Abstract
Pulmonary fibrosis is a relentlessly progressive disease for which the etiology can be idiopathic or associated with environmental or occupational exposures. There is not a clear explanation for the chronic and progressive nature of the disease, leaving treatment and prevention options limited. However, there is increasing evidence of an autoimmune component, since fibrotic diseases are often accompanied by production of autoantibodies. Because exposure to silicates such as silica and asbestos can lead to both autoantibodies and pulmonary/pleural fibrosis, these exposures provide an excellent tool for examining the relationship between these outcomes. This study explored the possibility that autoantibodies induced by asbestos exposure in mice would affect fibroblast phenotype. L929 fibroblasts and primary lung fibroblasts were treated with serum IgG from asbestos- or saline-treated mice, and tested for binding using cell-based ELISA, and for phenotypic changes using immunofluorescence, laser scanning cytometry and Sirius Red collagen assay. Autoantibodies in the serum of C57Bl/6 mice exposed to asbestos (but not sera from untreated mice) bound to mouse fibroblasts. The autoantibodies induced differentiation to a myofibroblast phenotype, as demonstrated by increased expression of smooth muscle α-actin (SMA), which was lost when the serum was cleared of IgG. Cells treated with purified IgG of exposed mice produced excess collagen. Using ELISA, we tested serum antibody binding to DNA topoisomerase (Topo) I, vimentin, TGFβ-R, and PDGF-Rα. Antibodies to DNA Topo I and to PDGF-Rα were detected, both of which have been shown by others to be able to affect fibroblast phenotype. The anti-fibroblast antibodies (AFA) also induced STAT-1 activation, implicating the PDGF-R pathway as part of the response to AFA binding. These data support the hypothesis that asbestos induces AFA that modify fibroblast phenotype, and suggest a mechanism whereby autoantibodies may mediate some of the fibrotic manifestations of asbestos exposure.
Introduction
Pulmonary diseases such as idiopathic pulmonary fibrosis (IPF) and cryptogenic fibrosing alveolitis (CFA) are progressive pathologies in which there is increasing evidence of a primary autoimmune etiology (Ihn et al., Citation2000; Youinou et al., Citation2006). In addition, the pathogenic role of anti-fibroblast antibodies (AFA) in the pulmonary fibrotic manifestations of systemic autoimmune diseases such as systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and rheumatoid arthritis (RA) is also supported by a growing literature (reviewed in Jindal and Agarwal, Citation2005). A novel and creative approach for filling knowledge gaps regarding mechanistic links between pulmonary fibrosis and autoimmunity is the study of the effects of silicate inhalation. Exposure to crystalline silica has long been associated with increased incidence of SSc and SLE, along with interstitial fibrosis (silicosis) (Haustein and Anderegg, Citation1998; Prakash, Citation1998). These diseases are associated with the production of a variety of autoantibodies (Conrad et al., Citation1997; Tan et al., Citation1999; Yang et al., Citation2003), and a role for these autoantibodies in the progression of fibrosis has been hypothesized. Additionally, the fibrous silicate asbestos is associated with chronic pulmonary fibrosis as well as autoimmune responses, with evidence that positive autoantibody tests are associated with severity or progression of lung disease (Tamura et al., Citation1996; Pfau et al., Citation2005).
Despite differences in the histology and location of fibrotic lesions in asbestosis, silicosis, and other forms of fibrosis such as SSc, all are characterized by the presence of myofibroblasts, cells that actively express smooth muscle α-actin (SMA) and collagen (Sappino et al., Citation1990; Perdue and Brody, Citation1994; Kirk et al., Citation1995; Mariani et al., Citation1996; Kaarteenaho-Wiik et al., Citation2000). Therefore, any autoimmune process that increases differentiation or activation of myofibroblasts would likely contribute to the pathology. Taken together, these data suggest that the inhalation of crystalline silica or asbestos can lead to autoimmune responses that may play a role in the pulmonary pathology.
Asbestos-related lung diseases (ARD), including fibrosis, pleural plaques, and cancer, continue to present a serious and significant problem despite increasing awareness of health hazards of asbestos inhalation. Although the mechanisms leading to the progression of these conditions have not been fully explained, there is evidence that some of the lung pathologies seen with both asbestos and silica exposures are immunologically mediated (Perkins et al., Citation1993; Hamilton et al., Citation1996; Holian et al., Citation1997). We have recently shown higher frequencies of positive antinuclear antibody (ANA) tests in a population exposed to amphibole asbestos (Libby, MT), than in a control population (Pfau et al., Citation2005). In addition, a higher mean titer of the ANAs was seen with increasing severity of lung disease in the Libby cohort. We therefore wondered whether amphibole asbestos leads to autoimmune responses that may play a role in ARD progression. This is a fundamentally novel approach to understanding the etiology of chronic fibrotic diseases, in which the failure to resolve the initial damage would be attributed, at least in part, to an autoimmune process.
In order to explore the mechanisms of asbestos-induced autoimmune responses, we have developed a mouse model using C57Bl/6 mice, which have been described as non-autoimmune prone (Hudson et al., Citation2003). They have been characterized as having a primarily TH1 type of immune responsiveness to a range of infectious and non-infectious stimuli, and they are susceptible to silica- and asbestos-induced lung fibrosis (Davis et al., Citation1998; Warshamana et al., Citation2002). Following intratracheal asbestos instillation, female C57Bl/6 mice develop positive ANA tests and mild glomerulonephritis characterized by immune complex deposition, proliferation of mesangial cells, and focal glomerular sclerosis (Pfau et al., Citation2008). Nevertheless, the most obvious pathology in amphibole asbestos-exposed mice, as well as in asbestos-exposed humans, is pleural and interstitial lung fibrosis (Putnam et al., Citation2008). It therefore provides an excellent model to test whether the autoantibodies play a role in fibrotic processes.
Materials and methods
Mice
Female (8-week-old) C57Bl/6 mice used for the initial asbestos exposures were obtained from a barrier facility at The Jackson Laboratories (Bar Harbor, ME) and were maintained in microisolator cages. All animal studies were approved by the UM and/or Idaho State University Institutional Animal Care and Use Committee and were conducted in accordance with guidelines established by the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. For the exposures, mice were housed under specific pathogen-free conditions with a 12-h light/12-h dark cycle, constant temperature, and free access to food and water. A dirty bedding exposure sentinel program was used to ensure exclusion of a variety of mouse viruses and other pathogens. Euthanasia was performed by intraperitoneal (IP) injection of a lethal dose of sodium pentobarbital (Nembutol®) or CO2 asphyxiation, consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Treatment of mice
As previously described (Pfau et al., Citation2008), saline alone, Korean tremolite (NIST), or Libby amphibole (6-Mix, USGS) were intratracheally instilled with a total of two doses, each dose being 60 µg of the fibers sonicated in sterile phosphate-buffered saline (PBS, pH 7.4), given 1 week apart in the first 2 weeks of the 6–7 month experiments. Six-Mix asbestos is a mixture of amphibole fibers collected at the Libby MT vermiculite mine site. Tremolite is one of the major components of the 6-Mix from Libby MT (Meeker et al., Citation2003; Webber et al., Citation2008). For asbestos instillations, mice were anesthetized IP with ketamine and xylazine, and a 1-cm longitudinal incision was made in a shaved area from below the chin. Suspensions of 30 µl were injected into the trachea via 25 gauge needles, and the incision was closed with 3 M Vetbond tissue adhesive. At the end of the experiment, the mice were euthanized by lethal overdose of Nembutol® (5 mg/mouse, in 100 µl, IP). Blood was harvested by cardiac puncture after euthanization, and serum was collected from the blood by centrifugation following clotting at room temperature (RT). Serum samples were stored at minus 20°C until used in experiments.
Serum immunoglobulin purification or clearance
Total serum IgG was purified by Melon™ Gel IgG purification kits (Pierce, Rockford, IL), and then concentrated to a stock of 1 mg/ml using Microcon YM-10 centrifugal filter devices (Millipore Corp., Bedford, MA). Protein was quantified spectrophotometrically using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE). Alternatively, mouse serum was cleared of IgG using Protein G Agarose beads, with a modified protocol from package insert (KPL, Gaithersburg, MD). Serum was first diluted 1:3 in a binding buffer (pH 7.4) comprised of 0.1 M sodium phosphate and 0.15 M sodium chloride, then diluted again with the Protein G beads, at a 2:1 (bead to serum) ratio. These mixtures were then incubated for 2 h at 4°C with intermittent vortexing every 10–15 min for 30 s. The tubes were then put on a rotator for an overnight incubation at 4°C. The following day the beads were pelleted by centrifugation, and the cleared serum (supernatant) was placed in a clean tube. The diluted cleared serum was concentrated back to its original volume using Microcon YM-10 filters (Millipore) at 14,000 × g for 20 min. Cleared sera were shown to lack all detectable heavy and light chain IgG by SDS PAGE stained with Coomassie Blue. These samples were banked at −20°C until analysis.
Mouse fibroblasts
All cells were cultured at 37°C in a 5% CO2 incubator (Thermo Forma, Waltham, MA) in complete media: DMEM with L-glutamine (Mediatech, Herndon, VA) supplemented with 10% FBS (fetal bovine serum), 1% sodium pyruvate, 1% penicillin/streptomycin (Gibco BRL, Bethesda MD), and 0.1% 2-mercaptoethanol. L929 mouse fibroblasts were obtained from ATCC (Manassas, VA). Primary skin fibroblasts were obtained from skin tissue blocks (≈ 5 mm square) of euthanized healthy C57Bl/6 mice. The tissue blocks were dipped in 70% ethanol then washed in sterile PBS, then cultured in 6-well plates in complete media for 1 week, held down with a sterile coverslip. The fibroblasts that grew out of the skin blocks were then cultured to confluence and checked for fibroblast morphology and staining with anti-fibroblast antibody (anti-prolyl 4-hydroxylase, Abcam, Cambridge, MA). Primary lung fibroblasts were collected from untreated C57Bl/6 mice by finely mincing the lungs and then incubating the tissue for 2 h in 5 ml of collagenase buffer (1 mg/ml collagenase IA (Sigma, St. Louis, MO) in PBS with 1 µl DNAse I (Gibco, Carlsbad, CA)). The fibroblasts were located in the top interface layer following gradient centrifugation. They were washed and plated, resulting in nearly pure cultures (> 90%; based on anti-prolyl 4-hydroxylase staining) after growing to confluence and washing off nonadherent cells and debris.
AFA cell-binding assay
An AFA cell-binding assay was performed, slightly modified according to Ihn et al. (Citation2000). Briefly, fibroblasts were grown to confluence in a 96-well culture plate (Nalge Nunc, Naperville IL). The cells were then fixed by overnight incubation in 1% paraformaldehyde. After blocking the cells (1 h at RT) with 3% bovine serum albumin (BSA)/PBS, serum from mice exposed to either asbestos or saline (each diluted 1:100 in 3% BSA/PBS with 0.5% Tween-20) was added to appropriate wells in quadruplicate. A mouse anti-fibroblast (anti-prolyl 4-hydroxylase) antibody was used as a positive control. After a primary incubation of 2 h, the cells were washed three times with 3% BSA/PBS. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibodies (Jackson ImmunoResearch Labs, West Grove, PA) were then added to the wells (at 1:5000) and the cells incubated 1 h at RT before being thoroughly but gently washed with PBS. The wells then each received 100 µl chromogenic substrate (TMB) (3,3,5,5-tetramethylbenzidine) solution; after a fixed period of time, the reaction was stopped by addition of 50 µl 2 N H2SO4 to each well and the absorbance in each well was read at 450 nm on a Spectra-Max plate reader (Molecular Devices, Sunnyvale, CA). Subsequently, the experiment was repeated using live cells and wherein the incubation steps were performed at 37°C for 30 min. To prevent loss of the unfixed cells, the plates in these experiments were centrifuged briefly after each washing step.
Immunofluorescent detection of SMA
Cultured primary skin or lung fibroblasts were plated to 8-well chamber slides (Nalge Nunc) and grown to confluence. They were treated with serum or cleared serum (each diluted 1:50 from original volume in 1% FBS/DMEM) from asbestos or saline-treated mice for 24 h. The cells were fixed with ice cold acetone at -20°C for 5-10 min, rinsed twice with PBS, blocked for 1–2 h with 5% nonfat milk in PBS, then stained with mouse anti-SMA (Abcam) diluted 1:50 in blocking buffer for 1 h. After staining, the cells were washed 3x with PBS and then stained with Alexa 488-conjugated anti-mouse IgG secondary antibodies (Jackson ImmunoResearch Labs, West Grove, PA) diluted 1:200 for 1 h at RT. The wells were washed again and then counterstained with DAPI (Molecular Probes/Invitrogen, Carlsbad, CA), rinsed and cover-slipped. Pictures were taken with a Leica Fluorescence microscope using the 40× objective and IPLAB acquisition software (Biomedical Imaging Group, Lausanne, Switzerland), and all original images were saved as tiff files. Fluorescence intensity was measured using a laser scanning cytometer (LSC, CompuCyte, Cambridge, MA). For LSC, the cells were segmented as events, and counted, using nuclear contours of the DAPI staining, followed by analysis of Max Pixel green fluorescence using peripheral contours with background contour subtraction. A scan area of at least 5000 cell events was used in each analysis, and counting equal numbers of cells for each treatment group.
Collagen production
Extracellular collagen deposition by the fibroblasts was analyzed by LSC. L929 cells were grown in chamber slides to confluence and then treated with 4 µg/ml purified IgG from serum of saline or asbestos-treated mice for 3 days in media containing 5–10% FBS. The cells were washed, fixed with acetone, blocked with 5% goat serum, and then stained with rabbit anti-mouse collagen Type I antibody (Fitzgerald Inc., Concord, MA) followed by anti-rabbit secondary antibodies conjugated to Alexa 594 (Jackson ImmunoResearch Labs, West Grove, PA) diluted 1:200 in PBS with 5% goat serum. The slides were counterstained with DAPI to allow detection of nuclei for cell enumeration, and fluorescence quantified by LSC using phantom contours over equal numbers of cells for each treatment.
In addition, secreted collagen was measured using the Sircol™ Soluble Collagen Assay kit (Accurate Chemical Co., Westbury, NY) according to the manufacturer’s instructions. The fibroblasts were cultured in 6-well plates 3 days after confluence was reached, in 2 ml of complete media. Media was then changed to 5% FCS (fetal calf serum; Hyclone/ThermoScientific, Logan, UT) and the cells were treated with purified IgG from the sera of C57Bl/6 mice at 4 µg/ml for 3 days. In some experiments, asbestos, sonicated in sterile PBS, was added to the fibroblasts at 40 µg/cm2, a concentration previously shown not to be cytotoxic to these cells (data not shown). The medium was removed from the wells to 15 ml conical tubes and the collagen was precipitated using 4 M NaCl. The pelleted collagen samples and collagen standard curve dilutions were then suspended in 100 µl of 0.5 M acetic acid. The rest of the assay followed the kit protocol and the final color development was carried out in a 96-well microtiter plate, and read at 540 nm on a Spectra-Max microtiter plate reader.
ELISA for TGFβ in serum samples and in culture supernatants from treated fibroblasts
Active transforming growth factor (TGF)-β was measured using an ELISA kit from eBiosciences (San Diego, CA), following the manufacturer’s protocol. This kit was specific for active TGFβ and had a sensitivity of 60 pg/ml. We also pretreated aliquots of the serum samples with acid (1 N HCl for 5 min, followed by neutralization with NaOH) to activate latent TGFβ, in order to measure total TGFβ using the same kit. Primary mouse lung fibroblasts were cultured as above and treated with 1:50 dilution of mouse serum from saline- or asbestos-treated mice for 2 days. The culture supernatants were collected and centrifuged to remove cells and debris. A sample of cell culture media alone was used to determine the background level of TGFβ from the FBS in the media. In addition, samples from the mouse sera themselves were also similarly treated and tested for total TGFβ.
ELISA for antibodies to vimentin, TGFβ-R, Topo 1, and PDGF-Rα in mouse sera
Levels of anti-Topo 1 antibody in mouse sera were determined using a Mouse Anti-Scl-70 ELISA kit (Alpha Diagnostic International, San Antonio, TX). ELISA assay was performed according to the manufacturer’s instructions; the kit had a sensitivity of 10 ng/ml. Briefly, 100 µl of diluted mouse sera (1:100 in sample/conjugate diluent) were added to commercially coated wells and incubated for 1 h at RT. After washing the wells three times with 300 µl of kit washing buffer, 100 µl diluted goat anti-mouse IgG-HRP conjugate was added to each well and incubated for 30 min at RT. After another five washing steps, 100 µl TMB was added to each well and incubated for 15 min at RT. The reaction was terminated by adding 100 µl of Stop Solution to all wells. Absorbance was then measured on a microtiter plate reader at 450 nm (Molecular Devices). Anti-Scl-70 concentration in the samples was determined by extrapolation from the standard curve. Each sample was run in duplicate. Measurements of the amount of binding of anti-Topo I antibodies (Abcam) to L929 fibroblasts were performed by cell-based ELISA as described above for surface binding of AFA. To further demonstrate that this was surface binding only, anti-Ro52 antibody (M20, Santa Cruz Biotech, Santa Cruz, CA) was used as a ubiquitous cytoplasmic target in parallel binding experiments.
To detect the presence of antibodies to vimentin and to receptors for TGFβ (TGFβ-RII), and platelet-derived growth factor (PDGF; PDGF-Rα) in the sera, ELISA plate wells were coated overnight with 100 ng purified recombinant protein/well (R&D Systems, Minneapolis, MN) in citrate coating buffer (pH 8.0). The wells were washed and then blocked with 5% nonfat milk for 2 h. The diluted serum was added as above, and the ELISA performed as above, using 50 µl of 2 N H2SO4 as Stop Solution. Antibodies to PDGF-Rα, vimentin, or TGFβ-RII (R&D Systems) were used as positive controls at appropriate dilutions to yield 1 µg/ml final concentrations per well.
STAT-1 and SMAD2/3 nuclear translocation
To determine whether the autoantibodies had an effect on signal transduction in fibroblasts, L929 cells were plated to 8-well chamber slides and treated with pooled serum from asbestos-treated mice or pooled cleared serum (IgG removed using Protein G) for 2 h. The cells were then washed, fixed with ice cold acetone, blocked as above, and stained with rabbit antibodies to STAT-1 (H95) or SMAD2/3 (FL-425) (both Santa Cruz Biotech) followed by Alexa 488-conjugated anti-rabbit IgG secondary antibodies (Jackson ImmunoResearch Labs, West Grove, PA). Wells were then washed again and counterstained with DAPI, rinsed and cover-slipped. Fluorescence intensity (Max Pixel) in the nucleus vs. cytoplasm was measured using the LSC by segmenting events using nuclear contours based on the DAPI staining, followed by analysis of Max Pixel green fluorescence using peripheral contours with background contour subtraction. A scan area of at least 5000 cell events was used in each analysis, counting equal numbers of cells for each treatment group. Following analysis, all values were standardized to the background fluorescence (secondary antibody only), and the median fluorescence intensity (MFI) of nuclear contours was divided by MFI of peripheral contours. Translocation to the nucleus was defined as a ratio of nuclear/peripheral fluorescence that was > 1.0.
Statistical analyses
Data are expressed as raw counts within discrete categories, and expressed graphically as mean ± SEM. Comparisons of means were evaluated by an unpaired t-test. Scale-level data analysis involved analysis of variance (ANOVA) followed by Bonferroni’s correction for multiple pair-wise mean comparisons. Statistical significance was determined with the probability of Type I error at P < 0.05. Experimental designs with directional hypotheses used one-tailed P-values, whereas designs with nondirectional hypotheses used two-tailed P-values. A Grubb’s test was used to remove one statistical outlier. Data analysis was performed using PRISM software (GraphPad; San Diego, CA), and graphics were generated in Excel. All experiments were repeated at least twice, and representative data are shown.
Results
Serum antibodies from asbestos-instilled mice bind to mouse fibroblasts
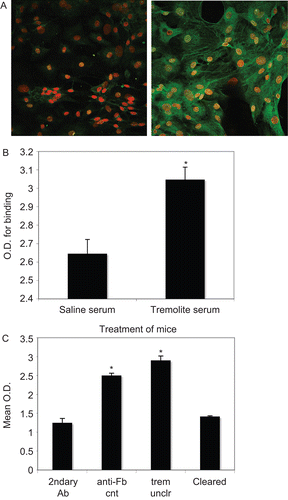
In order to test the hypothesis that serum antibodies from the asbestos-instilled mice target and affect fibroblasts, the presence of AFA in the sera of these mice was demonstrated (). shows an image of mouse primary skin fibroblasts stained with serum from saline- or tremolite asbestos-exposed mice, followed by Alexa 488 secondary antibody. Staining of fibroblasts was minimal with serum from saline-instilled mice, whereas virtually all of the cells stained brightly with serum from asbestos-instilled mice. The binding was quantified in a separate experiment using L929 mouse fibroblast cells by cell-based ELISA (). When the serum was cleared of IgG using Protein G precipitation, the optical density in the cell-based ELISA was reduced to background levels, demonstrating that the staining was due to IgG (). These results were reproduced using serum from 6-Mix exposed mice (data not shown). An alternate cell-based ELISA protocol using live mouse lung fibroblasts produced similar results, in which binding by serum antibodies from asbestos-instilled mice was nearly three times that of saline mice (OD for tremolite serum = 2.05 (SEM = 0.03); for saline serum OD = 0.72 (SEM = 0.07); secondary antibody only OD = 0.5; data not shown). Confirmation of the effect using the L929 cells allowed us to use the cell line for subsequent experiments that required larger numbers of cells.
Figure 1. Binding of serum antibodies to mouse fibroblasts indicates presence of anti-fibroblast antibodies in serum from asbestos-instilled mice. (A) Mouse primary skin fibroblasts were fixed with paraformaldehyde and stained using serum from (left) saline- or (right) tremolite-exposed mice as the primary antibody, followed by incubation with Alexa-488-conjugated anti-mouse IgG secondary Ab. Nuclei are stained with propidium iodide. Images (at 400×) are representative of multiple experiments. (B) A cell-based ELISA was performed using L929 mouse fibroblasts, to measure the binding of serum antibodies to the cells. As in A, binding of serum antibodies was compared between sera from saline- or tremolite-treated mice. N = 7, *P < 0.05 by unpaired 2-tailed t-test. (C) The cell-based ELISA was repeated using L929 cells, and including a positive control anti-fibroblast antibody (anti-Fb, anti-prolyl-4-hydroxy-lase), as well as serum from tremolite-exposed mice, either uncleared of IgG (trem unclr) or that had been cleared of IgG by Protein G precipitation (cleared). N = 4, *P < 0.05 compared to secondary antibody-only control.
Serum antibodies from asbestos-instilled mice induced expression of SMA
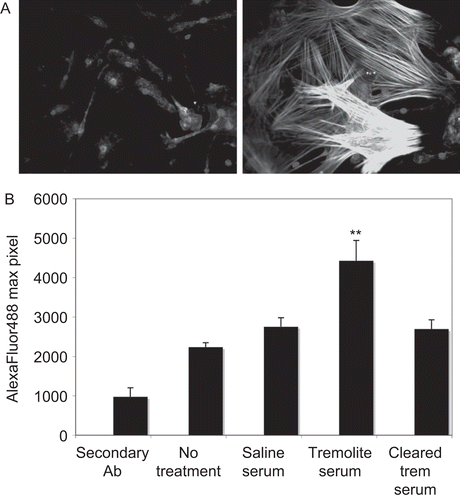
We next tested the hypothesis that these AFA would alter the phenotype of the cells to a myofibroblast phenotype by staining for expression of SMA. First, using primary mouse skin fibroblasts there was a dramatic induction of SMA expression when the cells were treated with serum from asbestos-instilled mice for 24 h (). The left panel shows fibroblasts treated with serum from saline-treated mice, with normal fibroblast morphology and no actin stress fibers surrounding the nuclei. The right panel shows fibroblasts treated with serum from asbestos-treated mice, where actin stress fibers are highly expressed and the cells are enlarged and extended. To confirm the effect in primary lung fibroblasts, the expression of SMA in response to serum treatment was quantified by LSC, and the fluorescence intensity of SMA expression in these cells was significantly increased compared to untreated cells and those treated with serum from saline-instilled mice (). To demonstrate that this effect was due to serum immunoglobulin and not another serum factor, SMA expression was measured after treatment with serum that was cleared of IgG using Protein G precipitation. In this case, the increased expression of SMA was abrogated (). Data shown here were generated using serum from tremolite-exposed mice, but the serum from 6-Mix exposed mice yielded similar results.
Figure 2. Expression of smooth muscle α-actin (SMA) is induced by treatment with sera from asbestos-exposed mice. (A) Mouse primary skin fibroblasts treated with sera from (left panel) saline- or (right panel) tremolite-exposed mice for 24 h were then fixed, permeabilized, and stained with anti-SMA. The secondary antibody was Alexa 488-conjugated and fluorescence was visualized at 400×. (B) The experiment was repeated using C57Bl/6 primary lung fibroblast cells, and fluorescence from the SMA expression was quantified by LSC as described in Materials and methods section. N = 3, **P < 0.01 comparing SMA expression induced by tremolite mouse serum to expression by untreated cells, using one-way ANOVA with Bonferroni’s Multiple Comparison Test.
Serum antibodies from asbestos-instilled mice induced production of Type I collagen from L929 cells
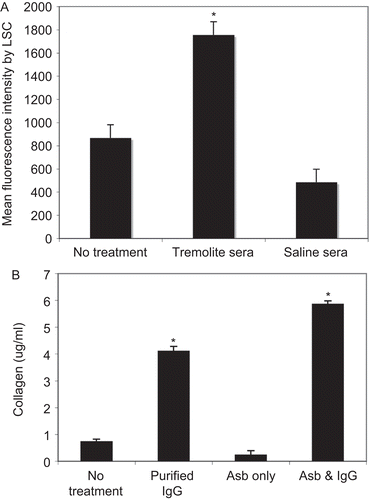
In order to assess the functional activity of the differentiating fibroblasts, the extracellular production of collagen was measured. For this experiment, purified IgG from the serum was used in place of the whole serum in order to further demonstrate that the effect was not due to other serum factors. shows that serum IgG from tremolite-instilled, but not saline-instilled mice, induced expression of collagen as shown by significantly increased fluorescence staining with anti-collagen Type I antibodies, measured by LSC. Collagen production by the L929 cells was next measured via Sirius Red staining and quantification against a standard curve. Although asbestos (tremolite) itself did not increase the collagen production by L929 fibroblasts, purified serum IgG, with or without asbestos, led to significantly elevated collagen production above that of untreated fibroblasts (). There was also a slight increase in this production above the IgG-alone treatment value when cells were treated with both IgG and asbestos.
Figure 3. Secretion of Type I collagen was induced by purified IgG from asbestos-exposed mice. (A) L929 cells were grown to confluence, treated with purified IgG from saline- or tremolite-exposed mice for 3 days, and then collagen production was detected using anti-collagen Type I antibody followed by Alexa 488-conjugated secondary antibody. Fluorescence was quantified by LSC. (B) Collagen production by L929 cells was also measured by Sircol assay, as described in Materials and methods section. Purified IgG = IgG from tremolite-exposed mice, Asb = tremolite asbestos alone at 40 µg/cm2, Asb and IgG = combined treatment with both tremolite and purified IgG from tremolite-instilled mice. N = 4 in each group, *P < 0.05 using one-way ANOVA with Bonferroni’s Multiple Comparison Test.
TGFβ was not significantly increased in sera of asbestos-treated mice
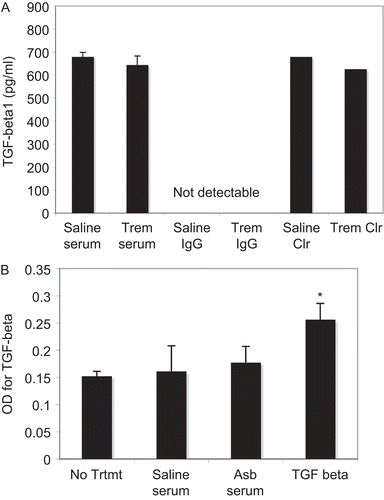
None of the serum samples from the asbestos-treated mice contained increased levels of total or active TGFβ, compared to the serum from saline-treated mice. illustrates that all of the samples, including the cleared serum samples, had < 1 ng/ml of total TGFβ. Therefore, it was not the presence of increased TGFβ in the serum that induced the effects on fibroblast phenotype. also shows that no TGFβ was detectable in the purified IgG samples. In addition, primary mouse lung fibroblasts did not produce TGFβ in response to treatment with serum antibodies from asbestos-exposed mice (). Therefore, the effect of the serum antibodies on SMA and collagen in our assays were not due to autocrine responses involving TGFβ released from the treated fibroblasts.
Figure 4. ELISA for TGFβ in mouse serum and fibroblast culture supernatants. (A) Sera from saline- or asbestos-exposed mice were assayed for total TGFβ1 using an ELISA kit. N = 8 mice in each group. (B) Total TGFβ1 was also measured in supernatant samples from cultures of primary mouse lung fibroblasts exposed to serum from saline- and asbestos-exposed mice or to recombinant TGFβ at 4 ng/ml. N = 3, *P < 0.05 compared to No Treatment group.
Asbestos-treated mice had developed antibodies to DNA Topo 1 and PDGF-Rα, but not to vimentin or the murine receptor for TGFβ (TGFβ-RII)
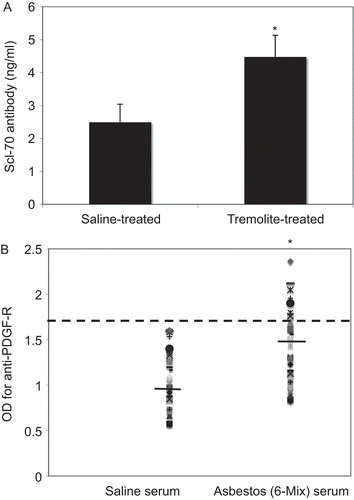
We hypothesized that the AFA antibody might be targeting any of several fibroblast proteins for which antibodies have been shown to activate fibroblasts, so we used ELISA assays to detect specific antibodies in the sera of the treated mice. There was no evidence of antibodies to TGFβ-RII or to vimentin, comparing sera from saline- and asbestos-treated mice (data not shown). Antibodies to DNA Topo 1 were detected at a significantly higher level in asbestos-treated mouse sera, compared to saline-treated mice (). Because it has been hypothesized that the binding of anti-Topo 1 antibodies to fibroblasts is to a molecular mimic on the surface of cells (Hénault et al., Citation2004), we tested the binding of commercial anti-Topo 1 antibodies to the surface of L929 fibroblasts. On fixed but non-permeabilized cells, there was a high level of staining by anti-Topo 1 antibodies; however, the binding did not appear to saturate even at high concentrations of antibody (100 µg/ml; data not shown). The fact that there was no staining of the ubiquitous cytoplasmic protein, Ro52, suggests that this anti-Topo 1 binding was on the exterior of the cell (data not shown). In addition, antibodies to PDGF-Rα were detected in sera from asbestos-treated mice, showing a significantly higher mean OD in serum from the asbestos-treated mice, with ≈25% of the asbestos-treated mice having an absorbance value that exceeded two standard deviations above the mean for the saline-treated group (). There was no statistically significant difference in the mean OD for mice instilled with 6-Mix compared to tremolite-treated mice (data not shown).
Figure 5. ELISA for antibodies to DNA topoisomerase I and PDGF-Rα. (A) The presence of anti-Topo I antibodies (Scl-70) were detected at a significantly higher level in the sera of asbestos-instilled mice by Scl-70 ELISA. N = 5 mice, *P < 0.05 by unpaired t-test. (B) ELISA was used to detect antibodies to PDGF-Rα. Scatter graph shows the OD values for individual mice in each group, with the mean for that groups indicated by a solid line. A hypothetical cut-off for positive/negative (dotted line) was calculated as two standard deviations above the mean for the saline group. *P < 0.05, N = 24 mice (combined mice from tremolite and 6-mix exposures).
Activation of STAT-1 with serum from asbestos-treated mice
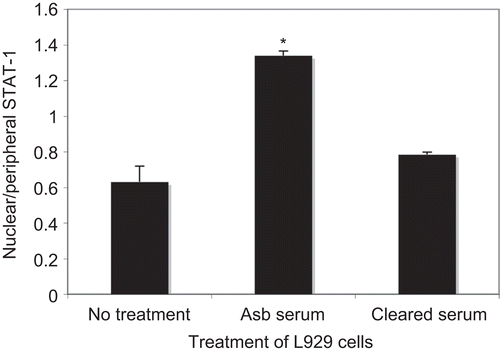
Treatment of L929 cells with serum from asbestos-treated mice led to activation of STAT-1 with translocation to the nucleus (). Cleared serum lost the ability to activate STAT-1, implicating AFA in this activation. However, SMAD2/3 was not shown to translocate following the same treatment (data not shown).
Figure 6. STAT-1 translocates to the nucleus of treated L929 fibroblasts following treatment with asbestos-treated mouse serum. STAT-1 translocation was measured by LSC as described in the Materials and methods section. L929 cells were treated for 2 h with serum or cleared serum (1:50) from asbestos-treated mice, then fixed and stained for STAT-1. Max Pixel fluorescence intensity was determined in nuclear and peripheral contours, and data are expressed as the ratio of nuclear/peripheral staining intensity. N = 3 wells per treatment, *P < 0.05.
Discussion
The possible role of autoantibodies to fibroblasts, endothelial, and epithelial cells in vascular and fibrotic disorders is receiving considerable attention as the evidence of their pathogenicity expands. Antibodies to endothelial cells have been implicated in vasculitis (Del Papa et al., Citation1994), SSc (Ihn et al., Citation2000), and SLE (Renaudineau et al., Citation2002). Anti-epithelial cell antibodies are being studied in CFA (Singh and du Bois, Citation2001) and nonallergic asthma (Nahm et al., Citation2002). Serum autoantibodies in scleroderma patients have been shown to bind to fibroblasts and activate differentiation to myofibroblasts, which are pro-fibrogenic (Chizzolini et al., Citation2002). Therefore, our central hypothesis was that asbestos exposure induces autoimmune responses that produce AFA. By binding to target proteins on fibroblasts, these antibodies could then exacerbate chronic inflammatory conditions responsible for asbestos-related fibrosis by increasing fibroblast pro-fibrotic and pro-inflammatory activities. This study has direct relevance to understanding the common mechanisms in other autoimmune diseases, as well as disease processes not previously identified as autoimmune. Further understanding of the complex cellular etiology of pulmonary fibrosis will have the potential to provide an approach to prevention and development of therapeutic modalities for fibrotic disorders.
Exposure to asbestos and silica lead to patterns of illness that are very similar to systemic autoimmune diseases including both pulmonary fibrosis and serum ANA (Nigam et al., Citation1993; Cooper et al., Citation2002; Pfau et al., Citation2005). Although the mechanisms leading to the progression of these conditions have not been fully explained, there is evidence that some of the lung pathologies seen with both asbestos and silica exposures are immunologically mediated (Perkins et al., Citation1993; Hamilton et al., Citation1996; Holian et al., Citation1997). Although many of the cells and mediators involved in the initiation of silicate-associated pulmonary fibrosis have been studied, the reasons for the chronic and progressive nature of silicosis and asbestosis remain unclear. Despite differences in the histology and location of fibrotic lesions in asbestosis and silicosis, both have been associated with the presence of myofibroblasts characterized by an expression of SMA and active collagen production (Perdue and Brody, Citation1994; Mariani et al., Citation1996; Kaarteenaho-Wiik et al., Citation2000). In addition, scleroderma has long been characterized by the presence of SMA-expressing myofibroblasts (Sappino et al., Citation1990; Kirk et al., Citation1995). Because of the potential role of AFA in SSc fibrosis, we hypothesized that there may be a similar mechanism driving the pulmonary fibrosis from asbestos exposure.
These data showing the binding of serum antibodies from asbestos-exposed mice to mouse fibroblasts and their ability to induce a phenotypic change from normal fibroblasts to myofibroblasts suggest that asbestos-induced fibrosis could have an autoimmune component. The importance of this study is evident from other data related to the occupational and environmental asbestos exposures that have occurred in Libby, MT as a result of asbestos-contaminated vermiculite mining near the community. The ARD seen in Libby includes interstitial fibrosis, pleural disease, carcinoma, and mesothelioma, with apparently rapid progression in at least a subset of the patients (Whitehouse, Citation2004). Also, an increased frequency of positive ANA tests was found in the Libby serum samples compared to a matched control population (Missoula, MT) (Pfau et al., Citation2005). Interestingly, analysis of the data showed a significant association of positive ANA tests in subjects with severe lung disease. These data support a study by Tamura et al., in which positive ANAs were more frequent in asbestos-exposed subjects who had pulmonary lesions (Tamura et al., Citation1993). A 3-year follow-up study of those patients demonstrated that positive ANAs were correlated with the progression of asbestosis (Tamura et al. Citation1996). Similar trends indicating a rapid progression of pulmonary fibrosis in ANA-positive asbestosis patients were also reported by Turner-Warwick in two separate studies (Turner-Warwick and Parkes, Citation1970; Ronda et al., Citation2002). Taken together, these data suggested that there might be an autoimmune component to these pulmonary pathologies, emphasizing the importance of mechanistic studies.
The data reported here clearly show that AFA are present in the serum of asbestos-treated C57Bl/6 mice, binding both permeabilized and non-permeabilized cells. It was important to include assessment of the intracellular presence of the antibodies since Ronda et al. (Citation2002) have shown that AFA are internalized within an hour by a non-Fc receptor dependent mechanism. Therefore, using only live unfixed cells for the binding assay might underestimate the amount of binding since the secondary antibodies may not be similarly internalized. Concerns about nonspecific and heterophile binding of serum autoantibodies have led to several studies that have fine-tuned the methods for detecting these autoantibodies that are not generally measurable by standard autoimmune tests such as ANA assays (Revelen et al., Citation2000, Citation2002; Youinou et al., Citation2006), and the methods used in this study were consistent with these reports. In addition, our study further demonstrated the specificity of binding by comparing to control sera from saline-exposed mice. In all functional studies, the treatment was performed in the presence of 5–10% FCS, thereby blocking heterophile binding.
Dermal fibroblasts in SSc have a unique pro-inflammatory phenotype including expression of SMA, IL-1α and β, and IL-6 (Kawaguchi, Citation1994; Kadono et al., Citation1998; Jelaska and Korn, Citation2000). These myofibroblast cells are implicated in fibrosis through deposition of collagen (Jelaska and Korn, Citation2000); therefore, any factor that enhances or prolongs this phenotypic change could contribute to progressive fibrosis. Chizzolini et al. (Citation2002) and more recently Fineschi et al. (Citation2008) have shown that autoantibodies to fibroblasts can cause increased adhesion molecules and cytokine expression suggestive of pro-fibrotic fibroblast activation. Consistent with those studies, the AFA in sera of asbestos-treated mice induced a myofibroblast phenotype as indicated by increased SMA expression, as well as collagen production.
Because serum cleared of IgG was unable to induce SMA, and purified IgG from the asbestos-treated mice did, we show that the AFA primarily involved in the fibroblast activation is IgG, and not some other serum factor. However, we also wanted to know whether high levels of TGFβ in the serum could also contribute to the potentiation of fibrosis following asbestos exposure in mice. TGFβ signaling is a major factor in the induction of fibrosis in both mice and humans. However, at 6 months after exposure, when AFA were present in the sera, the asbestos-exposed mice did not show any increased serum TGFβ. Although we did not test for local TGFβ production in the lung, we did treat primary mouse lung fibroblasts with either asbestos or the antibodies from the asbestos-exposed mice in vitro, and showed no increased production of TGFβ. Therefore, the activation of fibroblasts by the asbestos-induced AFA, at least in vitro, appears to be independent of TGFβ.
We also wondered whether a particular immunoglobulin subclass was responsible for the binding. In the present study, the secondary antibody was specific for IgG and recognized most subclasses of mouse IgG, according to the manufacturer. In our previous study, characterizing the autoimmune response to asbestos in C57Bl/6 mice, we showed that there were no significant differences in the relative amounts of the various Ig classes and IgG subclasses between the saline- and asbestos-treated mice (Pfau et al., Citation2008). Because of the potential importance of IgA in the lung, we have also wondered whether the ANA in asbestos-exposed subjects were of the IgA isotype. Although the relative amounts of serum IgA was higher in our asbestos-exposed population compared to the control group, when the ANA screening was performed using an anti-IgA secondary antibody, none of the ANA tests were positive (Pfau et al., Citation2005). It would nevertheless be important to show the presence of these AFA in vivo and to determine the isotype of any AFA present in the lung.
Consistent with previous studies in SSc patients, in which some of the AFA appear to bind to DNA topoisomerase I, we showed that the asbestos-exposed mice had significantly increased reactivity to Scl-70, the active form of DNA Topo I. In addition, commercial anti-Topo 1 antibodies were used to test whether these antibodies could be binding to the surface of the cells. Although there appeared to be binding by anti-Topo 1 antibodies, the binding did not saturate even at very high concentrations. Although anti-Topo 1 antibodies from scleroderma patients have been shown to bind to fibroblasts (Hénault et al., Citation2004), these antibodies have not been shown to directly activate fibroblasts (Fineschi et al., Citation2007). It remains unclear what the anti-Topo 1 antibodies are binding to on fibroblasts, since normally DNA topoisomerase 1 is sequestered in the nucleus. Hénault showed that apoptotic cells release Topo 1 and that it binds to the surface of fibroblasts, thereby allowing anti-Topo 1 to bind (Hénault et al., Citation2006). Subsequently, those antibodies could recruit macrophages via Fc receptors, leading to localized inflammation (Hénault et al., Citation2006). Therefore, anti-Topo 1 antibodies may play an indirect role in activation of inflammatory and fibrotic events.
Other studies have explored possible antigenic targets for AFA, and have shown that various patient groups have antibodies to vimentin, and the receptors for TGFβ or PDGF-A. Vimentin is an intermediate filament that forms part of the cytoskeleton and plays active roles in migration, movement and cellular activation. Antibodies to vimentin have been found in patients with IPF (Yang et al., Citation2002). However, asbestos-exposed mice did not show any serum reactivity to vimentin. On fibroblasts, TGFβ1signals via TGFβ-RII and leads to SMA expression and collagen deposition (Rahimi and Leof, Citation2007). Therefore, antibodies to this receptor could conceivably mimic these effects. Although we hypothesized that TGFβ1 antibodies would be found in the asbestos-exposed mice, these results were also negative. Finally, antibodies to PDGF-Rα have been found in patients with scleroderma, and the antibodies were found to activate myofibroblast transformation (Baroni et al., Citation2006). At least 25% of the asbestos-treated mice had elevated levels of antibodies to PDGF-Rα, whereas none of the saline-treated mice did. Interestingly, asbestos exposure has been shown to significantly upregulate the expression of PDGF-R on the surface of fibroblasts in rats prior to the development of fibrosis (Lasky et al., Citation1998). This may help explain the apparent additive effects of tremolite asbestos with the purified IgG (see ) in that the asbestos may upregulate expression of the target receptor for the AFA. The AFA was bound to primary fibroblasts from both skin and lung, indicating that the target was not specific to the lung. However, upregulation of the target by concomitant exposure to asbestos might exacerbate local effects. Therefore, further study of the potential role of anti-PDGF-R antibodies in asbestos-induced fibrosis are clearly warranted. Because not all of the mice were positive for anti-PDGF-R, there are likely other AFA in the asbestos-treated mice that may contribute to the effects on lung fibroblasts. It will obviously be of critical importance to examine the sera of asbestos-exposed humans for the presence of these autoantibodies.
In order to determine a possible pathway by which the AFA could be activating the fibroblasts, two transcription factors known to be involved in activation of fibroblasts were tested for translocation to the nucleus following treatment with serum from the asbestos-treated mice. STAT-1 is used by the PDGF-R pathway, and SMAD2/3 is in the TGFβ pathway. The fact that STAT-1, but not SMAD2/3, was activated by the AFA suggests that the anti-PDGF-R antibodies may be active in binding and signaling through the receptor. Further research to define this pathway and the specific antibodies involved is needed.
Conclusion
These data show that asbestos exposure in mice induces autoantibodies that bind and activate normal fibroblasts to express a myofibroblast-like phenotype, suggesting a possible role of autoimmunity in the progression of asbestos-induced fibrosis. Antibodies to DNA Topo 1 and PDGF-Rα were shown to be components of the AFA in asbestos-exposed mice, both of which could potentially contribute to the pro-fibrotic fibroblast phenotype. While these data do not suggest that all pulmonary fibrosis has an autoimmune component, they do strongly support further exploration of the potential for autoantibodies to contribute to some of the relentlessly progressive nature of the disease.
Acknowledgments
The Authors acknowledge the support of Pamela Shaw and the CEHS Fluorescence Cytometry Core Facility, and thank Marvin J. Fritzler, MD, PhD for consultation on the project. We gratefully acknowledge contributions by undergraduate researchers Kip Katseanes and Jeremy Roe at Idaho State University. J.J. Sentissi is currently with Southern Research Institute, Birmingham AL.
Declaration of interest
This work was funded by NIH grants P20 NCRR 017670 (CoBRE) and R21 ES-012956, and ISU FRC Grant # 681-543-20, with support from NIH P20 (INBRE) NCRR 016454.
References
- Baroni, S. S., Santillo, M., Bevilacqua, F., Luchetti, M., Spadoni, T., Mancini, M., Fraticelli, P., Sambo, P., Funaro, A., Kazlauskas, A., Avvedimento, E. V., and Gabrielli, A. 2006. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 354:2667–2676.
- Chizzolini, C., Raschi, E., Rezzonico, R., Testoni, C., Mallone, R., Gabrielli, A., Facchini, A., Del Papa, N., Borghi, M. O., Dayer, J. M., and Meroni, P. L. 2002. Autoantibodies to fibroblasts induce a proadhesive and proinflammatory fibroblast phenotype in patients with systemic sclerosis. Arthritis Rheum. 46:1602–1613.
- Conrad, K., Tan, E. M., Humbel, R. L., and Shoenfeld, Y. 1997. Autoantibodies - diagnostic, pathogenic and prognostic relevance. Clin Exp Rheumatol. 15:457–465.
- Cooper, G. S., Miller, F. W., and Germolec, D. R. 2002. Occupational exposures and autoimmune diseases. Int Immunopharmacol. 2:303–313.
- Davis, G. S., Leslie, K. O., and Hemenway, D. R. 1998. Silicosis in mice: effects of dose, time, and genetic strain. J Environ Pathol Toxicol Oncol. 17:81–97.
- Del Papa, N., Conforti, G., Gambini, D., La Rosa, L., Tincani, A., D’Cruz, D., Khamashta, M., Hughes, G. R., Balestrieri, G., and Meroni, P. L. 1994. Characterization of the endothelial surface proteins recognized by anti-endothelial antibodies in primary and secondary autoimmune vasculitis. Clin Immunol Immunopathol. 70:211–216.
- Fineschi, S., Cozzi, F., Burger, D., Dayer, J. M., Meroni, P. L., and Chizzolini, C. 2007. Anti-fibroblast antibodies detected by cell-based ELISA in systemic sclerosis enhance the collagenolytic activity and matrix metalloproteinase-1 production in dermal fibroblasts. Rheumatology (Oxford) 46:1779–1785.
- Fineschi, S., Goffin, L., Rezzonico, R., Cozzi, F., Dayer, J. M., Meroni, P. L., and Chizzolini, C. 2008. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis Rheum. 58:3913–3923.
- Hamilton, R. F., Iyer, L. L., and Holian, A. 1996. Asbestos induces apoptosis in human alveolar macrophages. Am J Physiol. 271:L813–L819.
- Haustein, U. F., and Anderegg, U. 1998. Silica induced scleroderma - clinical and experimental aspects. J Rheumatol. 25:1917–1926.
- Hénault, J., Tremblay, M., Clément, I., Raymond, Y., and Senécal, J. L. 2004. Direct binding of anti-DNA topoisomerase I autoantibodies to the cell surface of fibroblasts in patients with systemic sclerosis. Arthritis Rheum. 50:3265–3274.
- Hénault, J., Robitaille, G., Senécal, J. L., and Raymond, Y. 2006. DNA topoisomerase I binding to fibroblasts induces monocyte adhesion and activation in the presence of anti-topoisomerase I autoantibodies from systemic sclerosis patients. Arthritis Rheum. 54:963–973.
- Holian, A., Uthman, M. O., Goltsova, T., Brown, S. D., and Hamilton, R. F., Jr.. 1997. Asbestos and silica-induced changes in human alveolar macrophage phenotype. Environ Health Perspect. 105 Suppl 5:1139–1142.
- Hudson, C. A., Cao, L., Kasten-Jolly, J., Kirkwood, J. N., and Lawrence, D. A. 2003. Susceptibility of lupus-prone NZM mouse strains to lead exacerbation of systemic lupus erythematosus symptoms. J Toxicol Environ Health Part A. 66:895–918.
- Ihn, H., Sato, S., Fujimoto, M., Igarashi, A., Yazawa, N., Kubo, M., Kikuchi, K., Takehara, K., and Tamaki, K. 2000. Characterization of autoantibodies to endothelial cells in systemic sclerosis (SSc): association with pulmonary fibrosis. Clin Exp Immunol. 119:203–209.
- Jelaska, A., and Korn, J. H. 2000. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 43:2230–2239.
- Jindal, S. K., and Agarwal, R. 2005. Autoimmunity and interstitial lung disease. Curr Opin Pulm Med. 11:438–446.
- Kadono, T., Kikuchi, K., Ihn, H., Takehara, K., and Tamaki, K. 1998. Increased production of interleukin 6 and interleukin 8 in scleroderma fibroblasts. J Rheumatol. 25:296–301.
- Kaarteenaho-Wiik, R., Lakari, E., Soini, Y., Pöllänen, R., Kinnula, V. L., Pääkkö, P. 2000. Tenascin expression and distribution in pleural inflammatory and fibrotic diseases. J Histochem Cytochem. 48:1257–1268.
- Kawaguchi, Y. 1994. IL-1 alpha gene expression and protein production by fibroblasts from patients with systemic sclerosis. Clin Exp Immunol. 97:445–450.
- Kirk, T. Z., Mark, M. E., Chua, C. C., Chua, B. H., and Mayes, M. D. 1995. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J Biol Chem. 270:3423–3428.
- Lasky, J. A., Tonthat, B., Liu, J. Y., Friedman, M., and Brody, A. R. 1998. Up-regulation of the PDGFα receptor precedes asbestos-induced lung fibrosis in rats. Am J Respir Crit Care Med. 157:1652–1657.
- Mariani, T. J., Roby, J. D., Mecham, R. P., Parks, W. C., Crouch, E., and Pierce, R. A. 1996. Localization of type I pro-collagen gene expression in silica-induced granulomatous lung disease and implication of transforming growth factor-β as a mediator of fibrosis. Am J Pathol. 148:151–164.
- Meeker, G., Bern, A., Brownfield, I., Lowers, H., Sutley, S., Hoefen, T., and Vance, J. 2003. The Composition and morphology of amphiboles from the Rainy Creek Complex, near Libby, Montana. Am Mineral. 88:1955–1969.
- Nahm, D. H., Lee, Y. E., Yim, E. J., Park, H. S., Yim, H., Kang, Y., and Kim, J. K. 2002. Identification of cytokeratin 18 as a bronchial epithelial autoantigen associated with non-allergic asthma. Am J Respir Crit Care Med. 165:1536–1539.
- Nigam, S. K., Suthar, A. M., Patel, M. M., Karnik, A. B., Dave, S. K., Kashyap, S. K., and Venkaiah, K. 1993. Humoral immunological profile of workers exposed to asbestos in asbestos mines. Indian J Med Res. 98:274–277.
- Perdue, T. D., and Brody, A. R. 1994. Distribution of transforming growth factor-β1, fibronectin, and smooth muscle actin in asbestos-induced pulmonary fibrosis in rats. J Histochem Cytochem. 42:1061–1070.
- Perkins, R. C., Scheule, R. K., Hamilton, R., Gomes, G., Freidman, G., and Holian, A. 1993. Human alveolar macrophage cytokine release in response to in vitro and in vivo asbestos exposure. Exp Lung Res. 19:55–65.
- Pfau, J. C., Sentissi, J. J., Weller, G., and Putnam, E. A. 2005. Assessment of autoimmune responses associated with asbestos exposure in Libby, MT, USA. Environ Health Perspect. 113:25–30.
- Pfau, J. C., Sentissi, J. J., Li, S., Calderon-Garciduenas, L., Brown, J. M., and Blake, D. J. 2008. Asbestos-induced autoimmunity in C57BL/6 mice. J Immunotoxicol. 5:129–137.
- Prakash, U. B. 1998. Respiratory complications in mixed connective tissue disease. Clin Chest Med. 19:733–46, ix.
- Putnam, E. A., Smartt, A., Groves, A., Schwanke, C., Brezinski, M., and Pershouse, M. A. 2008. Gene expression changes after exposure to six-mix in a mouse model. J Immunotoxicol. 5:139–144.
- Rahimi, R. A., and Leof, E. B. 2007. TGFβ signaling: A tale of two responses. J Cell Biochem. 102:593–608.
- Renaudineau, Y., Dugue, C., Dueymes, M., and Youinou, P. 2002. Anti-endothelial cell antibodies in systemic lupus erythematosus. Autoimmun Rev. 1:365–372.
- Revelen, R., Bordron, A., Dueymes, M., Youinou, P., and Arvieux, J. 2000. False positivity in a cyto-ELISA for anti-endothelial cell antibodies caused by heterophile antibodies to bovine serum proteins. Clin Chem. 46:273–278.
- Revelen, R., D’Arbonneau, F., Guillevin, L., Bordron, A., Youinou, P., and Dueymes, M. 2002. Comparison of cell-ELISA, flow cytometry and Western blotting for the detection of anti-endothelial cell antibodies. Clin Exp Rheumatol. 20:19–26.
- Ronda, N., Gatti, R., Giacosa, R., Raschi, E., Testoni, C., Meroni, P. L., Buzio, C., and Orlandini, G. 2002. Antifibroblast antibodies from systemic sclerosis patients are internalized by fibroblasts via a caveolin-linked pathway. Arthritis Rheum. 46:1595–1601.
- Sappino, A. P., Masouyé, I., Saurat, J. H., and Gabbiani, G. 1990. Smooth muscle differentiation in scleroderma fibroblastic cells. Am J Pathol. 137:585–591.
- Singh, S., and du Bois, R. 2001. Autoantibodies in cryptogenic fibrosing alveolitis. Respir Res. 2:61–63.
- Tamura, M., Liang, D., Tokuyama, T., Yoneda, T., Kasuga, H., Narita, N., Sada, K., Miyazaki, R., and Okada, S. 1993. [Study on the relationship between appearance of autoantibodies and chest X-ray findings of asbestos plant employees]. Sangyo Igaku. 35:406–412.
- Tamura, M., Tokuyama, T., Kasuga, H., Yoneda, T., Miyazaki, R., and Narita, N. 1996. [Study on correlation between chest X-P course findings and change in antinuclear antibody in asbestos plant employees]. Sangyo Eiseigaku Zasshi. 38:138–141.
- Tan, F. K., Arnett, F. C., Antohi, S., Saito, S., Mirarchi, A., Spiera, H., Sasaki, T., Shoichi, O., Takeuchi, K., Pandey, J. P., Silver, R. M., LeRoy, C., Postlethwaite, A. E., and Bona, C. A. 1999. Autoantibodies to the extracellular matrix microfibrillar protein, fibrillin-1, in patients with scleroderma and other connective tissue diseases. J Immunol. 163:1066–1072.
- Turner-Warwick, M., and Parkes, W. R. 1970. Circulating rheumatoid and antinuclear factors in asbestos workers. Br Med J. 3:492–495.
- Warshamana, G. S., Pociask, D. A., Sime, P., Schwartz, D. A., and Brody, A. R. 2002. Susceptibility to asbestos-induced and transforming growth factor-β1-induced fibroproliferative lung disease in two strains of mice. Am J Respir Cell Mol Biol. 27:705–713.
- Webber, J. S., Blake, D. J., Ward, T. J., and Pfau, J. C. 2008. Separation and characterization of respirable amphibole fibers from Libby, Montana. Inhal Toxicol. 20:733–740.
- Whitehouse, A. C. 2004. Asbestos-related pleural disease due to tremolite associated with progressive loss of lung function: serial observations in 123 miners, family members, and residents of Libby, Montana. Am J Ind Med. 46:219–225.
- Yang, J. M., Hildebrandt, B., Luderschmidt, C., and Pollard, K. M. 2003. Human scleroderma sera contain autoantibodies to protein components specific to the U3 small nucleolar RNP complex. Arthritis Rheum. 48:210–217.
- Yang, Y., Fujita, J., Bandoh, S., Ohtsuki, Y., Yamadori, I., Yoshinouchi, T., and Ishida, T. 2002. Detection of anti-vimentin antibody in sera of patients with idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. Clin Exp Immunol. 128:169–174.
- Youinou, P., Le Dantec, C., Bendaoud, B., Renaudineau, Y., Pers, J. O., and Jamin, C. 2006. Endothelium, a target for immune-mediated assault in connective tissue disease. Autoimmun Rev. 5:222–228.