Abstract
Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, possesses potent anti-tumor activity against a variety of malignant cells. However, its action on lymphocytes and the underlying mechanism are not completely understood. In this study, we aimed to analyze the effects of VPA on the proliferation, activation, and apoptosis of murine lymphocytes activated with concanavalin A (ConA). Our results showed that VPA inhibited the proliferation of ConA-activated lymphocytes in a dose-dependent manner. Low-dose VPA (≤1.1 mM) enhanced CD69 expression on the activated lymphocytes, whereas at high doses (≥ 3.3 mM) it decreased CD69 expression. Furthermore, VPA reduced activation-induced apoptotic cell death at low doses, but at high doses it promoted apoptotic cell death of activated lymphocytes dramatically. It was found that the Bax/Bcl-2 ratio and phosphorylation of histone H2A.X was decreased at low doses of VPA but was increased at high doses. The phosphorylation of STAT3 was also differentially regulated by different doses of VPA. VPA, at 5 mM induced the phosphorylation of p38 but not JNK and extracellular signal-regulated kinase (ERK)1/2. In addition, VPA induced a dose-dependent increase in the acetylation of histone H3. These results demonstrate that VPA exhibits dose-dependent biphasic effect on apoptosis of activated lymphocytes probably through differential modulation of several apoptosis-related signaling pathways.
Introduction
Valproic acid (VPA) has been used for decades in the treatment of epilepsy (Johannessen and Johannessen, Citation2003). It has also been found to be a histone deacetylase (HDAC) inhibitor (Phiel et al., Citation2001). Notably, a significant increase in HDAC activity is observed in almost all malignant cells with altered chromatin structure, gene transcription, and cell differentiation (Smith and Workman, Citation2009). As an HDAC inhibitor, VPA has been reported to inhibit the proliferation of several transformed cells (Cinatl et al., Citation2002; Fortunati et al., Citation2004) and to induce apoptosis in a variety of human cancers (Kawagoe et al., Citation2002; Takai et al., Citation2004). Although the underlying mechanism by which apoptosis of transformed cells in response to VPA treatment is not completely elucidated, it was reported that in endometrial cancer cells, VPA stimulated both caspase-dependent and -independent apoptotic signaling pathways (Michaelis et al., Citation2005).
Due to its use as an anti-epilepsy agent, many studies of VPA have focused on the neural system. It has been demonstrated that VPA has neuroprotective effects. For example, VPA could protect rat cortical neurons from glutamate-induced excitotoxicity (Hashimoto et al., Citation2002) and prolong the lifespan of cultured cortical neurons (Jeong et al., Citation2003). The neuronal protection afforded by VPA treatment might be mediated through the extracellular signal-regulated kinases (ERK) pathway and via the inhibition of pro-apoptotic molecules such as Bcl-2 (Pérez et al., Citation2003). Interestingly, VPA (1 mM) could inhibit apoptosis in endothelial cells through inducing ERK1/2 activation (Michaelis et al., Citation2005). In contrast, in microglia, VPA could induce apoptosis via p38 mitogen-activated protein kinase (MAPK) and mitochondrial apoptosis pathway (Xie et al., Citation2010). These studies suggest that the effects of VPA may depend on the cell types and dosages used in different investigations.
Recently, the immunosuppressive effects of VPA have also been noted by investigators. VPA weakens the cytotoxic activity of natural killer cells on human leukemia cells (Ogbomo et al., Citation2007) and exhibits strong immunosuppressive effects on the function of macrophages and dendritic cells (DC), as well as DC-mediated chemotaxis of monocytes (Brogdon et al., Citation2007; Nencioni et al., Citation2007). The fact that HDAC inhibitors increase the generation of regulatory T-cells provides an additional rationale for the use of these chemicals in the treatment of inflammation (Skov et al., Citation2003; Tao et al., Citation2007). These studies indicate that HDAC inhibitors can modulate both innate and adaptive immune responses. Yet, studies of the immunomodulatory effects of VPA on lymphocytes are still limited and the underlying mechanism needs further investigation.
In this study, we investigated the potential effects of VPA on the proliferation, activation, cell cycle distribution, and apoptosis of murine lymphocytes activated with concanavalin A (ConA). Our results showed that VPA exhibited biphasic effects on the activation and apoptosis of murine lymphocytes. The biphasic effects on activated lymphocytes were probably mediated through differential modulation of several apoptosis-related signaling pathways.
Materials and methods
Animals and reagents
Female BALB/c mice (6–8 weeks-of-age) were supplied by the Experimental Animal Center of Southern Medical University (Guangzhou, China). Animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Jinan University. The animals were housed in plastic cages in a facility maintained at 20–26°C with a 40–70% relative humidity and a 12-h light/12-h dark cycle. All animals were provided access to standard chow (Experimental Animal Center of Southern Medical University) and distilled water ad libitum.
VPA sodium salt, phorbol 12,13-dibutyrate (PDB), ionomycin (Ion) monensin, ConA, and dimethyl sulfoxide (DMSO) were purchased from Sigma (St. Louis, MO). VPA stock (2 M) was prepared in phosphate-buffered saline (PBS, pH 7.4) and stored at −20°C. RPMI-1640 and fetal bovine serum (FBS) were obtained from Gibco/Invitrogen (Carlsbad, CA). Carboxyfluorescein diacetate succinimidyl ester (CFSE) was obtained from Molecular Probes/Invitrogen (Carlsbad, CA).
Isolation and culture of mouse lymphocytes
Mice were sacrificed by cervical dislocation and the inguinal lymph nodes were isolated. A single-cell suspension was prepared by passing the tissue through a 200-µm nylon mesh screen. The cells were washed twice with PBS, counted, and resuspended in RPMI-1640 medium containing 10% FBS, 100 U penicillin/mL, 100 µg streptomycin/mL, 2 mM L-glutamine, and 50 µM 2-mercaptoethanol (complete medium). Lymphocytes were seeded at a density of 2 × 106 cells/mL in 24-well plates (500 µL/well) and incubated at 37°C in a humidified atmosphere of 5% CO2.
CFSE labeling assay
CFSE labeling of mouse lymphocytes was performed as described previously (Xu et al., Citation2009). In brief, cells at a density of 2 × 107 cells/mL were stained with CFSE (2 µM) for 10 min at 37°C, washed thoroughly with PBS, and resuspended in complete medium. Subsequently, 1 × 106 cells were seeded in each well of a 24-well plate and stimulated with ConA (5 µg/mL) in the presence or absence of various doses of VPA. The cells were harvested after 72 h incubation at 37°C. The fluorescence intensity of CFSE was then analyzed on a flow cytometer (FACSCalibur; Becton Dickinson, Mountain View, CA). A minimum of 10,000 events was collected per sample analyzed.
Analysis of CD69 cell surface expressions
After appropriate treatment, the cells were collected and washed twice with PBS-F (PBS containing 0.1% NaN3 and 1% FBS) and then stained with phycoerythrin (PE)-conjugated monoclonal anti-CD69 (BioLegend, San Diego, CA) for 20 min in the dark at 4°C. After washing with PBS-F, the cells were fixed with 4% paraformaldehyde in PBS and then analyzed on a flow cytometer.
Cell cycle analysis
After appropriate incubation, mouse lymphocytes were collected, washed once with PBS, and fixed with 70% ethanol at −20°C for at least 1.5 h. The fixed cells were washed with PBS again and stained for 1 h in the dark at room temperature with 1 mL staining solution containing 20 µg propidium iodide (PI) and 30 µg RNase A. The DNA contents were then analyzed by flow cytometry. All data were acquired and analyzed using CELLQuest software (Becton Dickinson).
Protein extraction and western blot analysis
Lymphocytes were stimulated with ConA (5 µg/mL) in the presence or absence of VPA at 37°C in a humidified incubator with 5% CO2. The cells were collected at appropriate times and washed twice with ice-cold PBS. After centrifugation (300 × g, 10 min, 4°C), the pelleted cells were resuspended in 150 µL of cell lysis buffer (50 mM Tris–HCl [pH 7.4], 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid [EDTA], 0.1 mM dithiothreitol [DTT], 25 mM sodium fluoride, 1 mM sodium orthovanadate, and 1 mM phenylmethanesulfonyl fluoride and protease inhibitor cocktail [Roche, Indianapolis, IN]). The protein concentration of each cell lysate was then determined using a Micro BCA assay kit (Pierce, Rockford, IL).
For the analysis of histone acetylation and phosphorylation, total proteins were obtained by lysing whole cells with 1X SDS–polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. Forty micrograms of total protein were loaded into each well of a 12% SDS-PAGE, followed by electrotransfer to polyvinylidene difluoride membranes (Amersham, Piscataway, NJ). After incubation with blocking buffer (1× Tris-buffered saline [TBS], 0.1% Tween-20 with 5% [w/v] non-fat dry milk) for 1 h at room temperature, the membranes were immunoblotted using antibodies against phospho-STAT3 (Tyr705), phospho-ERK1/2 (Thr202/Tyr204), phospho-JNK (Thr183/Tyr185), phospho-p38 (Thr180/Tyr182), acetyl-H3 (K9), H3, Bcl-2, Bax, phospho-H2A.X (Cell Signaling Technology, Danvers, MA), STAT3, and β-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA). After incubation with horseradish peroxidase (HRP)-labeled secondary antibody, specific bands were detected on X-ray film using a BeyoECL Plus Western Blotting detection System (Beyotime, Haimen, China), according to the manufacturer’s instructions. The level of immunoreactivity was measured as peak intensity using an image capture and analysis system (Alpha Innotech, San Leandro, CA).
Statistical analysis
Data are presented as mean ± standard deviation (SD). Unpaired Student’s t-tests were used for comparison of two groups. A value of P < 0.05 was considered significant.
Results
VPA inhibited the proliferation of mouse lymphocytes
The effect of VPA on the proliferation of mouse lymphocytes in response to ConA was monitored by CFSE staining and analyzed by flow cytometry. As shown in , no cell proliferation was observed in the control group without stimulation, whereas about 75% of the lymphocytes had undergone cell division at least once during the stimulation of ConA for 72 h. VPA inhibited the proliferation of ConA-stimulated lymphocytes in a dose-dependent manner with an IC50 value of 0.95 (±0.23) mM.
Figure 1. Flow cytometry analysis of carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled cells. Lymphocytes were labeled with CFSE before stimulation with concanavalin A (ConA) in the presence or absence of valproic acid (VPA). One of three independent experiments is shown. M1: Percentage of parent population; M2: percentage of divided cells.
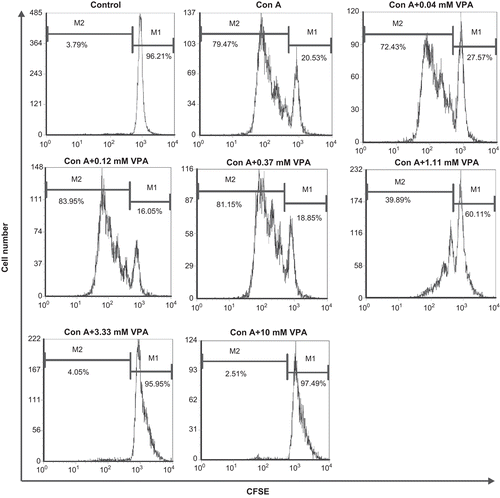
VPA exhibited biphasic effect on the expression of CD69 on lymphocytes
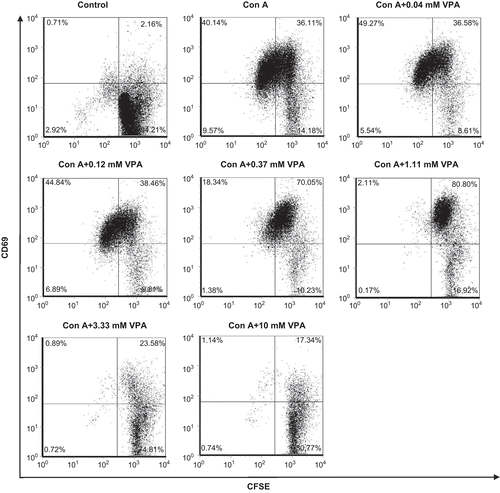
The expression of CD69, an early activation marker of lymphocytes, was low on resting lymphocytes. Upon the stimulation of ConA, CD69 expression was quickly up-regulated and the CD69+ lymphocytes were significantly increased as compared with the unstimulated group. Accordingly, the resting cells were mobilized to undergo cell division. Only those cells that expressed high level of CD69 could proliferate markedly (). In the presence of VPA, at a concentration between 0.04 and 1.11 mM, CD69 surface expression on ConA-stimulated lymphocytes was further increased (75.92 ± 0.50 vs. 2.92 ± 0.06, P < 0.01). But when VPA concentration was over 3.33 mM, this compound turned to inhibit the expression of CD69 on ConA-stimulated lymphocytes (CD69+ cells became <25%, P < 0.01) ( and ). These results demonstrated that the effect of VPA on the activation of lymphocytes in response to ConA stimulation was biphasic.
Table 1. Effect of valproic acid (VPA) on CD69 surface expression by activated lymphocytes stimulated with concanavalin A (ConA) for 72 h.
Figure 2. Effect of valproic acid (VPA) on CD69 expression on the cell surfaces of activated lymphocytes. Cells were stimulated with concanavalin A (ConA) for 72 h in vitro in the presence or absence of VPA. CD69 surface expression on the cells was determined by flow cytometry. Data are one of three independent experiments.
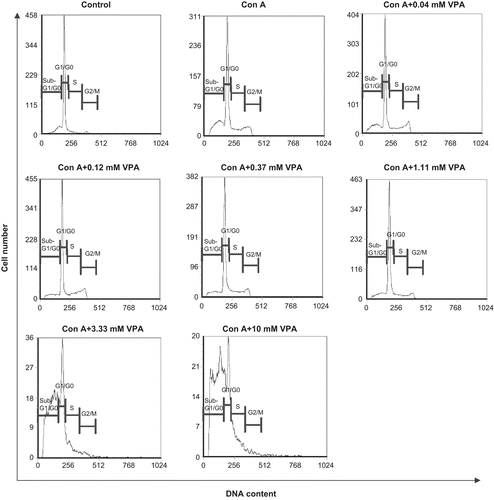
Effect of VPA on cell cycle distribution and apoptosis of activated lymphocytes
Next, we assessed the effect of VPA on cell cycle distribution and apoptosis of ConA-stimulated mouse lymphocytes. The results are presented in and . Most of the unstimulated lymphocytes stayed in G1/G0 phase (71%) and only a small number of cells in S (7%) or G2/M phases (4%). About 16% of cells were distributed in apoptotic peak (sub-G1/G0), a result consistent with others’ reports that mature resting lymphocytes gradually undergo spontaneous apoptosis in vitro (Perandones et al., Citation1993). After stimulation with ConA for 72 h, a large number of lymphocytes progressed into cell division and the apoptotic cells were increased to 26%, a phenomenon known as activation-induced cell death (AICD). Although in the presence of VPA (0.04–1.11 mM), the ConA-induced AICD was alleviated with the sub-G0/G1 ratios lower than 21% (P < 0.01). However, when VPA concentrations were ≥3.33 mM, the AICD was further aggravated and the proportions of apoptotic cells were markedly increased to >50% (P < 0.01) ( and ). These results suggested that VPA had biphasic effect on the apoptotic cell death in activated lymphocytes.
Table 2. Effect of valproic acid (VPA) on cell cycle distribution and apoptosis of activated lymphocytes stimulated with concanavalin A (ConA) for 72 h.
Figure 3. Effect of valproic acid (VPA) on cell cycle distribution and apoptosis of activated lymphocytes. Cells were stimulated by concanavalin A (ConA) in the presence or absence of VPA for 72 h, stained with propidium iodide, and analyzed by flow cytometry. Data are one of three independent experiments. Cell cycle phases are indicated above each mark.
To identify whether the mitochondria-related proteins in the apoptotic signaling pathway were involved in AICD in activated lymphocytes, we investigated the protein expression of pro-apoptotic Bax as well as anti-apoptotic Bcl-2 at 1 and 24 h by Western blot analysis (). After VPA treatment for 1 h, the expression of Bax was not changed significantly whereas Bcl-2 expression was slightly increased by 0.2 mM VPA and not changed by high doses when compared with the ConA-treated group. In contrast, after 24 h treatment of VPA, Bcl-2 remained almost stable, whereas Bax was decreased in the presence of 0.2 mM and 1 mM VPA, and markedly increased at 5 mM VPA when compared with ConA group. As the Bax/Bcl-2 ratio is a critical factor in the regulation of apoptosis (Mackey et al., Citation1998), this result suggested that VPA inhibited or promoted apoptosis in activated lymphocytes by dose-dependently modulating the Bax/Bcl-2 ratio.
Figure 4. Effect of valproic acid (VPA) on apoptosis-related proteins Bax and Bcl-2 expression in concanavalin A (ConA)-activated lymphocytes. Lymphocytes were stimulated with ConA in the presence or absence of VPA for 1 and 24 h. Bax and Bcl-2 levels were then assessed by western blotting. β-Tubulin was used as a loading control. Data, expressed as a ratio to the loading control, are from one of three independent experiments. The level of immunoreactivity was measured as peak intensity using AlphaEaseFC software.

VPA induced accumulation of acetylated H3 and exerted biphasic effects on phosphorylation of H2A.X in mouse lymphocytes
To determine the effect of VPA on HDAC activity in mouse lymphocytes, we determined the acetylation levels of histone H3. Western blotting analysis revealed a dose-dependent accumulation of acetylated histone H3 in lymphocytes after treatment with VPA for 24 h (), suggesting that HDAC activity was inhibited effectively. Furthermore, the formation of phosphorylated H2A.X (γH2A.X) was assessed to determine if VPA could affect DNA double-strand breaks (DSB). H2A.X is one of the earliest substrates to be phosphorylated in the presence of a DNA DSB (Lee et al., Citation2010). As shown in , ConA induced an increase in γH2A.X level in mouse lymphocytes. Interestingly, both 0.2 and 1 mM VPA reduced the level of γH2A.X when compared with the ConA-treated group, whereas the level of γH2A.X in the presence of 5 mM VPA was increased to a level comparable with ConA-treated group. These results suggested that low doses of VPA could reduce the amount of DNA DSBs in ConA-activated lymphocytes.
Figure 5. Effect of valproic acid (VPA) on the expression of acetylated histone H3 (acetyl-H3) and phosphorylated H2A.X (γH2A.X) in concanavalin A (ConA)-stimulated lymphocytes. Cells were stimulated with ConA in the presence or absence of VPA for 24 h. γH2A.X and acetyl-H3 levels were then assessed by Western blotting. Total histone H3 was used as a loading control. Data are expressed as a ratio to the loading control. All blots represent data from three independent experiments.

Effect of VPA on phosphorylation of STAT3 and MAPKs in ConA-stimulated lymphocytes
MAPKs have been shown to be involved in cell death and survival (Vacotto et al., Citation2008). To determine whether MAPKs were involved in VPA-mediated biphasic effect on the apoptosis of ConA-induced lymphocyte activation, we examined the activation of p38, ERK, and JNK in activated lymphocytes treated with VPA for 72 h by western blot analysis. As shown in , mM VPA dramatically induced the phosphorylation of p38 whereas both 0.2 and 1 mM VPA had no effect. In addition, no phosphorylation of ERK1/2 and JNK was found. Since the activation of ERK is generally associated with inhibition of apoptosis, and phosphorylation of p38 promotes apoptosis (Xie et al., Citation2010), this result suggested that high-dose VPA might enhance apoptotic cell death of activated lymphocytes by activation of p38 MAPK pathway.
Figure 6. Effect of valproic acid (VPA) on the phosphorylation of mitogen-activated protein kinases (MAPKs) (A) and STAT3 (B) in concanavalin A (ConA)-stimulated lymphocytes. Cells were stimulated with ConA in the presence or absence of VPA for 72 h. Whole-cell lysates were then analyzed by Western blotting for the levels of p38, ERK1/2, and JNK phosphorylation (P-p38, P-ERK1/2, and P-JNK) and phospho-STAT3 (P-STAT3). STAT3 and β-tubulin levels were used as the loading controls. Data are expressed as a ratio to the loading control. All blots represent data from one of three independent experiments.
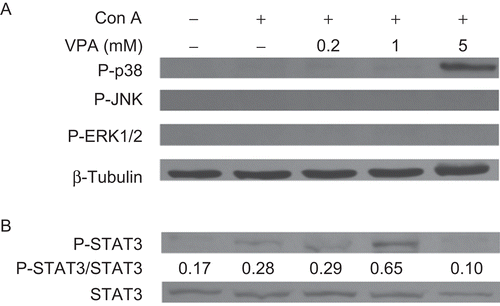
We next examined the status of phosphorylated STAT3 in ConA-activated lymphocytes. Western blot analyses showed that phosphorylated STAT3 was up-regulated in 1 mM VPA-treated groups when compared with the ConA group. However, 5 mM VPA markedly reduced the level of phosphorylated STAT3 (). Although phosphorylated STAT3 can promote cell proliferation and angiogenesis, inhibit apoptosis, and drive invasion and metastasis (Al Zaid Siddiquee and Turkson, Citation2008), our results suggested that low-dose VPA may attenuate AICD through activation of STAT3 and high-dose VPA may promote apoptotic cell death by inhibiting the phosphorylation of STAT3.
Discussion
In this study, we investigated the effect of VPA on the apoptosis of activated lymphocytes. Most of the lymphocytes became activated after ConA stimulation, as assessed by the surface expression of CD69, and underwent cell division. VPA inhibited the proliferation of activated lymphocytes and modulated the expression of CD69 dose-dependently. Moreover, VPA of low doses (≤1.1 mM) attenuated apoptotic cell death whereas high doses (≥3.3 mM) dramatically promoted apoptosis in activated lymphocytes. Our results suggest that VPA has biphasic effect on the apoptotic cell death of activated lymphocytes.
We were interested in the molecular mechanism of this biphasic effect of VPA on activated lymphocytes. Our data suggest that the biphasic effect of VPA might be due to differential modulations of the expression of cell signaling- and apoptosis-related genes. In the early phases, the modulation of Bax/Bcl-2 balance may play a role in regulating apoptosis induced by VPA; in the later phase, regulation of STAT3 and p38 is associated with the modulation of apoptosis. In normal cells, the Bax/Bcl-2 ratio is kept in balance to maintain cell survival. Disruption of this balance may lead to apoptosis or tumorigenesis (Mackey et al., Citation1998). In this study, low-dose VPA decreased the Bax/Bcl-2 ratio that seems to be associated with an anti-apoptotic effect, whereas high-dose VPA increased this ratio, which may be related to apoptosis-promoting activity.
STAT3 plays an important role in cell growth, survival, proliferation, differentiation, and apoptosis (Bill et al. Citation2010). It is evident that abnormal STAT3 activation promotes tumorigenesis in part by up-regulating the expression of anti-apoptotic proteins (such as Bcl-xL, Bcl-2, c-myc, etc.) and down-regulating pro-apoptotic genes, thus promoting the survival and proliferation of tumor cells (Al Zaid Siddiquee and Turkson, Citation2008). In this study, we found that the level of phosphorylated STAT3 was significantly elevated by low doses of VPA but reduced at high doses. This result suggests that low-dose VPA-enhanced phosphorylation of STAT3 may lead to the attenuation of activation-induced apoptosis whereas high-dose VPA-induced dephosphorylation of STAT3 may result in enhanced activation-induced apoptotic cell death. Thus, the phosphorylation status of STAT3 may partially be responsible for the biphasic effect on the apoptosis of activated lymphocytes.
MAPKs are involved in many cellular processes including development, differentiation, proliferation, and apoptosis (Vacotto et al., Citation2008). There are three main members of the MAPKs family: c-Jun N-terminal kinase (JNK), p38, and ERK (Xie et al., Citation2010). Studies have shown that VPA induces an increase in the level of phosphorylated p38 but has no effects on ERK and JNK, leading to apoptosis by mitochondrial pathways in both BV-2 microglia and mouse primary microglial cells (Xie et al., Citation2010). However, one study showed that VPA-induced ERK1/2 phosphorylation causes phosphorylation of anti-apoptotic Bcl-2 and inhibits serum starvation-induced HUVEC apoptosis and cytochrome c release from the mitochondria (Michaelis et al., Citation2005). In line with these studies, we found that high-dose VPA increased the level of phosphorylated p38 but had no effect on the other MAPKs, suggesting that activation of p38 MAPK may be involved in the VPA-enhanced apoptosis of activated lymphocytes.
Recent evidence indicates that VPA may contribute to genomic instability. The reason is because it can function as a HDAC inhibitor, leading to the acetylation of histone tails (Marchion et al., Citation2005) and the relaxing of chromatin, thus making it more susceptible to DNA damage, including DSBs (Coyle et al., Citation2005). It has been shown that phosphorylated histone H2A.X (γH2A.X) is an early marker of DNA DSBs that can lead to altered gene expression and induction of apoptosis (Coyle et al., Citation2005; Marchion et al., Citation2005). HDAC activities should be effectively blocked by VPA as revealed by histone H3 acetylation. Although VPA enhanced histone H3 acetylation in a dose-dependent manner, its effect on γH2A.X levels was biphasic, which may be relevant to the biphasic effect of VPA on apoptosis.
Since activated lymphocytes undergo AICD in order to maintain homeostasis, low-dose VPA may confer protection against AICD of the activated lymphocytes in vivo because the therapeutic plasma concentration of VPA is 0.3–0.6 mM via the oral route (Williams et al., Citation2002). Attenuation of normal AICD may interfere with the physiological contraction phase of immune responses, leading to abnormal sustaining of immune responses (Perandones et al., Citation1993). This immunomodulatory effect of VPA should be noticed when it is used in clinic.
In summary, our results demonstrated that VPA had biphasic effects on activated lymphocytes: at low doses it promoted the early activation of lymphocytes and reduced cell apoptosis whereas at high doses it attenuated lymphocyte activation and enhanced apoptosis. The results of this study suggest that the dose-dependent immunomodulatory activities of VPA should be considered when it is used as an anti-tumor agent.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No. 21609403) and Natural Science Fund of Guangdong Province (No. 8451063201000340).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Al Zaid Siddiquee, K., and Turkson, J. 2008. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 18:254–267.
- Bill, M. A., Fuchs, J. R., Li, C., Yui, J., Bakan, C., Benson, D. M. Jr, Schwartz, E. B., Abdelhamid, D., Lin, J., Hoyt, D. G., Fossey, S. L., Young, G. S., Carson, W. E., 3rd, Li, P. K., and Lesinski, G. B. 2010. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol Cancer. 9:165.
- Brogdon, J. L., Xu, Y., Szabo, S. J., An, S., Buxton, F., Cohen, D., and Huang, Q. 2007. Histone deacetylase activities are required for innate immune cell control of TH1 but not TH2 effector cell function. Blood 109:1123–1130.
- Cinatl, J., Jr, Kotchetkov, R., Blaheta, R., Driever, P. H., Vogel, J. U., and Cinatl, J. 2002. Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)-C cells by valproic acid: Enhancement by combination with interferon-alpha. Int J Oncol. 20:97–106.
- Coyle, T. E., Bair, A. K., Stein, C., Vajpayee, N., Mehdi, S., and Wright, J. 2005. Acute leukemia associated with valproic acid treatment: A novel mechanism for leukemogenesis? Am J Hematol. 78:256–260.
- Fortunati, N., Catalano, M. G., Arena, K., Brignardello, E., Piovesan, A., and Boccuzzi, G. 2004. Valproic acid induces the expression of the Na+/I− symporter and iodine uptake in poorly differentiated thyroid cancer cells. J Clin Endocrinol Metab. 89:1006–1009.
- Hashimoto, R., Hough, C., Nakazawa, T., Yamamoto, T., and Chuang, D. M. 2002. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: Involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 80:589–597.
- Jeong, M. R., Hashimoto, R., Senatorov, V. V., Fujimaki, K., Ren, M., Lee, M. S., and Chuang, D. M. 2003. Valproic acid, a mood stabilizer and anticonvulsant, protects rat cerebral cortical neurons from spontaneous cell death: A role of histone deacetylase inhibition. FEBS Lett. 542:74–78.
- Johannessen, C. U., and Johannessen, S. I. 2003. Valproate: Past, present, and future. CNS Drug Rev. 9:199–216.
- Kawagoe, R., Kawagoe, H., and Sano, K. 2002. Valproic acid induces apoptosis in human leukemia cells by stimulating both caspase-dependent and -independent apoptotic signaling pathways. Leuk Res. 26:495–502.
- Lee, J. H., Choy, M. L., Ngo, L., Foster, S. S., and Marks, P. A. 2010. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA. 107:14639–14644.
- Mackey, T. J., Borkowski, A., Amin, P., Jacobs, S. C., and Kyprianou, N. 1998. Bcl-2/bax ratio as a predictive marker for therapeutic response to radiotherapy in patients with prostate cancer. Urology 52:1085–1090.
- Marchion, D. C., Bicaku, E., Daud, A. I., Sullivan, D. M., and Munster, P. N. 2005. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 65:3815–3822.
- Michaelis, M., Suhan, T., Michaelis, U. R., Beek, K., Rothweiler, F., Tausch, L., Werz, O., Eikel, D., Zörnig, M., Nau, H., Fleming, I., Doerr, H. W., and Cinatl, J., Jr. 2005. Valproic acid induces extracellular signal-regulated kinase-1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 13:446–453.
- Nencioni, A., Beck, J., Werth, D., Grünebach, F., Patrone, F., Ballestrero, A., and Brossart, P. 2007. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 13:3933–3941.
- Ogbomo, H., Michaelis, M., Kreuter, J., Doerr, H. W., and Cinatl, J., Jr. 2007. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 581:1317–1322.
- Perandones, C. E., Illera, V. A., Peckham, D., Stunz, L. L., and Ashman, R. F. 1993. Regulation of apoptosis in vitro in mature murine spleen T-cells. J Immunol. 151:3521–3529.
- Pérez, M., Rojo, A. I., Wandosell, F., Díaz-Nido, J., and Avila, J. 2003. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J. 372:129–136.
- Phiel, C. J., Zhang, F., Huang, E. Y., Guenther, M. G., Lazar, M. A., and Klein, P. S. 2001. Histone deacetylase is a direct target of valproic acid, a potent anti-convulsant, mood stabilizer, and teratogen. J Biol Chem. 276:36734–36741.
- Skov, S., Rieneck, K., Bovin, L. F., Skak, K., Tomra, S., Michelsen, B. K., and Ødum N. 2003. Histone deacetylase inhibitors: A new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood 101:1430–1438.
- Smith, K. T., and Workman, J. L. 2009. Histone deacetylase inhibitors: Anticancer compounds. Int J Biochem Cell Biol. 41:21–25.
- Takai, N., Desmond, J. C., Kumagai, T., Gui, D., Said, J. W., Whittaker, S., Miyakawa, I., and Koeffler, H. P. 2004. Histone deacetylase inhibitors have a profound anti-growth activity in endometrial cancer cells. Clin Cancer Res. 10:1141–1149.
- Tao, R., de Zoeten, E. F., Ozkaynak, E., Chen, C., Wang, L., Porrett, P. M., Li, B., Turka, L. A., Olson, E. N., Greene, M. I., Wells, A. D., and Hancock, W. W. 2007. Deacetylase inhibition promotes the generation and function of regulatory T-cells. Nat Med. 13:1299–1307.
- Vacotto, M., Coso, O., and Fiszer de Plazas, S. 2008. Programmed cell death and differential JNK, p38, and ERK response in a prenatal acute hypoxic hypoxia model. Neurochem Int. 52:857–863.
- Williams, R. S., Cheng, L., Mudge, A. W., and Harwood, A. J. 2002. A common mechanism of action for three mood-stabilizing drugs. Nature 417:292–295.
- Xie, N., Wang, C., Lin, Y., Li, H., Chen, L., Zhang, T., Sun, Y., Zhang, Y., Yin, D., and Chi, Z. 2010. The role of p38 MAPK in valproic acid induced microglia apoptosis. Neurosci Lett. 482:51–56.
- Xu, W. B., Xu, L. H., Lu, H. S., Ou-Yang, D. Y., Shi, H. J., Di, J. F., and He, X. H. 2009. The immunosuppressive effect of gossypol in mice is mediated by inhibition of lymphocyte proliferation and by induction of cell apoptosis. Acta Pharmacol Sin. 30:597–604.