
Abstract
The importance of desmosomes in tissue homeostasis is highlighted by natural and engineered mutations in desmosomal genes, which compromise the skin or heart and in some instances both. Desmosomal gene mutations account for 45–50% of cases of arrhythmogenic right ventricular cardiomyopathy, and are mutated in an array of other disorders such as striate palmoplantar keratoderma, hypotrichosis with or without skin vesicles and lethal acantholytic epidermolysis bullosa. Recently, we reported loss-of-function mutations in the human ADAM17 gene, encoding for the ‘sheddase’ ADAM17, a transmembrane protein which cleaves extracellular domains of substrate proteins including TNF-α, growth factors and desmoglein (DSG) 2. Patients present with cardiomyopathy and an inflammatory skin and bowel syndrome with defective DSG processing. In contrast, the dominantly inherited tylosis with oesophageal cancer appears to result from gain-of-function in ADAM17 due to increased processing via iRHOM2. This review discusses the heterogeneity of mutations in desmosomes and their regulatory proteins.
DESMOSOMES – DYNAMIC ADHESION STRUCTURES IN THE SKIN
Desmosomes are complex macromolecular structures with a key role in maintaining collateral epidermal integrity. Discovered by Italian pathologist Giulio Bizzozero (1846–1901), desmosomes are expressed in a variety of tissue types exposed to mechanical stress, such as the intestinal mucosa and the epithelial cells of the nephron, but are most abundant in the skin and myocardium (CitationStaehelin, 1974; CitationKelly, 1966; CitationHolthofer et al., 2007; CitationFarquhar & Palade, 1963). A primary function of desmosomes is the anchoring of cytoskeletal keratin intermediate filaments in the epidermis, desmin intermediate filaments in the heart, and vimentin intermediate filaments in meningeal cells and the follicular dendritic cells of lymph nodes to the cell membrane (CitationGreen & Gaudry, 2000).
All desmosomes, independent of their distribution, are formed by three main classes of proteins divided into three parallel individual zones, arranged symmetrically on the cytoplasmic faces of the plasma membranes of bordering cells and separated by the extracellular domain. The five known desmosomal components are the desmosomal cadherins, represented by desmogleins (DSG1–DSG4) and desmocollins (DSC1–DSC3), the armadillo family members plakoglobin (PG)/ γ-catenin and the plakophilins (PKP1–PKP3), and the plakin linker protein desmoplakin (DSP), which anchors the intermediate keratin filaments. The key role of these proteins in the skin (and heart) is reflected by the spectrum of desmosomal mutations identified, which are associated with monogenic skin diseases with/ without cardiac involvement (; CitationBrooke et al., 2012a). Furthermore, mutations in the desmosome are a major genetic risk factor for arrhythmogenic right ventricular cardiomyopathy (ARVC); a hereditary disorder of the cardiac muscle characterised by ventricular arrhythmias, cardiac failure, and sudden cardiac death. The range of genetic disorders arising from mutations affecting the desmosomal genes, and indeed within the same gene, highlights the complexity and incomplete understanding of how desmosomal components interrelate with each other and with other compartments in a cell-type and differentiation-dependent manner.
Table 1. Examples of monogenic human disorders associated with desmosomal mutations.
To date, more than 850 human mutations have been reported in desmosomal genes (CitationAl-Jassar et al., 2013) leading to disorders such as palmoplantar keratoderma (PPK), woolly hair and ARVC (CitationNorgett et al., 2000; CitationMcKoy et al., 2000), non-syndromic striate PPK (SPPK; CitationArmstrong et al., 1999; CitationRickman et al., 1999) and hypotrichosis (CitationKljuic et al., 2003a). The next sections will focus on individual protein components of the desmosome, their basic biology and disease association.
DSP
DSP, the most abundant desmosomal protein, plays a key role as the linker between the plasma membrane and the intermediate filament complex (CitationDelva et al., 2009). The protein is predicted to form homodimers through an α-helical coiled-coil rod domain which also interconnects a globular amino-terminus, responsible for binding the arm proteins PG and PKPs, and a carboxy-terminus domain, responsible for the attachment of intermediate filaments (CitationHolthofer et al., 2007; CitationKowalczyk et al., 1994; CitationBornslaeger et al., 2001; CitationChoi et al., 2002; CitationYin & Green, 2004). Until recently, only two isoforms of DSP (DSPI and DSPII) have been known. DSPI and DSPII isoforms are produced as a result of alternative mRNA splicing, with DSPII the shorter isoform of the two. Both are widely expressed in numerous tissues, with DSPII absent from the heart and simple epithelia (CitationAngst et al., 1990). A minor DSP isoform derived from DSPI, named DSPIα, produced by the alternative splicing of DSPI mRNA has also been described, and is detectable in lower levels than those of the dominant isoforms; however, it presents a similar tissue distribution (CitationCabral et al., 2010b).
Using immunogold labelling of DSP, CitationFranke et al. (2006) observed that in normal heart muscle, DSP is located in all plaques of the desmosome-like and fascia-adherens-type junctions, with a very intense signal within the desmosome-like junctions. Several in vivo and in vitro studies support the importance of DSP in desmosome assembly and function, and show its pivotal role in the development of epidermis, neuroepithelium, heart and blood vessels (CitationGallicano et al., 2001; CitationVasioukhin et al., 2001).
The first reported link of DSP mutations with human skin disease was with autosomal dominant SPPK which presents with a longitudinal pattern of hyperkeratosis (CitationArmstrong et al., 1999). The loss-of-function mutations suggested that the disease mechanism was haploinsufficiency and that the dosage of DSP was critical in stressed areas of the skin such as the palm and sole. Indeed, histology of affected skin revealed loss of suprabasal keratinocyte adhesion. The first reported recessive DSP mutation was identified in Ecuadorian families with Carvajal syndrome: ARVC with dilated cardiomyopathy, woolly hair and SPPK, but also with hyperkeratosis at other stress sites in the skin including the flexures. The homozygous mutation truncates the DSP protein, losing a portion of the IF-binding site leading to loss of cell adhesion and a collapsed IF network (CitationNorgett et al., 2000; CitationGetsios et al., 2004b; CitationHuen et al., 2002). Subsequently, a variety of DSP-associated conditions such as cardiocutaneous syndromes have been reported with varying degrees of severity. As mentioned earlier, heterozygous premature termination codon mutations in exons common to all DSP isoforms (termed haploinsufficiency) underlie dominant non-syndromic SPPK (CitationArmstrong et al., 1999; CitationWhittock et al., 1999). In contrast, the homozygous mutation p.R1267X leads to complete absence of DSPI but to normal levels of DSPII (CitationUzumcu et al., 2006). This loss of expression of DSPI is associated with autosomal recessive mild epidermolytic PPK (but no obvious keratinocyte adhesion defect), woolly hair and aggressive arrhythmogenic dilated cardiomyopathy, leading to severe ventricular dysfunction and associated arrhythmia at an age in which the cardiac manifestations of desmosomal disease are very rarely encountered. Functional studies in keratinocytes suggest that DSPII plays a more significant role than DSPI in maintaining robust adhesion, supporting cell-type-specific functions for the DSP isoforms (CitationCabral et al., 2012).
Another example of DSP-associated disease includes compound heterozygosity of an amino-terminal missense mutation and a carboxy-terminal nonsense mutation leading to severe keratoderma, skin fragility and woolly hair, or alopecia with or without cardiac involvement (CitationAsimaki et al., 2007; CitationWhittock et al., 2002). Recessive DSP mutations can also underlie severe acantholytic epidermolysis bullosa, a lethal disorder which presents with complete alopecia, neonatal teeth, nail loss and death due to transcutaneous fluid loss as a result of extensive skin erosion (CitationJonkman et al., 2005). Furthermore, dominant DSP mutations are also linked to non-syndromic ARVC but with no obvious cutaneous phenotype (CitationRampazzo et al., 2002; CitationNorman, 2005). All these human genetic data suggest that DSP has critical functions in addition to those of cell adhesion and IF stability. It should be noted that DSP knockout mice show lethality in early embryonic stages, presumably due to loss of integrity of the embryonic ectoderm (CitationGallicano et al., 1998).
DESMOSOMAL CADHERINS
Desmosomal cadherins belong to the larger cadherin superfamily which also includes T-cadherin, FAT family cadherins (CitationAngst et al., 1990), seven-pass transmembrane cadherins, protocadherins and classical cadherins, all sharing an approximately 110 amino acid motif involved in adhesion and calcium binding (CitationTakeichi, 1990). DSGs and DSCs are the transmembrane components that bridge adjacent cells and are embedded in the cytoplasmic plaques, forming the dense extracellular midline seen in mature desmosomes. They share 30% amino acid identity with each other and classical cadherins (CitationGarrod et al., 2002), with DSCs being more closely related to the classical cadherins than to DSGs (CitationKljuic et al., 2004).
Desmosomal cadherins are composed of five extracellular cadherin repeats (EC1-5) containing Ca2+-binding sites and a cell-adhesion recognition (CAR) site (CitationTselepis et al., 1998; CitationRunswick et al., 2001). A unique characteristic of all DSCs is the alternative splicing that generates a complete DSCa form and a shortened DSCb form of the protein by the insertion of a mini-exon containing a stop codon, the shortened length of their carboxy-terminal domain being the only difference between the two isoforms (CitationCollins et al., 1991). DSGs contain an extended 500 amino acid tail, the function of which is not fully understood.
Desmosomal cadherins show complex developmental and differentiation-specific patterns of expression (CitationHolthofer et al., 2007), which implies that desmosomes within different tissues are biochemically and functionally distinct. The precise role of the tissue-specific expression patterns of desmosomal cadherins has yet to be elucidated, but manipulation of desmosomal cadherin expression suggests that tight regulation of their expression pattern is critical to tissue homeostasis (CitationBannon et al., 2001). Within the epidermis, these genes are differentially expressed as keratinocytes undergo terminal differentiation (CitationKottke et al., 2006; CitationHolthofer et al., 2007) as follows: DSG1 and DSC1 are strongly expressed in the granular and spinous layers, their levels decreasing in the lower levels of the epidermis (CitationKing et al., 1995; CitationShimizu et al., 1995; CitationNorth et al., 1996); DSG2 and DSC2 are expressed in all desmosome-bearing tissues. They represent the predominant isoforms in simple epithelia (CitationLegan et al., 1994; CitationSchafer et al., 1996), and are mainly expressed in the basal layer of stratified epidermis (CitationGarrod et al., 2002; CitationNorth et al., 1996). DSG4 is primarily expressed in the hair follicle and is restricted to the more differentiated layers in stratified epithelia (CitationDelva et al., 2009). DSGs 1, 3 and 4, and DSCs 1 and 3 are predominantly expressed in the epidermis, while DSG2 and DSC2 are highly expressed in the myocardium (CitationLi and Radice, 2010). Within the cornified layer of the epidermis (stratum corneum), desmosomes are modified into corneodesmosomes, structures that contain DSG1, DSC1 and corneodesmosin as their major extracellular constituents.
Like DSP, the first linkage of desmosomal cadherins with human disease came from the skin with DSG1 haploinsufficiency mutations associated with autosomal dominant SPPK (; CitationKljuic et al., 2003b; CitationRickman et al., 1999; CitationAwad et al., 2006a). Recently, homozygous loss-of-function DSG1 mutations were identified in an autosomal recessive syndrome characterised by severe dermatitis, allergies and metabolic wasting (SAM) (CitationSamuelov et al., 2013). The mutation carriers in these families presented with non-syndromic SPPK.
Figure 1. Striate palmoplantar keratoderma associated with an autosomal dominantly inherited loss of function mutation in DSG1.
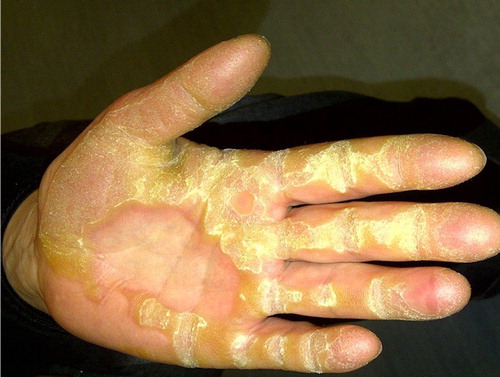
Mutations in other desmosomal cadherins have also been associated with monogenic human disorders (). For example, dominant DSC2 and DSG2 mutations have been associated with non-syndromic ARVC (CitationSyrris et al., 2006; CitationPilichou et al., 2006). A variety of mutations in DSG4 include frameshift, splice-site, missense and nonsense, responsible for the autosomal recessive hair conditions Monilethrix and hypotrichosis (CitationSchaffer et al., 2006; CitationZlotogorski et al., 2006; CitationShimomura et al., 2006). In mice, DSG4 deficiency presents with a lanceolate hair phenotype, characterised by sparse, fragile, broken hair shafts, follicular dystrophy and ichthyosiform dermatitis (CitationJahoda et al., 2004; CitationBazzi et al., 2005).
In humans, a homozygous nonsense DSC3 mutation was identified in a family with autosomal recessive hypotrichosis including absence of eyebrows and eyelashes plus generalised recurrent fluid–filled skin vesicle formation (CitationAyub et al., 2009). DSC3-deficiency in mice is lethal in the very early embryonic stages. Despite the lack of disease-causing mutations being identified in DSG3 and DSC1 in humans, a DSG3-knockout mouse presented with hair loss and loss of epithelial integrity (CitationKoch et al., 1997), while DSC1- deficient mice have skin defects which become more apparent 2 days after birth, and later on develop into localised lesions and epidermal fragility with localised hair loss (CitationChidgey, 2001).
THE ARMADILLO FAMILY
PG and the PKPs (CitationHatzfeld, 2005; CitationHatzfeld, 2007), all members of the armadillo family, are adaptor proteins with roles in facilitating the adhesion of DSP to intermediate filaments, in regulating clustering of desmosomal components, and in mediating important signal transduction pathways.
PG
PG is composed of 12 arm repeats that share 65% amino acid identity with β-catenin, the equivalent protein associated with adherens junctions. The central armadillo domain of PG interacts with DSP, which in turn tethers intermediate filaments to the desmosomal plaque. PG can also translocate to adherens junctions and bind E-cadherin in the same manner as β-catenin, but its higher affinity for DSP may explain why PG and not β-catenin locates to desmosomes (CitationChoi et al., 2009). The critical role of PG in desmosome assembly was demonstrated using knockout studies in mice. Impaired cell cohesion was observed, indicative of compromised desmosome function. Most knockout mice were embryonic lethal although in some cases mouse pups were born but presented epidermal fragility, heart defects and died shortly after birth (CitationAcehan et al., 2008; CitationBierkamp et al., 1996; CitationRuiz et al., 1996).
Human genetic studies in individuals from the Greek island of Naxos affected with an autosomal recessive condition known as ‘Naxos disease’, which includes ARVC alongside woolly hair and mild epidermolytic PPK, identified homozygous truncating mutations in JUP encoding PG as the underlying cause of this syndrome (CitationMcKoy et al., 2000; CitationProtonotarios & Tsatsopoulou, 2004; CitationDelmar & McKenna, 2010). Another recent study described a recessive missense mutation in JUP in a patient who presented with PPK and total alopecia with a cardiac phenotype (CitationErken et al., 2011). Pigors et al. reported a lethal phenotype caused by a homozygous nonsense mutation in JUP leading to severe congenital skin fragility with generalised epidermolysis, massive transcutaneous fluid loss and no apparent cardiac dysfunction. The complete loss of PG in patient skin led to fewer desmosomes and no adhesion structures between keratinocytes (CitationPigors et al., 2011). To add further complexity to the disease mechanisms associated with PG, CitationCabral et al. (2010a) identified loss of function JUP mutations with a recessive syndrome of skin fragility, diffuse PPK, and woolly hair but no signs of ARVC. Little or no PG expression was detected in the skin of these patients. Again like with DSP, these human genetic findings support both overlapping and distinct roles for PG in the epidermis and heart. Recently, Li et al., created an epidermal conditional Jup-knockout mouse model with a skin phenotype of perturbed cell proliferation, apoptosis and differentiation and also compromised immune defence (CitationLi et al., 2012).
PKPs
PKPs play a role in the clustering of desmosomal proteins during the formation of desmosomes. The N-terminal head domain of PKP1 can associate with DSG1, PG, keratin and actin filaments, and ultimately with DSP through what appears to be a robust association that drives DSP recruitment to cell–cell junctions (CitationKowalczyk et al., 1999; CitationHatzfeld et al., 2000; CitationWahl, 2005; CitationHofmann et al., 2000). PKP3 interacts with the largest number of desmosomal proteins, including DSP, PG, DSG1–DSG3, DSC3a and DSC3b, and DSC1a and DSC2a (CitationHatzfeld, 2007). PKP2 plays an important role in transporting DSP to the plasma membrane during desmosome assembly, but does so less efficiently than PKP1 (CitationGreen et al., 2010; CitationChen et al., 2002). The mechanism behind PKP1- and PKP3-mediated desmosomal assembly is not yet fully determined, although it appears that PKP2 functions as a scaffold for protein kinase C alpha (PKCα) and regulates DSP association with intermediate filaments (CitationGreen et al., 2010; CitationGodsel et al., 2005; CitationBass-Zubek et al., 2008).
Both PKP1 and 2 exist in two isoforms, a shorter ‘a’ form and a longer ‘b’ form (CitationMertens et al., 1996; CitationSchmidt et al., 1997). The short ‘a’ form is predominant, while the ‘b’ form is found exclusively in the nucleus. The presence of PKP2 in the nucleus is regulated by the 14-3-3 protein and contributes to the RNA polymerase III holoenzyme complex (CitationDesai et al., 2009). PKPs show tissue- and differentiation-specific patterns of expression similar to those of the desmosomal cadherins. It has been observed that while PKP3 shows expression throughout simple epithelia and all layers of stratified epithelia apart from hepatocytes, PKP1 is mostly expressed in the suprabasal layers of stratified epithelia. PKP2 expression extends to simple epithelia, lower layers of stratified epithelia and non-epithelial tissues such as lymph nodes and cardiac muscle, where it is the only isoform present (CitationHeid et al., 1994; CitationMertens et al., 1996, Citation1999; CitationSchmidt et al., 1997; CitationFranke et al., 2007; CitationBonne et al., 1999).
A variety of PKP1 mutations including missense, splice-site and nonsense are linked with phenotypes ranging from skin fragility to severe autosomal recessive ectodermal dysplasia, including perioral cracking and inflammation, scant hair, reduced sweating and astigmatism (CitationBoyce et al., 2012; CitationMcGrath et al., 1997; CitationPieperhoff et al., 2010; CitationTanaka et al., 2009; CitationZheng et al., 2005; CitationErsoy-Evans et al., 2006). PKP2 mutations are a major genetic cause of non-syndromic autosomal dominant ARVC (CitationGerull et al., 2004). Pkp2-null mice display mid-gestational embryonic lethality caused by cardiac patterning defects and fragility of the myocardium (CitationGrossmann et al., 2004), alongside retraction of intermediate filaments from the plasma membrane, demonstrating the importance of PKPs in DSP recruitment and intermediate filament tethering to desmosomes (CitationDelva et al., 2009). While no disease-causing mutations have been reported in humans for PKP3, ablation of this isoform in mice results in defective hair follicle morphogenesis, increased keratinocyte proliferation and DSP mislocalisation, leading to susceptibility to dermatitis and secondary alopecia (CitationSklyarova et al., 2008).
MODULATION OF DESMOSOMAL ARCHITECTURE AND ADHESION
Desmosomes are not just static structures that keep cells together; instead, they are very dynamic and adaptable complexes as shown by their ability to adopt different conformations with different adhesive affinities, suppressing pathways important for establishing cell polarity and determining the balance between proliferation and differentiation, all through interactions with signalling cascades.
The hyper-adhesiveness of desmosomes is regulated by the presence or absence of Ca2+, but there are other factors that mediate desmosome assembly and intercellular adhesion. Some of these factors are PKC, proteolytic processing through proteins such as ADAMs and cathepsins/cystatins, EGFR signalling, raft regulation and the yet unclear mechanism of the ubiquitin-proteasome system (UPS; CitationNekrasova & Green, 2013; CitationYin et al., 2005; CitationStahley et al., 2014; CitationLoffek et al., 2012).
DSGs and DSCs are the proteins that play key roles in Ca2+ regulation, required for strong cell–cell adhesion (CitationGetsios et al., 2004a), by their interaction across the intercellular space, in both a homophilic and a heterophilic manner. Via several binding motifs within their structure, DSGs and DSCs bind Ca2+, and assume a rigidified functional conformation (CitationPokutta & Weis, 2007), increasing the level of adhesion between neighbouring cells and creating what is described as the dense midline of desmosomes. In low-Ca2+ conditions, the desmosomal plaque components and membrane proteins are transported to the plasma membrane, together or in separate compartments, but when desmosomal assembly is triggered, cadherins and DSP complexes do not associate as in normal Ca2+ conditions and remain separate (CitationCirillo et al., 2010). During the early stages of desmosome formation, the assembly can reverse between the young and mature phases but ultimately desmosomes mature and can no longer be dissociated by Ca2+ depletion (CitationWatt et al., 1984). This is referred to as hyper-adhesion, the result of high-affinity and stable adhesive binding of desmosomal components into mature structures; it has not been observed in adherens or tight junctions (CitationKimura et al., 2007).
DARIER’S DISEASE
Patients with the autosomal-dominant condition Darier's disease present papules and plaques in seborrhoeic areas, acantholysis, blistering, mucosal lesions and abnormal keratinisation, aggravated by heat, friction, infections and UVB irradiation (CitationBeck et al., 1977; CitationSakuntabhai et al., 1999). Impaired adhesion, a feature of the disease, is likely to result from changes in DSP trafficking (CitationCelli et al., 2012; CitationDhitavat et al., 2003; CitationHobbs et al., 2011). The desmosomal proteins DSP, DSG3 and DSC3 appeared disorganised with a thicker and punctuated distribution under the plasma membrane, while DSG3 and DSC3 also presented perinuclear staining, suggesting that they had been retained in the endoplasmic reticulum (ER).
Genetic analysis of patients with Darier's disease found that mutations in the ATP2A2 gene coding for the SERCA2A Ca2+ pump are responsible for this condition (CitationSakuntabhai et al., 1999). In vitro studies using Darier keratinocytes showed a decreased ER Ca2+ concentration (CitationDode et al., 2003; CitationFoggia et al., 2006; CitationMiyauchi et al., 2006; CitationPani et al., 2006). Upon Ca2+ depletion, misfolded proteins accumulated in the ER leading to induction of the unfolded protein response (UPR; CitationYoshida, 2007). In Darier's disease, keratinocytes show constant activation of the UPR-signallingd pathway (CitationSavignac et al., 2014), enhanced further by treatment with Thapsigargin, a specific inhibitor of SERCA pumps (CitationThastrup et al., 1989). Treatment with Miglustat, an inhibitor of α-glucosidase, reversed the phenotype observed in Darier keratinocytes and allowed normal formation of adherens junctions and desmosomes (CitationSavignac et al., 2014).
ADAM17
ADAM17 (a disintegrin and metalloprotease 17) is responsible for shedding a number of ligands by cleavage of substrates in the juxtamembrane region. Substrates include a number of ligands for the EGF receptor (EGFR) and TNF-α, giving ADAM17 its alternative name TNF-α-converting enzyme. ADAM17 has additionally been shown to play a key role in ligand- independent Notch signalling, and in the shedding of cell adhesion molecules including DSG2 (CitationGschwind et al., 2003; CitationKlessner et al., 2009; CitationPruessmeyer & Ludwig, 2009; CitationSahin et al., 2004; CitationBech-Serra et al., 2006).
Homozygous loss-of-function ADAM17 mutations are associated with a syndrome of severe skin inflammation, increased susceptibility to infection, bowel inflammation and cardiomyopathy (CitationBlaydon et al., 2011a). Adam17 knockout in mice is lethal (CitationPeschon et al., 1998). Immunohistochemistry with an antibody against DSG1 and DSG2 showed increased staining intensity (CitationBlaydon et al., 2011a) implying a reduction in DSG2 shedding by ADAM17. Thus, the cardiomyopathy and other phenotypes in this syndrome may be related, in part, to impaired ADAM17-mediated DSG2 processing (CitationBlaydon et al., 2011a).
In contrast, tylosis with oesophageal cancer (TOC) results in increased processing and activity of ADAM17 (CitationBrooke et al., 2014). TOC patients suffer from PPK, follicular papules, oral keratosis, and up to a 95% lifetime risk of developing oesophageal cancer (CitationEllis et al., 1994; CitationHennies et al., 1995; CitationStevens et al., 1996). TOC results from mutations in a specific region of the long N-terminus of the inactive rhomboid protein iRHOM2, encoded by the gene RHBDF2 (CitationBlaydon et al., 2012; CitationSaarinen et al., 2012). iRHOM2 (and its homologue iRHOM1) traffic ADAM17 from the ER to the Golgi, where the inhibitory pro-domain is cleaved by furin (CitationAdrain et al., 2012; CitationMcIlwain et al., 2012). A dramatic increase in the iRHOM2-mediated processing of ADAM17 in TOC cutaneous keratinocytes compared to that of control cells was observed (CitationBrooke et al., 2014).
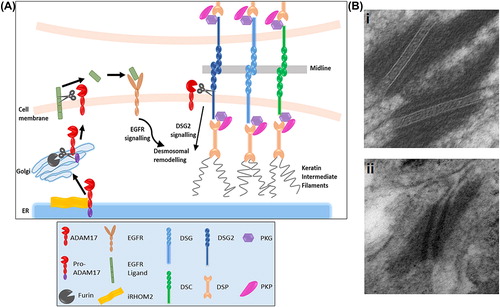
Strikingly, electron microscopy of the skin of TOC patients () revealed desmosomes lacking the electron-dense midlines seen in non-TOC skin (CitationBrooke et al., 2014), consistent with desmosomes in a wound-healing state (CitationGarrod, 2005). Furthermore, there appeared to be increased processing of DSG2 in TOC cutaneous keratinocytes (CitationBrooke et al., 2014). DSG2 is reported to be expressed at lower levels in epidermis so other cadherins may also be affected. Alternatively, desmosomal remodelling may result from the increased EGFR signalling in TOC keratinocytes shown by increased migration, insensitivity to exogenous EGF (CitationBlaydon et al., 2012) and increased shedding of EGFR ligands (CitationBrooke et al., 2014). EGFR signalling results in phosphorylation of desmosomal cadherins followed by cleavage and endocytosis (CitationKlessner et al., 2009; CitationLorch et al., 2004), and EGFR inhibition increases adhesion between cells (CitationLorch et al., 2004), so it is plausible that alterations in desmosomal composition may result from this altered EGFR signalling ().
Figure 2. ADAM17 and EGFR-mediated regulation of desmosomes. (A) Schematic showing the possible mechanisms of desmosomal regulation by the metalloproteinase enzyme ADAM17. Desmosomes may be disrupted indirectly through ADAM17-mediated shedding of EGFR ligands, leading to desmosomal remodelling through EGFR signalling; or regulated directly via ADAM17-mediated cleavage of DSG2 by ADAM17. Loss-of-function ADAM17 mutations lead to accumulation of cell-surface desmosomal proteins, while increased processing of ADAM17 by iRHOM2 in tylosis with oesophageal cancer causes increased shedding of EGFR ligands and DSG2. (B) In electron microscopy images, a clear electron-dense midline is seen in the desmosomes of normal skin (i) indicating desmosomes in a hyperadhesive state, while desmosomes in TOC skin lack this midline (ii) suggesting that the desmosomes are in a calcium-dependent wound-healing state. EM was carried out by Prof Akemi Ishida-Yamamoto and Graham McPhail.
FINAL DISCUSSION
A striking aspect of the desmosomal disorders is the wide spectrum of inherited conditions that can arise from a variety of dominant or recessive mutations in genes encoding the desmosomal proteins DSP and PG, in contrast to the more limited phenotypes resulting from mutations in the PKPs and the cadherin superfamily. Despite the increasing number of desmosomal mutations being identified in patients with phenotypes ranging from PPK to acantholysis, hair disruption and/or sudden cardiac death, still the mechanism of protein trafficking to the plasma membrane, desmosome assembly, regulation and signalling is less known. Furthermore, in autosomal dominant disorders such as Darier's and Hailey Hailey's disease (CitationHu et al., 2000), mutations affecting desmosomal regulation do not affect desmosomal proteins directly, but do so through mutations in the Ca2+ transport ATPases ATP2A2 and ATP2C1, therefore affecting the essential state of hyper-adhesiveness. More recently, our attention has turned towards protease inhibitors and their target proteases, not towards their protection against allergens in the upper layers of the epidermis, but their direct role in desmosomal homeostasis. In exfoliative ichthyosis, loss-of-function mutations in the protease inhibitor cystatin A (CSTA) destabilise the basal-suprabasal connection in the epidermis (CitationBlaydon et al., 2011b), emphasising a possible role in desmosome regulation through its target proteases. Furthermore, homozygous loss-of-function mutations of ADAM17 lead to a reduction in shedding of DSG2 (CitationBlaydon et al., 2011a), while in TOC patients harbouring iRHOM2 mutations, there is an increased processing and activity of ADAM17, with enhanced DSG2 and also EGFR ligand shedding (, CitationBrooke et al., 2014).
In summary, the human disorders of the desmosome suggest focussing our attention towards the complexity of regulatory signalling pathways in which desmosomal proteins play orchestrating roles.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Acehan D, Petzold C, Gumper I, Sabatini DD, Muller EJ, Cowin P, Stokes DL (2008). Plakoglobin is required for effective intermediate filament anchorage to desmosomes. J Invest Dermatol, 128: 2665–2675.
- Adrain C, Zettl M, Christova Y, Taylor N, Freeman M (2012). Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science.335: 225–228.
- Al-Jassar C, Bikker H, Overduin M, Chidgey M (2013). Mechanistic basis of desmosome-targeted diseases. J Mol Biol.425: 4006–4022.
- Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T (2003). A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Card.42: 319–327.
- Angst BD, Nilles LA, Green KJ (1990). Desmoplakin II expression is not restricted to stratified epithelia. J Cell Sci.97: 247–257.
- Armstrong D, Mckenna K, Purkis P, Green K, Eady R, Leigh I, Hughes A (1999). Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet.8: 143–148.
- Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, Mckenna WJ (2007). A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Hum Genet.81: 964–973.
- Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russell SD, Bluemke DA, Dietz HC, Calkins H, Judge DP (2006a). DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet.79: 136–142.
- Awad MM, Dalal D, Tichnell C, James C, Tucker A, Abraham T, Spevak PJ, Calkins H, Judge DP (2006b). Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum Mutat.27: 1157–1167.
- Ayub M, Basit S, Jelani M, Rehman FU, Iqbal M, Yasinzai M, Ahmad W (2009). A homozygous nonsense mutation in the human desmocollin-3 (DSC3) gene underlies hereditary hypotrichosis and recurrent skin vesicles. Am J Hum Genet.85: 515–520.
- Bannon LJ, Cabrera BL, Stack MS, Green KJ (2001). Isoform-specific differences in the size of desmosomal cadherin/catenin complexes. J Invest Dermatol.117: 1302–1306.
- Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, Wahl JK, iii DENNING, MF, Green KJ (2008). Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol.181: 605–613.
- Bazzi H, Martinez-Mir A, Kljuic A, Christiano AM (2005). Desmoglein 4 mutations underlie localized autosomal recessive hypotrichosis in humans, mice, and rats. J Invest Dermatol Symp Proc.10: 222–224.
- Bech-Serra JJ, Santiago-Josefat B, Esselens C, Saftig P, Baselga J, Arribas J, Canals F (2006). Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol Cell Biol.26: 5086–5095.
- Beck AL, Jr., Finocchio AF, White JP (1977). Darier's disease: a kindred with a large number of cases. Br J Dermatol.97: 335–339.
- Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R (1996). Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol.180: 780–785.
- Blaydon DC, Biancheri MD, Di WL, Plagnol V, Cabral RM, Brooke MA, Van Heel DA, Ruschendorf F, Toynbee M, Walne A, O'toole EA, Martin JE, Lindley K, Vulliamy T, Abrams DJ, Macdonald TT, Harper JI, Kelsell DP (2011a). Inflammatory skin and bowel disease linked to ADAM17 Deletion. N Eng J Med.365: 32–38.
- Blaydon DC, Nitoiu D, Eckl KM, Cabral RM, Bland P, Hausser I, Van Heel DA, Rajpopat S, Fischer J, Oji V, Zvulunov A, Traupe H, Hennies HC, Kelsell DP (2011b). Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am J Hum Genet.89: 564–571.
- Blaydon DC, Etheridge SL, Risk JM, Hennies HC, Gay LJ, Carroll R, Plagnol V, Mcronald FE, Stevens HP, Spurr NK, Bishop DT, Ellis A, Jankowski J, Field JK, Leigh IM, South AP, Kelsell DP (2012). RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am J Hum Genet.90: 340–346.
- Bonne S, Van Hengel J., Nollet F, Kools P, Van Roy F (1999). Plakophilin-3, a novel armadillo-like protein present in nuclei and desmosomes of epithelial cells. J Cell Sci.112 (Pt 14): 2265–2276.
- Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, Green KJ (2001). Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci.114: 727–738.
- Boyce AE, McGrath JA, Techanukul T, Murrell DF, Chow CW, Mcgregor L, Warren LJ (2012). Ectodermal dysplasia-skin fragility syndrome due to a new homozygous internal deletion mutation in the PKP1 gene. Australas J Dermatol.53: 61–65.
- Brooke MA, Etheridge SL, Kaplan N, Simpson C, O'toole E, Ishida-Yamamoto A, Marches O, Getsios S, Kelsell DP (2014). iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum Mol Genet. Epub ahead of print, doi:.10.1093/hmg/ddu120
- Brooke MA, Nitoiu D, Kelsell DP (2012). Cell-cell connectivity: desmosomes and disease. J Pathol.226: 158–171.
- Cabral RM, Liu L, Hogan C, Dopping-Hepenstal PJC, Winik BC, Asial RA, Dobson R, Mein CA, Baselaga PA, Mellerio JE, Nanda A, Boente MDC, Kelsell DP, McGrath JA, South AP (2010a). Homozygous mutations in the 5′ region of the JUP gene result in cutaneous disease but normal heart development in children. J Invest Dermatol.130: 1543–1550.
- Cabral RM, Wan H, Cole CL, Abrams DJ, Kelsell DP, South AP (2010b). Identification and characterization of DSPIa, a novel isoform of human desmoplakin. Cell Tissue Res.341: 121–129.
- Cabral RM, Tattersall D, Patel V, Hatzimasoura E, Abrams DJ, South AP, Kelsell DP (2012). The DSPII splice variant is critical for desmosome-mediated HaCaT keratinocyte adhesion. J Cell Sci. 125: 2853–2861.
- Celli A, Mackenzie DS, Zhai Y, Tu CL, Bikle DD, Holleran WM, Uchida Y, Mauro TM (2012). SERCA2-controlled Ca(2)+-dependent keratinocyte adhesion and differentiation is mediated via the sphingolipid pathway: a therapeutic target for Darier's disease. J Invest Dermatol.132: 1188–1195.
- Chen X, Bonne S, Hatzfeld M, Van Roy F, Green KJ (2002). Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem.277: 10512–10522.
- Chidgey M (2001). Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J Cell Biol.155: 821–832.
- Choi HJ, Gross JC, Pokutta S, Weis WI (2009). Interactions of plakoglobin and beta-catenin with desmosomal cadherins: basis of selective exclusion of alpha- and beta-catenin from desmosomes. J Biol Chem.284: 31776–31788.
- Choi HJ, Park-Snyder S, Pascoe LT, Green KJ, Weis WI (2002). Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat Struct Biol.9: 612–620.
- Cirillo N, Lanza A, Prime SS (2010). Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res.316: 580–592.
- Collins JE, Legan PK, Kenny TP, Macgarvie J, Holton JL, Garrod DR (1991). Cloning and sequence analysis of desmosomal glycoproteins 2 and 3 (desmocollins): cadherin-like desmosomal adhesion molecules with heterogeneous cytoplasmic domains. J Cell Biol.113: 381–391.
- Delmar M, McKenna WJ (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res.107: 700–714.
- Delva E, Tucker DK, Kowalczyk AP (2009). The desmosome. Cold Spring Harb Perspect Biol.1: a002543.
- Desai BV, Harmon RM, Green KJ (2009). Desmosomes at a glance. J Cell Sci.122: 4401–4407.
- Dhitavat J, Cobbold C, Leslie N, Burge S, Hovnanian A (2003). Impaired trafficking of the desmoplakins in cultured Darier's disease keratinocytes. J Invest Dermatol.121: 1349–1355.
- Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A (2003). Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses. J Biol Chem.278: 47877–47889.
- Ellis A, Field JK, Field EA, Friedmann PS, Fryer A, Howard P, Leigh IM, Risk J, Shaw JM, Whittaker J (1994). Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family–a review of six generations. Eur J Cancer B Oral Oncol.30B: 102–112.
- Erken H, Yariz KO, Duman D, Kaya CT, Sayin T, Heper AO, Tekin M (2011). Cardiomyopathy with alopecia and palmoplantar keratoderma (CAPK) is caused by a JUP mutation. Br J Dermatol. 165: 917–921
- Ersoy-Evans S, Erkin G, Fassihi H, Chan I, Paller AS, Surucu S, McGrath JA (2006). Ectodermal dysplasia-skin fragility syndrome resulting from a new homozygous mutation, 888delC, in the desmosomal protein plakophilin 1. J Am Acad Dermatol.55: 157–161.
- Farquhar MG, Palade GE (1963). Junctional complexes in various epithelia. J Cell Biol.17: 375–412.
- Foggia L, Aronchik I, Aberg K, Brown B, Hovnanian A, Mauro TM (2006). Activity of the hSPCA1 Golgi Ca2+ pump is essential for Ca2+-mediated Ca2+ response and cell viability in Darier disease. J Cell Sci.119: 671–679.
- Franke WW, Borrmann CM, Grund C, Pieperhoff S (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol.85: 69–82.
- Franke WW, Schumacher H, Borrmann CM, Grund C, Winter-Simanowski S, Schlechter T, Pieperhoff S, Hofmann I (2007). The area composita of adhering junctions connecting heart muscle cells of vertebrates - III: assembly and disintegration of intercalated disks in rat cardiomyocytes growing in culture. Eur J Cell Biol.86: 127–142.
- Gallicano GI, Bauer C, Fuchs E (2001). Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development.128: 929–941.
- Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998). Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol.143: 2009–2022.
- Garrod DR (2005). Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci.118: 5743–5754.
- Garrod DR, Merritt AJ, Nie Z (2002). Desmosomal cadherins. Curr Opin Cell Biol.14: 537–545.
- Gerull B, Heuser A, Wichter T, Paul M, Basson CT, Mcdermott DA, Lerman BB, Markowitz SM, Ellinor PT, Macrae CA, Peters S, Grossmann KS, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L (2004). Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet.36: 1162–1164.
- Getsios S, Amargo EV, Dusek RL, Ishii K, Sheu L, Godsel LM, Green KJ (2004a). Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation.72: 419–433.
- Getsios S, Huen AC, Green KJ (2004b). Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol.5: 271–281.
- Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-Mcgillicuddy LT, Huen AC, Thorne ME, Gaudry CA, Park JK, Myung K, Goldman RD, Chew TL, Green KJ (2005). Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol.171: 1045–1059.
- Green KJ, Gaudry CA (2000). Are desmosomes more than tethers for intermediate filaments?Nat Rev Mol Cell Biol.1: 208–216.
- Green KJ, Getsios S, Troyanovsky S, Godsel LM (2010). Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol.2: a000125.
- Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004). Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol.167: 149–160.
- Gschwind A, Hart S, Fischer OM, Ullrich A (2003). TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J.22: 2411–2421.
- Hatzfeld M (2005). The p120 family of cell adhesion molecules. Eur J Cell Biol.84: 205–214.
- Hatzfeld M (2007). Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion?Biochim Biophys Acta.1773: 69–77.
- Hatzfeld M, Haffner C, Schulze K, Vinzens U (2000). The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol.149: 209–222.
- Heid HW, Schmidt A, Zimbelmann R, Schafer S, Winter-Simanowski S, Stumpp S, Keith M, Figge U, Schnolzer M, Franke WW (1994). Cell type-specific desmosomal plaque proteins of the plakoglobin family: plakophilin 1 (band 6 protein). Differentiation.58: 113–131.
- Hennies HC, Hagedorn M, Reis A (1995). Palmoplantar keratoderma in association with carcinoma of the esophagus maps to chromosome 17q distal to the keratin gene cluster. Genomics.29: 537–540.
- Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, Macrae CA, Gerull B (2006). Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet.79: 1081–1088.
- Hobbs RP, Amargo EV, Somasundaram A, Simpson CL, Prakriya M, Denning MF, Green KJ (2011). The calcium ATPase SERCA2 regulates desmoplakin dynamics and intercellular adhesive strength through modulation of PKCalpha signaling. FASEB J.25: 990–1001.
- Hofmann I, Mertens C, Brettel M, Nimmrich V, Schnolzer M, Herrmann H (2000). Interaction of plakophilins with desmoplakin and intermediate filament proteins: an in vitro analysis. J Cell Sci.113: 2471–2483.
- Holthofer B, Windoffer R, Troyanovsky S, Leube RE (2007). Structure and function of desmosomes. Int Rev Cytol.264: 65–163.
- Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, Ogawa H, Ikeda S, Mauro T, Epstein EH, J. R (2000). Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nature Genet. 24: 61–65.
- Huen AC, Park JK, Godsel LM, Chen X, Bannon LJ, Amargo EV, Hudson TY, Mongiu AK, Leigh IM, Kelsell DP, Gumbiner BM, Green KJ (2002). Intermediate filament-membrane attachments function synergistically with actin-dependent contacts to regulate intercellular adhesive strength. J Cell Biol.159: 1005–1017.
- Jahoda CA, Kljuic A, O'shaughnessy R, Crossley N, Whitehouse CJ, Robinson M, Reynolds AJ, Demarchez M, Porter RM, Shapiro L, Christiano AM (2004). The lanceolate hair rat phenotype results from a missense mutation in a calcium coordinating site of the desmoglein 4 gene. Genomics.83: 747–756.
- Jonkman MF, Pasmooij AM, Pasmans SG, Van Den Berg MP, Ter Horst HJ, Timmer A, Pas HH (2005). Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet.77: 653–660.
- Kelly DE (1966). Fine structure of desmosomes., hemidesmosomes, and an adepidermal globular layer in developing newt epidermis. J Cell Biol.28: 51–72.
- Kimura TE, Merritt AJ, Garrod DR (2007). Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol.127: 775–781.
- King IA, Sullivan KH, Bennett R, Jr, Buxton RS (1995). The desmocollins of human foreskin epidermis: identification and chromosomal assignment of a third gene and expression patterns of the three isoforms. J Invest Dermatol.105: 314–21.
- Klessner JL, Desai BV, Amargo EV, Getsios S, Green KJ (2009). EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol Biol Cell.20: 328–337.
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O'shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, Gordon D, Uitto J, Whiting D, Ott J, Fischer S, Gilliam TC, Jahoda CA, Morris RJ, Panteleyev AA, Nguyen VT, Christiano AM (2003a). Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 113: 249–260.
- Kljuic A, Gilead L, Martinez-Mir A, Frank J, Christiano AM, Zlotogorski A (2003b). A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol.12: 523–527.
- Kljuic A, Bauer RC, Christiano AM (2004). Genomic organization of mouse desmocollin genes reveals evolutionary conservation. DNA Seq.15: 148–152.
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR (1997). Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol.137: 1091–1102.
- Kottke MD, Delva E, Kowalczyk AP (2006). The desmosome: cell science lessons from human diseases. J Cell Sci.119: 797–806.
- Kowalczyk AP, Hatzfeld M, Bornslaeger EA, Kopp DS, Borgwardt JE, Corcoran CM, Settler A, Green KJ (1999). The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J Biol Chem.274: 18145–18148.
- Kowalczyk AP, Stappenbeck TS, Parry DA, Palka HL, Virata ML, Bornslaeger EA, Nilles LA, Green KJ (1994). Structure and function of desmosomal transmembrane core and plaque molecules. Biophys Chem.50: 97–112.
- Legan PK, Yue KK, Chidgey MA, Holton JL, Wilkinson RW, Garrod DR (1994). The bovine desmocollin family: a new gene and expression patterns reflecting epithelial cell proliferation and differentiation. J Cell Biol.126: 507–518.
- Li D, Zhang W, Liu Y, Haneline LS, Shou W (2012). Lack of Plakoglobin in Epidermis Leads to Keratoderma. J Biol Chem.287: 10435–10443.
- Li J, Radice GL (2010). A new perspective on intercalated disc organization: implications for heart disease. Dermatol Res Pract.2010: 207835.
- Loffek S, Bruckner-Tuderman L, Magin TM (2012). Involvement of the ubiquitin-proteasome system in the stabilization of cell-cell contacts in human keratinocytes. Exp Dermatol.21: 791–793.
- Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, Stack MS, Green KJ (2004). Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem.279: 37191–37200.
- McGrath JA, Mcmillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RA (1997). Mutations in the plakophilin 1 gene result in ectodermal dysplasia/ skin fragility syndrome. Nat Genet.17: 240–244.
- McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC, Lang KS, Haussinger D, Wakeham A, Itie-Youten A, Khokha R, Ohashi PS, Blobel CP, Mak TW (2012). iRhom2 Regulation of TACE Controls TNF-Mediated Protection Against Listeria and Responses to LPS. Science.335: 229–232.
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, Mckenna WJ (2000). Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet.355: 2119–2124.
- Mertens C, Kuhn C, Franke WW (1996). Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol.135: 1009–1025.
- Mertens C, Kuhn C, Moll R, Schwetlick I, Franke WW (1999). Desmosomal plakophilin 2 as a differentiation marker in normal and malignant tissues. Differentiation.64: 277–290.
- Miyauchi Y, Daiho T, Yamasaki K, Takahashi H, Ishida-Yamamoto A, Danko S, Suzuki H, Iizuka H (2006). Comprehensive analysis of expression and function of 51 sarco(endo)plasmic reticulum Ca2+-ATPase mutants associated with Darier disease. J Biol Chem.281: 22882–22895.
- Nekrasova O, Green KJ (2013). Desmosome assembly and dynamics. Trends Cell Biol.23: 537–546.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000). Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet.9: 2761–2766.
- Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP (2006). Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol.126: 1651–1654.
- Norman M (2005). Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation.112: 636–642.
- North AJ, Chidgey MA, Clarke JP, Bardsley WG, Garrod DR (1996). Distinct desmocollin isoforms occur in the same desmosomes and show reciprocally graded distributions in bovine nasal epidermis. Proc Natl Acad Sci USA.93: 7701–7705.
- Pani B, Cornatzer E, Cornatzer W, Shin DM, Pittelkow MR, Hovnanian A, Ambudkar IS, Singh BB (2006). Up-regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell.17: 4446–4458.
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA (1998). An essential role for ectodomain shedding in mammalian development. Science.282: 1281–1284.
- Pieperhoff S, Barth M, Rickelt S, Franke WW (2010). Desmosomal molecules in and out of adhering junctions: normal and diseased States of epidermal, cardiac and mesenchymally derived cells. Dermatol Res Pract.2010: 139167.
- Pigors M, Kiritsi D, Krumpelmann S, Wagner N, He Y, Podda M, Kohlhase J, Hausser I, Bruckner-Tuderman L, Has C (2011). Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum Mol Genet.20: 1811–1819.
- Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A (2006). Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation.113: 1171–1179.
- Pokutta S, Weis WI (2007). Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol.23: 237–261.
- Protonotarios N, Tsatsopoulou A (2004). Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol.13: 185–194.
- Pruessmeyer J, Ludwig A (2009). The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin Cell Dev Biol.20: 164–174.
- Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA (2002). Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J of Hum Genet.71: 1200–1206.
- Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RA, Leigh IM, Arnemann J, Magee AI, Kelsell DP, Buxton RS (1999). N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet.8: 971–976.
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W (1996). Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol.135: 215–225.
- Runswick SK, O'hare MJ, Jones L, Streuli CH, Garrod DR (2001). Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol.3: 823–830.
- Saarinen S, Vahteristo P, Lehtonen R, Aittomäki K, Launonen V, Kiviluoto T, Aaltonen L (2012). Analysis of a finnish family confirms RHBDF2 mutations as the underlying factor in tylosis with esophageal cancer. Fam Cancer.11: 525–528.
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP (2004). Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol.164: 769–779.
- Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O'donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T, Hovnanian A (1999). Mutations in ATP2A2, encoding a Ca2 + pump, cause Darier disease. Nat Genet.21: 271–277.
- Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, Koetsier JL, Gat A, Goldberg I, Bergman R, Spiegel R, Eytan O, Geller S, Peleg S, Shomron N, Goh CSM, Wilson NJ, Smith FJD, Pohler E, Simpson MA, Mclean WHI, Irvine AD, Horowitz M, McGrath JA, Green KJ, Sprecher E (2013). Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet.45: 1244–1248.
- Savignac M, Simon M, Edir A, Guibbal L, Hovnanian A (2014). SERCA2 Dysfunction in Darier disease causes endoplasmic reticulum stress and impaired cell-to-cell adhesion strength: rescue by Miglustat. J Invest Dermatol. Epub ahead of print. doi:.10.1038/jid.2014.8.
- Schafer S, Stumpp S, Franke WW (1996). Immunological identification and characterization of the desmosomal cadherin Dsg2 in coupled and uncoupled epithelial cells and in human tissues. Differentiation.60: 99–108.
- Schaffer JV, Bazzi H, Vitebsky A, Witkiewicz A, Kovich OI, Kamino H, Shapiro LS, Amin SP, Orlow SJ, Christiano AM (2006). Mutations in the desmoglein 4 gene underlie localized autosomal recessive hypotrichosis with monilethrix hairs and congenital scalp erosions. J Invest Dermatol.126: 1286–1291.
- Schmidt A, Langbein L, Rode M, Pratzel S, Zimbelmann R, Franke WW (1997). Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue Res.290: 481–499.
- Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T (1995). Pemphigus vulgaris and pemphigus foliaceus sera show an inversely graded binding pattern to extracellular regions of desmosomes in different layers of human epidermis. J Invest Dermatol.105: 153–159.
- Shimomura Y, Sakamoto F, Kariya N, Matsunaga K, Ito M (2006). Mutations in the desmoglein 4 gene are associated with monilethrix-like congenital hypotrichosis. J Invest Dermatol.126: 1281–1285.
- Simpson MA, Mansour S, Ahnood D, Kalidas K, Patton MA, Mckenna WJ, Behr ER, Crosby AH (2009). Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology.113: 28–34.
- Sklyarova T, Bonne S, D'hooge P, Denecker G, Goossens S, De Rycke R., Borgonie G, Bosl M, Van Roy F, Van Hengel J (2008). Plakophilin-3-deficient mice develop hair coat abnormalities and are prone to cutaneous inflammation. J Invest Dermatol.128: 1375–1385.
- Staehelin LA (1974). Structure and function of intercellular junctions. Int Rev Cytol.39: 191–283.
- Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP (2014). Desmosome assembly and disassembly are membrane raft-dependent. PLoS One.9: e87809.
- Stevens HP, Kelsell DP, Bryant SP, Bishop DT, Spurr NK, Weissenbach J, Marger D, Marger RS, Leigh IM (1996). Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Literature survey and proposed updated classification of the keratodermas. Arch Dermatol.132: 640–651.
- Syrris P, Ward D, Asimaki A, Evans A, Sen-Chowdhry S, Hughes SE, Mckenna WJ (2007). Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J.28: 581–588.
- Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, Mckenna WJ (2006). Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet.79: 978–984.
- Takeichi M (1990). Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem.59: 237–252.
- Tanaka A, Lai-Cheong JE, Cafe ME, Gontijo B, Salomao PR, Pereira L, McGrath JA (2009). Novel truncating mutations in PKP1 and DSP cause similar skin phenotypes in two Brazilian families. Br J Dermatol.160: 692–697.
- Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK, Bjerrum PJ, Christensen SB, Hanley MR (1989). Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions.27: 17–23.
- Tselepis C, Chidgey M, North A, Garrod D (1998). Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci USA.95: 8064–8069.
- Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H, Sahin SE, Dupont E, Severs NJ, Leigh IM, Yuksel-Apak M, Kelsell DP, Wollnik B (2006). Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet.43: e05.
- Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E (2001). Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol.3: 1076–1085.
- Wahl JK, III (2005). A role for plakophilin-1 in the initiation of desmosome assembly. J Cell Biochem.96: 390–403.
- Watt FM, Mattey DL, Garrod DR (1984). Calcium- induced reorganization of desmosomal components in cultured human keratinocytes. J Cell Biol.99: 2211–2215.
- Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, McGrath JA (1999). Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol.113: 940–946.
- Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, Mclean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA (2002). Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol.118: 232–238.
- Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JCR, Green KJ (2005). Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci U S A.102: 5420–5425.
- Yin T, Green KJ (2004). Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol.15: 665–677.
- Yoshida H (2007). ER stress and diseases. FEBS J.274: 630–658.
- Zheng R, Bu DF, Zhu XJ (2005). Compound heterozygosity for new splice site mutations in the plakophilin 1 gene (PKP1) in a Chinese case of ectodermal dysplasia-skin fragility syndrome. Acta Derm Venereol.85: 394–399.
- Zlotogorski A, Marek D, Horev L, Abu A, Ben-Amitai D, Gerad L, Ingber A, Frydman M, Reznik-Wolf H, Vardy DA, Pras E (2006). An autosomal recessive form of monilethrix is caused by mutations in DSG4: clinical overlap with localized autosomal recessive hypotrichosis. J Invest Dermatol.126: 1292–1296.