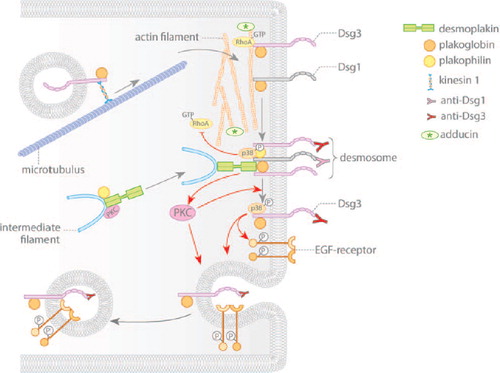
Few data are available on the steady-state dynamics and degradation of desmosomal proteins. FRAP studies performed with fluorescent Dsc2 molecules suggest that the desmosomal cadherins are not “trapped” in desmosomes but are exchanged over a timescale of minutes under the steady-state conditions (CitationWindoffer et al., 2002). In contrast, the entire desmosomal complex appears to be stable for hours indicating that two pools of the desmosomal cadherins exist on the cell surface; (a desmosomal-bound pool) and a second fraction outside of desmosomes (extradesmosomal pool) which is freely diffusible. The latter may form a reservoir for desmosomes or represent an intermediate state of molecules undergoing internalization. As membranes initially separate between desmosomes in response to pemphigus autoantibodies (CitationSchulze et al., 2012; CitationTakahashi et al., 1985), it may be concluded that extradesmosomal molecules are necessary for maintaining cell adhesion. Additionally, these molecules may be relevant for intracellular signaling, because some signaling molecules such as active Rho GTPases are specifically coupled to extradesmosomal Dsg3 (CitationTsang et al., 2012a).
Pathways for degradation of desmosomal cadherins under non-pathologic conditions have not been thoroughly investigated. It is known for Dsg2 that the Dsg unique region (DUR) in the cytoplasmic tail is necessary to stabilize the molecule at the membrane and to prevent internalization (CitationChen et al., 2012). Additional conclusions may be drawn from the events following incubation with pemphigus autoantibodies. Many studies clearly showed a depletion of cellular Dsg3 levels following pemphigus autoantibody binding (). It was demonstrated that bound PV-IgG was rapidly internalized into keratinocytes (CitationIwatsuki et al., 1989; CitationPatel et al., 1984). Depletion of Dsg3 has been demonstrated in mouse models (CitationShu et al., 2007; CitationSpindler et al., 2011; CitationTakahashi et al., 1985) and in an array of cultured keratinocytes (CitationAoyama & Kitajima, 1999; CitationCalkins et al., 2006; CitationCirillo et al., 2010; CitationYamamoto et al., 2007). This indicates that internalization and degradation of Dsg3 is a principal mechanism of antibody-induced loss of cell adhesion. The reduction of Dsg3 levels may also arise from autoantibody interference with desmosomal assembly (CitationMao et al., 2009). For both mechanisms it is necessary that cells respond specifically to autoantibody binding, which supports the concept that in most cases direct inhibition by autoantibodies alone is not sufficient to induce loss of cell cohesion.
Internalization and depletion in response to PV-IG seems to be specific for the desmoglein isoforms targeted by the autoantibodies. For example, PV-IgG were shown to cause depletion of Dsg3, whereas Dsg2 and desmoplakin levels remained largely unaffected (CitationCalkins et al., 2006). Similarly, PF-IgG reduced proteins levels of Dsg1 (CitationBrennan et al., 2010; CitationSpindler et al., 2007).
As rapidly as 20–60 minutes after autoantibody addition, Dsg3 is depleted from the Triton-X-100-soluble pool, that is, the pool that presumably represents extradesmosomal Dsg3 molecules () (CitationAoyama & Kitajima, 1999; CitationYamamoto et al., 2007). At later time points, Dsg3 reduction also is evident in the Triton-X-100-insoluble desmosomal pool. These data support the idea that Dsg3 needs to get uncoupled from desmosomes in order to become internalized. Alternatively, as a consequence of rapid depletion of the extradesmosomal pool, the supply for maintenance of desmosomes is depleted and slowly Dsg3 becomes reduced within desmosomes. The latter mechanism has been described as desmosomal non-assembly depletion hypothesis (CitationOktarina et al., 2011).
The internalization of Dsg3 is independent of clathrin and dynamin. Instead, this process likely requires lipid rafts since cholesterol-binding compounds prevented endocytosis (CitationDelva et al., 2008). Dsg3, together with plakoglobin, is transported to early and finally to late endosomes. Additionally, the tyrosine kinase EGF-R, which is activated following pemphigus autoantibody binding at least under certain conditions, is co-internalized together with Dsg3 () (CitationBektas et al., 2013). Internalization of Dsg3 can be prevented by p38MAPK inhibition and the latter also reduced EGF-R phosphorylation in response to pemphigus autoantibodies, suggesting that p38MAPK controls EGF-R activity in this context (CitationBektas et al., 2013; CitationJolly et al., 2010). Finally, inhibiting tyrosine kinase activity with genistein prevented Dsg3 internalization and blocked loss of cell cohesion in cultured keratinocytes (CitationDelva et al., 2008). Thus, a pathway is conceivable by which p38MAPK-dependent EGF-R phosphorylation drives Dsg3 internalization and degradation.
Depletion in response to PV-IgG binding depends on the maturity of desmosomes. Keratinocytes that were cultured for 3d in high Ca2+ medium were more prone to depletion compared to cells cultured for 8d (CitationSpindler et al., 2011). Moreover, PKC activation and PKC-mediated Dsg3 depletion in response to PV-IgG were only found in cells cultured in high Ca2+ for the first three days. As desmosomes become hyper-adhesive by long-time culture in high Ca2+ and by inhibition of PKC (CitationKimura et al., 2007), it is possible that PKC regulates hyper-adhesion and Dsg3 depletion by the same mechanisms. PKC activation observed in response to PV-IgG (CitationOsada et al., 1997) may revert the hyper-adhesive phenotype and may be a prerequisite for desmosomal Dsg3 exclusion. Although it was suggested that desmosomes are hyper-adhesive throughout all layers of the epidermis (CitationGarrod, 2010), it is possible that desmosomes of basal keratinocytes are less stable and thus more susceptible for depletion compared to desmosomes from superficial layers (CitationSpindler et al., 2011). This mechanism may contribute to the suprabasal blister localization in PV patients.
The data discussed so far suggest that PV-IgG-induced signaling events interfere with desmosome turnover either by depletion of extradesmosomal Dsg3 required for incorporation into desmosomes or by depletion of Dsg3 from desmosomes after uncoupling from the intermediate filament cytoskeleton. However, signaling events also may affect the shift from extradesmosomal Dsg3 into desmosomes by disturbing the dynamics of actin filaments. As outlined above, proper actin network organization regulated by the GTPase RhoA is required for assembly of desmosomes. As pemphigus IgG reduces the activity of RhoA (CitationWaschke et al., 2006) and reorganization of the actin network contributes to loss of cell cohesion (CitationGliem et al., 2010) pemphigus autoantibody binding may disturb the assembly pathways. In line with this, knockdown of adducin, an actin-binding protein necessary for the organization of the cortical actin meshwork, also reduced Dsg3 levels (CitationRoetzer et al., 2013).
CONCLUSIONS
Taken together, the data gathered by experiments using pemphigus autoantibodies which specifically target desmogleins indicate that desmosomal cadherins exhibit receptor functions to induce desmosome remodeling via a set of intracellular signaling molecules. It may be concluded from the similarity of the molecules involved that regulation of cytoskeletal anchorage on one hand and of desmosomal turnover on the other are not independent mechanisms but rather should be regarded as closely coupled events. Signaling molecules may regulate desmosomal maturation by modification of keratin filaments and desmosomal plaque proteins which finally results in Ca2+-insensitivity. These hyper-adhesive desmosomes undergo reduced remodeling, which in case of pemphigus, protects against autoantibody-induced Dsg3 depletion (CitationKitajima, 2013). The physiologic function of Dsg3-regulated p38MAPK and PKC modulation may be to adapt cell cohesion when epidermal integrity is compromised and keratinocytes need to migrate. This is consistent with the observation that wound re-epithelialization is dependent on PKC-mediated modulation of desmosomal adhesiveness (CitationThomason et al., 2012; CitationWallis et al., 2000) To further clarify these important and exciting functions, further studies on the mechanisms involved in outside-in signaling of the desmosomal cadherins are needed.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Amagai M, Stanley JR (2012). Desmoglein as a Target in Skin Disease and Beyond. J Invest Dermatol. 132: 776–784.
- Aoyama Y, Kitajima Y (1999). Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. 112: 67–71.
- Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, Wahl JK III, Denning MF, Green KJ (2008). Plakophilin 2: a critical scaffold for PKCα that regulates intercellular junction assembly. J Cell Biol. 181: 605–613.
- Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS (2013). A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem. 288: 9447–9456.
- Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS (2008). Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. 173: 1628–1636.
- Berkowitz P, Diaz LA, Hall RP, Rubenstein DS (2007). Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 128: 738–740.
- Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS (2005). Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 280: 23778–23784.
- Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS (2006). p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 103: 12855–12860.
- Brennan D, Hu Y, Medhat W, Dowling A, Mahoney MG (2010). Superficial Dsg2 Expression limits epidermal blister formation mediated by pemphigus foliaceus antibodies and exfoliative toxins. Dermatol Res Pract. 2010: 410278.
- Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E (2001). A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 153: 823–834.
- Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP (2006). Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 281: 7623–7634.
- Chen J, Nekrasova OE, Patel DM, Klessner JL, Godsel LM, Koetsier JL, Amargo EV, Desai BV, Green KJ (2012). The C-terminal unique region of desmoglein 2 inhibits its internalization via tail–tail interactions. J Cell Biol. 199: 699–711.
- Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA (2007). Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem. 282: 13804–13812.
- Cirillo N, Lanza A, Prime SS (2010). Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res. 316: 580–592.
- Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP (2008). Pemphigus Vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J Biol Chem. 283: 18303–18313.
- Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, Didona B, Cianchini G, Hertl M, Eming R Amagai M, Ohyama B, Hashimoto T, Sloostra J, Sallusto F, Zambruno G, Lanzavecchia A (2012). Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. 122: 3781–3790.
- Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y (1995). Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. 105: 329–333.
- Frusic-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y (2006). Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis–possible involvement of the EGF receptor. Autoimmunity. 39: 563–575.
- Garrod D (2010). Desmosomes In Vivo. Dermatol Res Pract. 2010: 212439.
- Garrod DR, Berika MY, Bardsley WF, Holmes D, Tabernero L (2005). Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci. 118: 5743–5754.
- Getsios S, Waschke J, Borradori L, Hertl M, Muller EJ (2010). From cell signaling to novel therapeutic concepts: international pemphigus meeting on advances in pemphigus research and therapy. J Invest Dermatol. 130: 1764–1768.
- Gil MP, Modol T, España A, López-Zabalza MJ (2012). Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol. 21: 254–259.
- Gliem M, Heupel W-M, Spindler V, Harms GS, Waschke J (2010). Actin reorganization contributes to loss of cell adhesion in pemphigus vulgaris. Am J Physiol Cell Physiol. 299: C606–C613.
- Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, Chen X, Green KJ (2010). Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol. Biol. Cell. 21: 2844–2859.
- Grando S (2012). Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. 45: 7–35.
- Heupel W-M, Engerer P, Schmidt E, Waschke J (2009a). Pemphigus vulgaris IgG cause loss of desmoglein-mediated adhesion and keratinocyte dissociation independent of epidermal growth factor receptor. Am J Pathol. 174: 475–485.
- Heupel W-M, Müller T, Efthymiadis A, Schmidt E, Drenckhahn D, Waschke J (2009b). Peptides targeting the desmoglein 3 adhesive interface prevent autoantibody-induced acantholysis in pemphigus. J Biol Chem. 284: 8589–8595.
- Heupel WM, Zillikens D, Drenckhahn D, Waschke J (2008). Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 181: 1825–1834.
- Hobbs RP, Green KJ (2012). Desmoplakin regulates desmosome hyperadhesion. J Invest Dermatol. 132: 482–485.
- Iwatsuki K, Takigawa M, Imaizumi S, Yamada M (1989). In vivo binding site of pemphigus vulgaris antibodies and their fate during acantholysis. J Am Acad Dermatol. 20: 578–582.
- Jolly PS, Berkowitz P, Bektas M, Lee H-E, Chua M, Diaz LA, Rubenstein DS (2010). p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 285: 8936–8941.
- Kimura TE, Merritt AJ, Garrod DR (2007). Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol. 127: 775–781.
- Kitajima Y (2013). New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci. 29: 1–13.
- Kneisel A, Hertl M (2011). Autoimmune bullous skin diseases. Part 1: Clinical manifestations.J Dtsch Dermatol Ges. 9: 844–857.
- Kröger C, Loschke F, Schwarz N, Windoffer R, Leube RE, Magin TM (2013). Keratins control intercellular adhesion involving PKC-α–mediated desmoplakin phosphorylation. J Cell Biol. 201: 681–692.
- Lanza A, Cirillo N, Rossiello R, Rienzo M, Cutillo L, Casamassimi A, de Nigris F, Schiano C, Rossiello L, Femiano F Gombos F, Napoli C (2008). Evidence of key role of Cdk2 overexpression in pemphigus vulgaris. J Biol Chem. 283: 8736–8745.
- Li N, Zhao M, Wang J, Liu Z, Diaz LA (2009). Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol. 182: 711–717.
- Mao X, Choi EJ, Payne AS (2009). Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. 129: 908–18.
- Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS (2013). MAPKAP Kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Invest Dermatol.
- Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, Ishii N, Hashimoto T, Amagai M, Zone JJ, Payne AS (2010). Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. 177: 2724–2730.
- Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA (2010). Antimitochondrial autoantibodies in pemphigus vulgaris. J Biol Chem. 285: 3695–3704.
- Meng J-J, Bornslaeger EA, Green KJ, Steinert PM, Ip W (1997). Two-hybrid analysis reveals fundamental differences in direct interactions between desmoplakin and cell type-specific intermediate filaments. J Biol Chem. 272: 21495–21503.
- Müller EJ, Williamson L, Kolly C, Suter MM (2008). Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation?J Invest Dermatol. 128: 501–516.
- Nekrasova O, Green KJ (2013). Desmosome assembly and dynamics. Trends Cell Biol. 23: 537–546.
- Nekrasova OE, Amargo EV, Smith WO, Chen J, Kreitzer GE, Green KJ (2011). Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J Cell Biol. 195: 1185–1203.
- Nguyen B, Dusek RL, Beaudry VG, Marinkovich MP, Attardi LD (2009). Loss of the desmosomal protein perp enhances the phenotypic effects of pemphigus vulgaris autoantibodies. J Invest Dermatol. 129: 1710–1718.
- Oktarina DAM, van der Wier G, Diercks GFH, Jonkman MF, Pas HH (2011). IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol. 165: 552–562.
- Osada K, Seishima M, Kitajima Y (1997). Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol. 108: 482–487.
- Patel HP, Diaz LA, Anhalt GJ, Labib RS, Takahashi Y (1984). Demonstration of pemphigus antibodies on the cell surface of murine epidermal cell monolayers and their internalization. J Invest Dermatol. 83: 409–415.
- Pretel M, España A, Marquina M, Beatriz Pelacho López-Picazo J, López-Zabalza J (2009). An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol. 18: 771–780.
- Rafei D, Müller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, Eming R (2011). IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol. 178: 718–723.
- Roetzer V, Waschke J, Spindler V (2013). Adducin modulates intercellular keratinocyte adhesion. FASEB J. 27: 650.15.
- Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, Amagai M, Kowalczyk AP (2012). Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One. 7: e50696.
- Sato M, Aoyama Y, Kitajima Y (2000). Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest. 80: 1583–1592.
- Schmidt E, Waschke J (2009). Apoptosis in pemphigus. Autoimmun Rev. 8: 533–537.
- Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, Siffert M, Zenhausern D, Bolli R, Koch PJ Garrod D, Suter MM, Müller EJ (2012). An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J Invest Dermatol. 132: 346–355.
- Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y (1995). Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. 104: 33–37.
- Shu E, Yamamoto Y, Aoyama Y, Kitajima Y (2007). Intraperitoneal injection of pemphigus vulgaris-IgG into mouse depletes epidermal keratinocytes of desmoglein 3 associated with generation of acantholysis. Arch Derm Res. 299: 165–167.
- Spindler V, Drenckhahn D, Zillikens D, Waschke J (2007). Pemphigus IgG causes skin splitting in the presence of both desmoglein 1 and desmoglein 3. Am J Pathol. 171: 906–16.
- Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J (2011). The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting?Am J Pathol. 179: 1905–16.
- Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, Hartlieb E, Harms GS, Schmidt E, Waschke J (2013). Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest. 123: 800–811.
- Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ (1985). Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol. 84: 41–6.
- Thomason HA, Cooper NH, Ansell DM, Chiu M, Merrit AJ, Hardman MJ, Garrod DR (2012). Direct evidence that PKCα positively regulates wound re-epithelialization: correlation with changes in desmosomal adhesiveness. J Pathol. 227: 346–356.
- Thomason HA, Scothern A, McHarg S, Garrod DR (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem J. 429: 419–433.
- Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A, Wan H (2012a). Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp Cell Res. 318: 2269–2283.
- Tsang SM, Brown L, Lin K, Liu L, Piper K, O’Toole EA, Grose R, Hart IR, Garrod DR, Fortune F, Wan H (2012b). Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol. 227: 81–93.
- Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D (2000). The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell. 11: 1077–1092.
- Waschke J (2008). The desmosome and pemphigus. Histochem Cell Biol. 130: 21–54.
- Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D (2005). Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 115: 3157–3165.
- Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D (2006). Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol. 175: 721–727.
- Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Muller EJ (2006). Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 25: 3298–309.
- Windoffer R, Borchert-Stuhltrager M, Leube RE (2002). Desmosomes: interconnected calcium-dependent structures of remarkable stability with significant integral membrane protein turnover. J Cell Sci. 115: 1717–1732.
- Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR (2010). Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Invest. 120: 4111–4117.
- Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y (2007). Anti-desmoglein 3 (Dsg3) Monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem. 282: 17866–17876.