Abstract
Lipopolysaccharide (LPS) is a known inducer of acute respiratory distress syndrome (ARDS) in humans and animals. In this study, ARDS was developed in rats by intratracheal instillation of LPS and the effect of two types of surfactant (natural vs. synthetic) was examined to determine their potential corrective roles in general, as well as to compare the two surfactants against one another in particular, in endotoxin-induced lung injury. Sprague–Dawley male rats were divided into four groups, i.e., rats given: buffer controls; 055:B5 E. coli LPS only; LPS and then porcine surfactant (P-SF); or, LPS and then synthetic surfactant (S-SF). In vivo administration of LPS led to an increase in expression of the cytokines tumor necrosis factor-α, interleukin (IL)-1β, IL-2, IL-4, interferon-γ, monocyte chemotactic protein-1, and macrophage inflammatory protein-1β in the lungs of rats. These effects were confirmed by immunofluorescence in lung tissue sections and/or by protein (Western immunoblot) and mRNA expression (reverse transcription polymerase chain reaction) analyses of tissue samples. Apart from IL-4, concentrations of each of these cytokines in bronchoalveolar lavage fluid recovered from the animals were significantly increased in the LPS-treated hosts. Instillation of either surfactant (70 h after the LPS) into the airways diminished the expression of each of the inducible-cytokines, with the porcine (natural) form seeming having the greater inhibitory effect. These data suggest that surfactant can play an important role in the treatment of endotoxin-induced lung injury and might possess robust anti-inflammatory effects. Further, it seems that both the natural and synthetic surfactants prevent inflammatory outcomes in the lungs by controlling cytokine(s) production by various inflammatory cells. Last, the studies here clearly indicated that in this aspect, natural surfactant appears to be more beneficial compared to synthetic surfactant.
Keywords::
Introduction
Acute respiratory distress syndrome (ARDS) is an inflammatory lung disorder characterized by severe hypoxemia, non-cardiogenic pulmonary edema, decreased pulmonary compliance, and diffuse lung infiltration (Lin et al., Citation2010), with mortality rates that exceed 50% (Temmesfeld-Wollbruck et al., Citation1995). Neutrophil accumulation in the pulmonary microcirculation is also observed (Kinoshita et al., Citation1999). In humans, pneumonia and sepsis are two common predisposing conditions for the development of ARDS. Though the mechanisms underlying its evolution are causal-related, there are certain pathologies that become evident and ultimately lead to the characteristic interstitial/alveolar edema, alveolar failure, and hypoxemia associated with ARDS (Tomashefski, Citation2000; Matute-Bello et al., Citation2008). Some of these include: damage to epithelial and endothelial cells; increased vascular permeability, over-recruitment/infiltration of leukocytes; cyto-/chemokine over-production, and a dysfunction of surfactant. As there are currently no specific effective therapies for ARDS, novel therapeutics are clearly needed.
Although animal models are useful tools to explore mechanisms underlying ARDS (and other human diseases) and to help discover\validate novel drugs that could mitigate or cure these pathologies, such models should sufficiently replicate the mechanisms, pathophysiology, and consequences of the given disease. In both animals and humans, exposure to lipopolysaccharide (LPS; a Gram-negative bacterial cell wall constituent) gives rise to several adverse outcomes in the lungs, including microvascular injury, leukocyte accumulation, edema, severe inflammation, and an increase in release of key pro-inflammatory cytokines (i.e., interleukin (IL)-1 and tumor necrosis factor (TNF)-α) and other toxic mediators, such as reactive oxygen species (ROS), pro-teases, and arachidonic acid metabolites. Each of these LPS-induced changes are likely to significantly contribute to development of lung injury in general—even potentially ARDS (Pugin et al., Citation1996; Germann and Hafner, Citation1998; Beck-Schimmer et al., Citation2005; Mirzapoiazova et al., Citation2007; Bastarache and Blackwell, Citation2009; Lin et al., Citation2010; Warren et al., Citation2010)); these outcomes are, of course, dose-dependent. As correctly noted in a recent review by Chen et al. (Citation2010), the LPS-induced injury animal model is a very useful experimental model closely resembling human ARDS. However, in discussing advantages of LPS-induced ARDS models, the Authors clearly indicated that users of such models “consider that different species/strains of animals have various genetic susceptibilities to LPS challenge, related to the LPS sources, the location and time of challenge, and the parameters measured”.
Regardless of whether the sources of information are human ARDS patients or induced animal models, there has been intense interest in understanding the involvement of cytokines in the pathogenesis of ARDS (Martin, Citation1997). Cytokines are low molecular weight soluble proteins (generally <30 kda) that transmit signals between cells and are produced in “cascades” in which the initial cytokine signals are amplified many-fold by target cells, such as epithelial cells, fibroblasts, and endothelial cells (Pittet et al., Citation1997). Cytokines function in “networks” in which feedback occurs at many points to coordinate and regulate cytokine and cellular responses (Michell, Citation1999). Studies of single cytokines have shown that no single cytokine consistently predicts either the onset or the outcome of ARDS, despite promising early results. Instead, it is now recognized that a balance of pro-inflammatory and anti-inflammatory factors influences the net inflammatory response in the lung (Martin, Citation1997). Experimental studies suggest that cytokine responses normally are compar-tmentalized in the lungs, which is lost to some extent during severe inflammatory responses (Ma et al., Citation2010). Thus, measurements of cytokines in the alveolar fluids or by BAL are likely to be more valuable than measurements in plasma or serum and provide the best current assessment of cytokine concentrations in the alveolar spaces.
Another important modulator of the molecular entities underlying ARDS is pulmonary surfactant. Alteration in the synthesis or composition of surfactant seems to have an important role in the pathogenesis of ARDS (Galani et al., Citation2010). Aside from the mechanical consequences of lower surfactant levels (decreased compliance), it has also been found that both surfactant lipids and surfactant-associated protein A (SP-A) and D, have non-surfactant-related functions and are actively associated with various defense functions in the lung (McCormack, Citation1997). In epidemiological observation several studies of surfactant replacement for ARDS have been performed in adults with respiratory failure (Gregory et al., Citation1991). Surfactant-associated proteins have been used as markers of Type II pneumocyte function, and the concentrations of the surfactant-associated proteins SP-A and SP-B are low at the onset of ARDS (Gregory et al., Citation1991). The lipid components of surfactant appear to have predominantly immunosuppressive effects as in the synthetic surfactant, adsurf and exosurf. In contrast, the surfactant-associated proteins have been reported to exhibit both pro-inflammatory (Arias-Diaz et al., Citation2000) and anti-inflammatory activity (Mittal and Sanyal, Citation2010a) as seen with curosurf, BLES and alveofact.
In view of the above background, we hypothesized that surfactants have a potential therapeutic effect on ARDS, in part, via modulation of the formation/release of certain select cytokines. To validate this hypothesis, we investigated the therapeutic effect of both a natural (porcine) and synthetic surfactant, and also compared the effects between the two surfactants, on expression of pro-inflammatory cytokines in an LPS-generated ARDS model. The results revealed that both surfactants have a potential therapeutic effect on ARDS via modulation of cytokines, and that porcine surfactant seems to be more the more potent agent.
Materials and methods
Surfactant preparation
Surfactant was isolated from porcine lung homogenate (P-SF) by a sucrose density gradient method described in Mittal and Sanyal (Citation2010b). Protein-free synthetic surfactant (S-SF) was prepared with dipalmitoylphosphohatidyl choline (DPPC), hexadecanol, and tyloxapol as described in Mittal and Sanyal (Citation2010b).
Experimental design
In these studies, control animals (rats) were administered 500 µl vehicle or LPS endotoxin (055:B5 Escherichia coli; Sigma, St. Louis, MO) by intratracheal instillation (procedure described below). At 70 h post-instillation, subsets of the LPS-treated rats were instilled with 500 µl of porcine or synthetic surfactant. At 72 h post-instillation of vehicle or LPS, all rats were euthanized by ether overdose and had their lungs either removed en bloc and prepared for immunohistochemical analyses/RNA isolation/lysate generation (see specific protocols below) or lavaged using standard procedures (Mittal and Sanyal, Citation2010c) to obtain bronchoalveolar lavage fluid (BALF) for cytokine analyses.
Animal model
Male SD rats (8–10 weeks-of-age, weighing 150–200 g) were obtained from the Central Animal House of Panjab University and placed in polypropylene cages located in rooms maintained at 25°C and under a 12 h photoperiod of light and darkness, respectively. All rats had access to a standard rodent chow pellet diet and fresh drinking water ad libitum. All animal procedures reported here were carried out in accordance with guidelines approved by the Panjab University Ethical Committee on the Use of Experimental Animals for Biomedical Research (Mittal and Sanyal, Citation2009).
Rats were anesthetized with ketamine (130 mg/kg, intraperitoneally) so that they would remain unconscious throughout the entire instillation procedure and had no cough reflex upon intubation. A small incision was made in the ventral region of the neck and the trachea carefully exposed. The rat was then placed on a slight incline, intubated with a 26-gauge needle and instilled with either 500 µl vehicle (50 mM Tris-HCl [pH-7.4], 150 mM NaCl, 1 mM sodium azide [NaN3], and 0.2 mM phenylmethylsulfonyl fluoride [PMSF] or LPS (suspended in vehicle to yield 300 µg/ml concentration). In each case, this step was followed by introduction of 2–3 boluses of 1 ml air to facilitate fluid distribution. Shortly thereafter, when normal spontaneous breathing was apparent, the neck incision was closed with silk sutures. To avoid any infection, betadine and neosporin powder were applied to the wound area. At 70 h post-instillation, subsets of the LPS-treated rats were re-anesthetized and instilled with 500 µl aliquots of porcine ([0.5 mg protein + 4.6 mg lipid]/500 µl buffer) or synthetic surfactant ([4.6 mg lipid]/500 µl buffer), and allocated into groups (each with four rats) designated P-SF and S-SF, respectively.
BALF isolation
At the end of each experiment, bronchoalveolar lavage was performed using 5 ml phosphate-buffered saline (PBS, pH-7.4), as reported earlier (Mittal and Sanyal, Citation2010c). The average fluid recovery was >90%. Each recovered volume was centrifuged at 1000 rpm for 10 min at 4°C and the supernatants were stored at−20°C until analysis. Furthermore, the cell pellet that remained was re-suspended in PBS and, subsequently, the number of total cells was determined using a Neubayer’s hemocytometer. In addition, the fraction of neutrophils present was determined using differential counts performed on 200 cells that had been stained with Wright Giemsa (Chabot et al., Citation2003)
Immunofluorescence studies
Lung tissues that had been removed en bloc were initially fixed in 10% formalin for 48 hr and subsequently embedded in paraffin wax. Sections (5-µm thickness) of each lung were then deparaffinized in three changes of xylene for 5 min each. The sections were then gradually hydrated in 100% and 95% ethanol for 10 min each and washed in deionized water for 1 min with stirring. The slides were placed in a container, covered with glycine-HCl buffer (pH-3.5) containing 0.01% (w/v) EDTA, and heated for 10 min at 95°C (with continuous topping off with fresh buffer) for antigen retrieval. The slides were cooled 20 min, then washed in distilled water.
Any non-specific binding by the samples was blocked by incubating the sections with 10% bovine serum albumin solution (BSA in phosphate-buffered saline [PBS, pH 7.2]). The sections were then incubated with polyclonal antibody (1:1000 dilution in PBS with 1.5% BSA) against SP-B, IL-1β, TNFα, IL-2, IL-4, monocyte chemotactic protein (MCP)-1, or macrophage inflammatory protein (MIP)-1β-in a moist chamber for 1 h at 37°C. After incubation, the sections were washed three times in PBS successively for 5 min each. The sections were then incubated with FITC-conjugated secondary antibody (1:10,000 dilution in PBS with 1.5-3% BSA) in a moist dark chamber for 1h at 37°C. Sections were washed again in the same manner as described above and were counterstained with DAPI for 20 min at 37°C, followed by a washing in PBS as noted above. Sections were then covered with glass coverslips after being mounted with an aqueous mounting medium (glycerol:PBS, 1:9); each slide was then sealed with transparent nail polish. All samples were then examined using an Axioscope A1, fluorescent microscope (Carl Zeiss, Germany) at excitation/emission wavelengths of 490 nm/525 nm for the FITC fluorophore and 350 nm/470 nm for the DAPI. For the analyses, four fields were counted in three different slides from each group; from this, quantitative data in terms of percentage of cytokine-positive cells was derived. Data were reported in terms of total cytokine-positive cells counted in a particular field as a percentage of all cells analyzed/field; a minimum of 100 cells/field were scored each time.
Cytokine measurements in BALF
IL-2 and IL-4 levels in BALF recovered from the rats were measured using a commercial ELISA kit (Bender MedSystems, San Diego, CA). The sensitivities of the IL-2 and IL-4 kits were 9.6 and 0.2 pg/ml, respectively. The optical density in each kit well was read at 450 nm using a MIOS mini microplate reader (Merck, Whitehouse Station, NJ).
Western blot analysis
To generate protein extracts, lungs were removed from the different treatment group rats and lysates prepared in fresh ice-cold protein lysis buffer [10 mM Tris, 100 mM NaCl, 5 mM EDTA, 1% Triton-X100, 1 mM PMSF and 2 mM DTT (pH 8)]. The extracts were cleared by centrifugation at 10,000 × g for 10 min at 4°C. Resultant supernatants were collected as ‘total lysate’. Protein concentration in each lysate was determined by the Bradford (Citation1976) method.
Protein samples (100 µg) from each treatment group were then separated via 10% SDS-PAGE and the resolved proteins electrophoretically transferred to nitrocellulose membranes (Genei, Bangalore, India). Immunoblots were prepared using primary antibodies (SP-B, IL-1β, TNF-α, IL-2, IL-4, interferon (IFN)-γ, MCP-1, MIP-1β, and β-actin-each at 1:1000 dilution) from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and their respective alkaline phosphatase-conjugated secondary antibodies (each at 1:10,000 dilution; Genei, Bangalore, India). A BCIP-NBT detection system was used to develop the blots. Bands obtained were densitometrically analyzed using Image J software; density was expressed as gray values (in densitometric units).
RNA isolation
Total RNA was isolated from rat lung tissue (50 µg starting tissue/rat) using RaFlex Total RNA Isolation Kit (Genei). Purity of each isolated RNA sample was checked by measuring its absorbance (at 260 and 280 nm). In addition, all samples were subjected to 1.2% formaldehyde agarose gel electrophoresis (denaturing gel) and staining with ethidium bromide to confirm material integrity and sample concentration.
Reverse transcription polymerase chain reaction procedure
Reverse transcription polymerase chain reactions were performed using specific primers for the respective genes. Primers were custom synthesized from Bangalore Genie (India). Length of the primers chosen was ~20 bp (Beck-Schimmer et al., Citation2005); primer sequences are shown in . A reaction for β-actin was also performed to rule out any experimental errors. For each rat, an aliquot of 3 µg total RNA was used in a PCR reaction performed in a thermal cycler (Eppendorf N.A., Hauppauge, NY). Reverse transcription was performed at 50°C for 50 min and activation at 95°C for 15 min, followed by PCR of 35 cycles: 94°C (denaturation) for 45 s, variable* (annealing) for 45 s, 72°C (extension) for 1 min (*annealing temperatures were for: IL-1β = 70.5°C; TNFα = 63.4°C; MCP-1 = 63.5°C; MIP-1β = 64°C; and, β-actin = 65.5°C). Lastly, the products were incubated at 72°C for 10 min to extend any incomplete single strands. Final PCR products (cDNA) were analyzed using 1.5% agarose gel electrophoresis; all band densitometric analyses were performed using Image J software.
Table 1. Primers used for RT-PCR analyses.
Statistical analysis
Statistical analysis was performed using SPSS Version 10.0 software. One-way analysis of variance (ANOVA) was done to compare means between the different treatments using post hoc comparison by the Least Significant Difference (LSD) method. A p < 0.05 was considered significant. All data were expressed as the mean (±SD) of four animals for each group.
Results
Cytokine and chemokine expression by Immunofluorescence
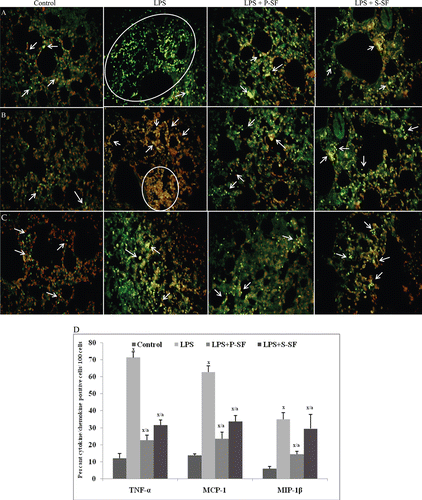
and show the localization of cytokines/chemokines in lung tissue sections using immunofluorescence; corresponding quantitative results are shown in and in control animals. An IL-1β expression of 12.0% cells was seen in the respiratory compartment that, upon LPS stimulation, was increased to 52.0% (p < 0.001). However, this upsurge was attenuated to 24.5% (p < 0.001) by intratracheally-applied P-SF; S-SF reduced IL-1β+ cell levels to 27.7% (p < 0.001) ( and ). IL-2 content in the lungs was increased as a result of the LPS treatment. This outcome is reflected by a change from 10% IL-2+ cells in the lungs of control rats to a level of 35.5% IL-2+ cells in tissues of LPS-treated rats (p < 0.001) ( and ). As a result of the instillation of surfactant, the percentage of IL-2+ cells was 15.0% and 22.3%, respectively, in the P-SF (p < 0.001) and S-SF (p < 0.001) groups. As illustrated in and , no significant alterations were found with respect to expression of IL-4 in LPS- and LPS+SF-treated hosts. TNFα content in the lungs was also increased as a result of LPS treatment. The levels of TNFα+ cells increased from 12.0% in control rats to 71.2% in endotoxin-injured rats (p < 0.001). In the presence of intratracheally-applied exogenous surfactant, TNFα expression was reduced, i.e., values of 22.7% and 31.5% TNFα+ cells in, respectively, the P-SF and S-SF animals (p < 0.001) ( and ).
Figure 1. Photomicrographs of (A) IL-1β, (B) IL-2, and (C) IL-4 expression in lung tissue sections when analyzed using immunofluorescence. FITC-conjugated secondary antibodies were used and tissues were counterstained with propidium iodide (PI). Figure is a representive photo-array from among four rats/treatment group. Arrows indicate cell positive for the indicated cytokine. Circles/ovals are also used to indicate cluster of positive cells. (D) Quantitative analysis of cytokine(s) positive cells. 100 cells were counted in each of four different slides from each group and the percentage of positive cells was calculated. Values marked with the x or a were statistically different from the control group or LPS-only group, respectively (p < 0.001).
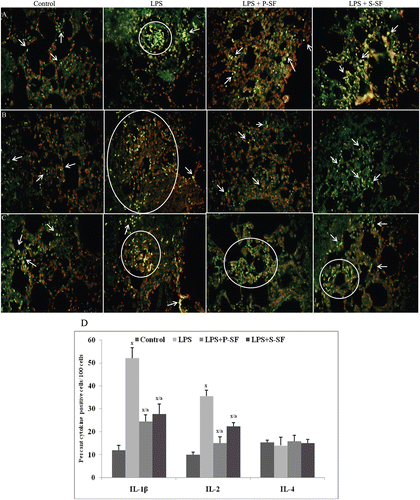
Figure 2. Photomicrographs of (A) TNFα, (B) MCP-1, and (C) MIP-1β expression in lung tissue sections when analyzed using immunofluorescence. FITC-conjugated secondary antibodies were used and tissues were counterstained with propidium iodide (PI). Figure is a representive photo-array from among four rats/treatment group. Arrows indicate cell positive for the indicated cytokine. Circles/ovals are also used to indicate cluster of positive cells. (D) Quantitative analysis of cytokine(s) positive cells. 100 cells were counted in each of four different slides from each group and the percentages of positive cells was calculated. Values marked with the x or a were statistically different from the control group or LPS-only group, respectively (p < 0.001).
MCP-1 plays a crucial role in neutrophil recruitment in endotoxin-induced lung injury. In control rat lungs, a level of 13.7% MCP-1+ cells was seen; this value was increased to 62.7% in LPS-injured lungs (p < 0.001). In the presence of instilled P-SF, this increase was reduced to just 23.5% (p < 0.001). As with P-SF, S-SF instillation also lowered the levels of MCP-1+ cells, i.e., final value of 33.7% (p < 0.001) ( and ). Endotoxin also caused an increase in MIP-1β levels in the tissue sections from 6% (control animals) to now 50% MIP-1β+ cells (p < 0.001). Surfactant treatment with P-SF and S-SF led, respectively, to decreases in MIP-1β levels, i.e., values of 14.5% and 29.5% MIP-1β+ cells (p < 0.001) ( and ).
Measurements of BALF IL-2 and IL-4
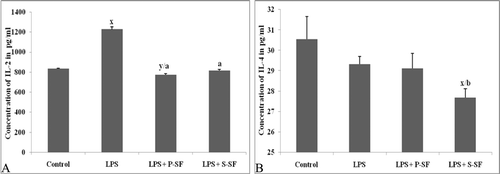
We measured the concentrations of IL-2 and IL-4 in the BAL fluid from animals of all the treatment groups. The median concentration of IL-2 was 833.5 pg/ml in the control group which was increased to 1226.0 pg/ml (p < 0.001) as seen in . Upon surfactant instillation, the IL-2 level was reduced to 774.0 pg/ml (p < 0.001) with P-SF and to 815.3 pg/ml (p < 0.001) with S-SF. No significant variations were seen in the IL-4 levels except in the LPS+SF treated group (. Results from a previous study in our laboratory (Mittal and Sanyal, Citation2010c) had already shown that levels of TNFα, IL-1β, IFNγ, MCP-1, and MIP-1β in the BALF were increased in response to LPS. Upon surfactant instillation, the ‘increased’ levels of these LPS-inducible cytokines were also reduced by both P-SF and S-SF.
Figure 3. (A) IL-2 and (B) IL-4 levels in BALF of treated animals. Bars indicate mean (±SD) of four animals. xp < 0.001, yp < 0.01 vs. control; ap < 0.001, bp < 0.01 vs. LPS-only rats (one-way ANOVA).
Protein expression of cytokines/chemokines in lung tissue
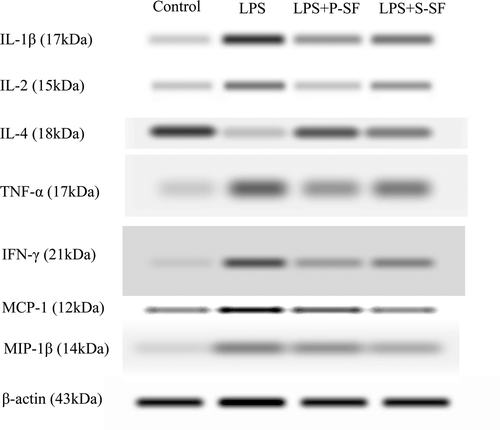
As indicated by immunofluorescence in and , expression of IL-1β, IL-2, and IL-4—as well as of TNFα, MCP-1, and MIP-1β—was clearly evident in the lung tissues. These results subsequently were subsequently found to agree with those obtained in the Western blot analyses (). Specifically, IL-1β, TNFα, and IL-2 protein expression in tissue increased upon LPS stimulation as compared to in control animal tissues; MCP-1 and MIP-1β protein concentration was also enhanced in LPS-treated rats. IFNγ expression was enhanced in the LPS-group rats as compared to in the control hosts. Surfactant instillation caused a decline in the expression of all of the above cytokines (in both the surfactant-treated groups) in comparison to the levels in rats that received LPS only.
Figure 4. Representative Western immunoblots of cytokines (e.g., IL-1β, IL-2, IL-4, TNFα, IFNγ, MCP-1, and MIP-1β) in lung tissues of rats in the various treatment regimens.
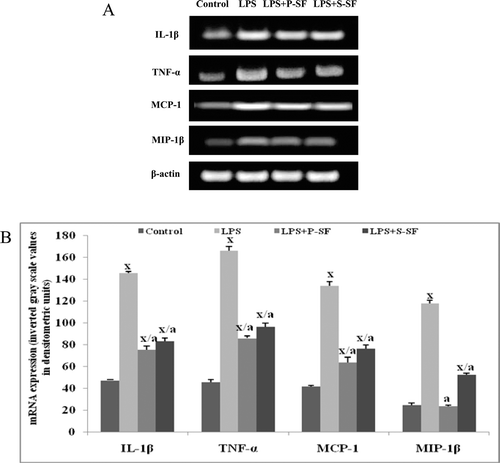
Cytokine/chemokine mRNA expression in rat lung
To obtain more information on cytokine/chemokine levels in the respiratory compartment of the entire lungs, mRNA was isolated from the lungs of rats 72 h after the LPS (or buffer) administration and the presence of cytokines/chemokines assessed by reverse transcription (RT)-PCR ( and ). Less mRNA expression was detectable in lung tissues of control rats that had received buffer only. After instillation (i.e., 72 h later) of LPS, significantly (p < 0.001) enhanced mRNA expression of the analyzed cytokines/chemokines (e.g., IL-1β, TNFα, MCP-1, MIP-1β) was noted. In surfactant-treated hosts, IL-1β and TNFα mRNA expression was significantly decreased (p < 0.001) in comparison to the expression in the LPS-stimulated animals, but these levels were still significantly (p < 0.001) greater than in the buffer-treated control rats. For MCP-1 and MIP-1β, mRNA expression of these chemokines was also attenuated with surfactant administration. Although expression levels were significantly (p < 0.001) decreased relative to values seen with rats that received LPS only, with MIP-1β, P-SF treatment led to an expression no longer different from that in control (buffer) rat tissues. In contrast, post-surfactant MCP-1 mRNA values were still significantly (p < 0.001) greater than those in buffer-treated control rats.
Figure 5. mRNA expression of genes for pro-inflammatory cytokines IL-1β, TNFα, MCP-1, and MIP-1β in lung tissues of rats in the various treatment regimens. (A) Representative reverse transcription polymerase chain reaction bands of rat lung tissues and (B) corresponding densitometric analyses. Values shown in (B) are mean (±SD) of three reverse transcription polymerase chain reaction results (i.e., from three rats)/treatment group. xp < 0.001 vs. control; ap < 0.001 vs. LPS-only rats (one-way ANOVA).
Discussion
We had hypothesized that surfactants have a potential therapeutic effect on ARDS, in part, via modulation of the formation/release of select cytokines. To validate this hypothesis, we investigated the therapeutic effect of both a natural (porcine) and synthetic surfactant, and also compared the effects between the two surfactants, on expression of pro-inflammatory cytokines in an LPS-generated ARDS model. The present study revealed that in fact each surfactant did impact on pro-/anti-inflammatory cytokine secretion in an LPS-induced ARDS rat model. The results also demonstrated that the porcine surfactant seemed to be the more potent agent with regard to preventing inflammatory outcomes in situ.
A hallmark of ARDS is widespread destruction of the alveolar epithelium and flooding of alveolar spaces with protein exudates containing large numbers of neutrophils (polymorpho-nuclear leukocytes [PMN]) (Janssen et al., Citation2011). Because of evidence linking PMN and lung injury, and the critical involvement of cytokines in recruitment of PMN (and other inflammatory cells/leukocytes) into the tissues, it was hoped that an analyses of cytokines in lung lavage fluid would provide clues about the mechanisms that regulate development of ARDS and suggest to us potential targets for modulation/therapeutic application.
Though most studies have focused on the potential importance of single cytokines, it is now recognized that a complex balance exists between pro- and anti-inflammatory cytokines, and that “cytokine balance” is a key concept in understanding the biological activity of cytokines in situ (Tutor et al., Citation1994). With respect to ARDS, Li et al. (Citation2011) found significant levels of TNFα and IL-1β in the lung fluids of patients who were at the onset of ARDS. Jacobs et al. (Citation1989) was the first to show that alveolar macrophages from patients with ARDS spontaneously released IL-1β, suggesting that these macrophages had been activated in the alveolar spaces. With respect to the other key cytokines that were analyzed here, MCP-1 (also termed a chemokine) is detectable in BALF at syndrome onset and persists in the lungs of patients with sustained ARDS (Baggiolini et al., Citation1997). MIP cytokine (again also termed a chemokine) was identified in BALF from patients studied on the first day of ARDS (Donnelly et al., Citation1997) and immunoreactive MIP is detectable in macrophages recovered from airspaces of ARDS patients. Further, MIP is detectable in the BALF of patients at risk for ARDS, and that the concentration of MIP increases in the lungs of patients with sustained ARDS (Martin et al., Citation1998).
It was unremarkable that expression of TNFα, IL-1β, and IFNγ in the lungs was affected by LPS instillation; this has been reported in too numerous investigations. Some portion of these increments could be explained, in part, by the observed increases in lung levels of MCP-1 (a cyto-/chemokine that regulates recruitment of monocytes that can be subsequently activated), and of MIP-1β (a cyto-/chemokine that can activate granulocytes and induce synthesis\release of IL-1, IL-6, and TNFα from local fibroblasts and macrophages). Alveolar macrophages, a major source of MCP-1, likely responded directly to the LPS in the treated rats. It is also possible that other local cells also may have produced MCP-1, but mainly in response to increasingly-present TNFα and IL-1β (Yamada et al., Citation2011). Like MCP-1, the increased presence of lung MIP-1 was likely a result of production by LPS-activated macrophages; however, it is also possible airway epithelial cells released this product in response to the increased presence of the other induced cytokines, even potentially MCP-1 (Becker and Soukup, Citation1999). In general, the results of the current studies showing the increased levels of each of the measured cyto-/chemokines (except IL-4) in response to LPS build upon the earlier findings linking together LPS (models), cytokines, and ARDS.
Upon reflection, it should not have been a surprise to us that in the 72-h timeframe, induction of IL-4 did not appear to ‘yet’ become significant. It is possible this period was too long to have picked up on any LPS-induced increase in this one cytokine. Zhang et al. (Citation2004) noted that after LPS instillation, peak increases in IL-4 occurred within 2 h. Similarly, Yeh et al (Citation2007) found a small albeit significant increase in BAL IL-4 content 24 hr after LPS instillation into the lungs of mice. Though our study may have failed to detect the ‘early’ IL-4 increase, it is of interest to note that Zhang et al. (Citation2004) and Hocke et al. (Citation2006) each suggested that initial over-expression of this anti-inflammatory cytokine in fact led to its ultimate action as a promoter and/or amplifier of inflammatory processes (i.e., now pro-inflammatory) in situ. In their paper, Hocke et al. restated the fact that increases in IL-4 expression are correlated with extracellular matrix deposition and fibrosis, and the mast cell recruitment/activation and B-cell differentiation critical to allergic disease. These authors also noted that in the late phase of ARDS, remodeling processes often lead to unregulated fibro-proliferation and fibrosis; while IL-10 (another anti-inflammatory cytokine) was assumed to contribute to both, it was preseumed that IL-4 only acted to stimulate fibroblast collagen production. To our knowledge, no prior data are available on BALF IL-4 level in clinical ARDS or animal models, and a clear role for IL-4 in ARDS remains to be defined.
Taken together, it is clear that select cytokines/chemokines likely have a role in the onset, progression, and/or persistence of ARDS. Currently, there are no specific effective therapies for ARDS. We had summarized that one potential therapeutic approach could be through the use of surfactant as this product constitutes a unique local immunoregulatory system. Specifically, surfactant modulates several inflammation-associated processes, including cell proliferation and the release of inflammatory mediators. Both synthetic and natural surfactants have been shown to inhibit the production of IL-1β, IL-6, and prostaglandin-E2 by alveolar macrophages (Thomassen et al., Citation1996). Surfactant-associated protein A (SP-A) has been shown to inhibit LPS-induced TNFα production by animal (McIntosh et al., Citation1996) and human (Borron et al., Citation2000) alveolar macrophages. Lung fibroblasts are similarly affected by surfactants, strongly suggesting that surfactants modulate cytokine production, in part, by downregulating DNA synthesis and second-messenger production (Balibrea and Arias-Díaz, Citation2003). A role for SP-A in the down-regulation of lung inflammation in situ has also been suggested by a study that used SP-A-deficient mice; these hosts were found to have increased inflammatory cytokine formation and/or release in response to an infectious insult compared to levels seen in wild-type mice (Arias-Diaz et al., Citation2000).
The results of the study here indicate that the expression of all the pro-inflammatory cytokines studied here (except IL-4) were higher as a result of the LPS treatment and that these increments were suppressed by treatment with either natural (porcine) or synthetic surfactants. In recent studies (Mittal and Sanyal, Citation2009, Citation2010a,Citationc, Citation2011), we reported that changes in ROS generation, levels of apoptosis, mitochondrial membrane potential, and calcium homeostasis in the lungs were also increased by LPS treatment and, that these too were mitigated by treatment with natural or synthetic surfactant. Lastly, we note that the original LPS-induced changes in particular cytokines/chemokine expression here mirrored those in the presence of certain types of (undefined) effector cells. These outcomes with respect to individual cell type presence indicated that the findings here are suggestive of quantitative rather than qualitative changes that were induced by the surfactants. At this time, it is not possible to conclude if the surfactants induced changes in the expression of the cytokines/chemokines and this led to the changes in effector cell presence or if the surfactants caused effects on the cells that in turn led to their decreased abilitiy to release the pro-inflammatory proteins. Ongoing investigations in our laboratory will seek to clarify this important series of points.
Despite not knowing the precise mechanism of action, it can be noted here that among the two surfactants tested, porcine surfactant was found to be more effective form in mitigating the induction of inflammatory cytokines/chemokines by LPS. Although this infomration could help guide investigators that are seeking to develop therapeutic regimens for treatment of ARDS, there are other complications to consider first. One key problem is that during ARDS, there may be ongoing degradation of surfactant apoproteins in the areas of lung with the most intense inflammation. As such, supranormal quantities of the surfactant apoproteins may need to be included in any surfactant (replacement) mixture to act as a reserve and to supplement existing surfactant in those alveoli (Kesecioglu et al., Citation2001). In addition, on the basis of the results here, it could be that a non-apoprotein-containing surfactant would only have limited efficacy in ARDS. Last, this novel therapeutic approach could potentially also be hampered by costs, i.e., natural surfactants are very expensive. For this reason alone, more synthetic surfactants need to be developed that have significant anti-inflammatory effects on par with the natural form(s), in order to ultimately suitable and affordable for the treatment of ARDS.
In summary, cytokine measurements in patients before and after the onset of ARDS have provided valuable insights about the complexity of the inflammatory response that occurs in the lungs. The present investigation, which used preparations bearing surfactant apoproteins directly instilled into the lung to mitigate the inflammatory effects of LPS, suggests that such therapy may have a meaningful impact on the course and outcome of ARDS.
Acknowledgements
The present work is supported by Indian Council of Medical Research (ICMR) New Delhi, India (Ref. No. 61/5/2005).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Arias-Diaz, J., Garcia-Verdugo, I., Casals, C., Sanchez-Rico, N., Vara, E. and Balibrea, J. L. 2000. Effect of surfactant protein A (SP-A) on the production of cytokines by human pulmonary macrophages. Shock. 14:300–306.
- Baggiolini, M., Dewald, B. and Moser, B. 1997. Human chemokines: An update. Annu Rev Immunol. 15:675–705.
- Balibrea, J. L. and Arias-Díaz, J. 2003. Acute respiratory distress syndrome in the septic surgical patient. World J Surg. 27:1275–1284.
- Bastarache, J. A. and Blackwell, T. S. 2009. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech. 2:218–223.
- Becker, S. and Soukup, J. M. 1999. Exposure to urban air particulates alters the macrophage-mediated inflammatory response to respiratory viral infection. J Toxicol Environ Health Part A. 57:445–457.
- Beck-Schimmer, B., Schwendener, R., Pasch, T., Reyes, L., Booy, C. and Schimmer, R. C. 2005. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respir Res. 6:61.
- Borron, P., McIntosh, J. C., Korfhagen, T. R., Whitsett, J. A., Taylor, J. and Wright, J. R. 2000. Surfactant-associated protein A inhibits LPS-induced cytokine and nitric oxide production in vivo. Am J Physiol Lung Cell Mol Physiol. 278:L840–L847.
- Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72:248–254.
- Chabot, S., Salez, L., McCormack, F. X., Touqui, L. and Chignard, M. 2003. Surfactant protein A inhibits lipopolysaccharide-induced in vivo production of interleukin-10 by mononuclear phagocytes during lung inflammation. Am J Respir Cell Mol Biol. 28:347–353.
- Chen, H., Bai, C. and Wang, X. 2010. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev Respir Med. 4:773–783.
- Donnelly, S. C., Haslett, C., Reid, P. T., Grant, I. S., Wallace, W. A., Metz, C. N., Bruce, L. J. and Bucala, R. 1997. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 3:320–323.
- Galani, V., Tatsaki, E., Bai, M., Kitsoulis, P., Lekka, M., Nakos, G. and Kanavaros, P. 2010. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An up-to-date cell-specific review. Pathol Res Pract. 206:145–150.
- Germann, P. G. and Häfner, D. 1998. A rat model of acute respiratory distress syndrome (ARDS): Part 1. Time dependency of histological and pathological changes. J Pharmacol Toxicol Methods. 40:101–107.
- Gregory, T. J., Longmore, W. J., Moxley, M. A., Whitsett, J. A., Reed, C. R., Fowler, A. A. 3rd, Hudson, L. D., Maunder, R. J., Crim, C., and Hyers, T. M. 1991. Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest. 88:1976–1981.
- Hocke, A. C., Ermert, M., Althoff, A., Brell, B., N’Guessan, P. D., Suttorp, N. and Ermert, L. 2006. Regulation of interleukin IL-4, IL-13, IL-10, and their downstream components in lipopolysaccharide-exposed rat lungs. Comparison of the constitutive expression between rats and humans. Cytokine. 33:199–211.
- Jacobs, R. F., Tabor, D. R., Burks, A. W. and Campbell, G. D. 1989. Elevated interleukin-1 release by human alveolar macrophages during the adult respiratory distress syndrome. Am Rev Respir Dis. 140:1686–1692.
- Janssen, W. J., Barthel, L., Muldrow, A., Oberley-Deegan, R. E., Kearns, M. T., Jakubzick, C. and Henson, P. M. 2011. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med. (In press).
- Kinoshita, M., Mochizuki, H. and Ono, S. 1999. Pulmonary neutrophil accumulation following human endotoxemia. Chest. 116:1709–1715.
- Kesecioglu, J., Schultz, M. J., Lundberg, D., Lauven, P. M., and Lachmann, B. 2001. Treatment of acute lung injury (ALI/ARDS) with surfactant. Am J Respir Crit Care Med. 163:A819.
- Li, S. L., Wu, Z. H., Zhang, S. Q., Qin, C. P., Li, T., Chen, Z. T., Jin, J. S., MA, S. F. and Li, J. 2011. The changes in secretory function of pulmonary intravascular macrophages after challenge of lipopoly saccharide. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 23:176–178.
- Lin, W. C., Lin, C. F., Chen, C. L., Chen, C. W. and Lin, Y. S. 2010. Prediction of outcome in patients with acute respiratory distress syndrome by bronchoalveolar lavage inflammatory mediators. Exp Biol Med (Maywood.) 235:57–65.
- Ma, X., Xu, D., Ai, Y., Ming, G. and Zhao, S. 2010. Fas inhibition attenuates lipopolysaccharide-induced apoptosis and cytokine release of rat Type II alveolar epithelial cells. Mol Biol Rep. 37:3051–3056.
- Martin, T. R. 1997. Cytokines and the acute respiratory distress syndrome (ARDS): A question of balance. Nat Med. 3:272–273.
- Martin, T. R., Donnelly, S. C., Steinberg, K. P., Ruzinski, J. T., Metz, C. N., Goodman, R. B., Hudson, L. D., and Bucala, R. 1998. Macrophage inhibitory factor (MIF) in the lungs of patients with acute respiratory distress syndrome (ARDS). Am J Respir Crit Care Med. 157:A459.
- Matute-Bello, G., Frevert, C. W. and Martin, T. R. 2008. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 295:L379–L399.
- McCormack, F. 1997. The structure and function of surfactant protein-A. Chest. 111:114S–119S.
- McIntosh, J. C., Mervin-Blake, S., Conner, E. and Wright, J. R. 1996. Surfactant protein A protects growing cells and reduces TNF-α activity from LPS-stimulated macrophages. Am J Physiol. 271:L310–L319.
- Mitchell, R. S. 1999. Lung cytokines and ARDS. Chest. 116:2S–8S.
- Mittal, N. and Sanyal, S. N. 2009. Exogenous surfactant suppresses inflammation in experimental endotoxin-induced lung injury. J Environ Pathol Toxicol Oncol. 28:341–349.
- Mittal, N., and Sanyal, S. N. 2010a. Cyclooxygenase inhibition enhances the effects of surfactant therapy in endotoxin induced rat model of ARDS. Inflammation; 34:92–98.
- Mittal, N., and Sanyal, S. N. 2010b. Immunomodulatory properties of exogenous surfactant in adult rat alveolar macrophages. Immunopharm Immunotoxicol. 32:153–159.
- Mittal, N., and Sanyal, S. N. 2010c. Intratracheal instillation of surfactant inhibits LPS-induced acute respiratory distress syndrome in rats. Am J Biomed Sci. 2:190–201.
- Mittal, N., and Sanyal, S. N. 2011. Exogenous surfactant prevents the mitochondrial depolarization and changes in Ca2+ homeostasis due to oxidative stress leads to apoptosis in experimental lung injury. Inst Integr Omics Biotech. (In press).
- Mirzapoiazova, T., Kolosova, I. A., Moreno, L., Sammani, S., Garcia, J. G. and Verin, A. D. 2007. Suppression of endotoxin-induced inflammation by taxol. Eur Respir J. 30:429–435.
- Pittet, J. F., Mackersie, R. C., Martin, T. R. and Matthay, M. A. 1997. Biological markers of acute lung injury: Prognostic and pathogenetic significance. Am J Respir Crit Care Med. 155:1187–1205.
- Pugin, J., Ricou, B., Steinberg, K. P., Suter, P. M. and Martin, T. R. 1996. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med. 153:1850–1856.
- Temmesfeld-Wollbrück, B., Walmrath, D., Grimminger, F. and Seeger, W. 1995. Prevention and therapy of the adult respiratory distress syndrome. Lung. 173:139–164.
- Thomassen, M. J., Antal, J. M., Barna, B. P., Divis, L. T., Meeker, D. P. and Wiedemann, H. P. 1996. Surfactant down-regulates synthesis of DNA and inflammatory mediators in normal human lung fibroblasts. Am J Physiol. 270:L159–L163.
- Tomashefski, J. F. Jr. 2000. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 21:435–466.
- Tutor, J. D., Mason, C. M., Dobard, E., Beckerman, R. C., Summer, W. R. and Nelson, S. 1994. Loss of compartmentalization of alveolar tumor necrosis factor after lung injury. Am J Respir Crit Care Med. 149:1107–1111.
- Warren, H. S., Fitting, C., Hoff, E., Adib-Conquy, M., Beasley-Topliffe, L., Tesini, B., Liang, X., Valentine, C., Hellman, J., Hayden, D. and Cavaillon, J. M. 2010. Resilience to bacterial infection: Difference between species could be due to proteins in serum. J Infect Dis. 201:223–232.
- Yamada, M., Kubo, H., Kobayashi, S., Ishizawa, K., He, M., Suzuki, T., Fujino, N., Kunishima, H., Hatta, M., Nishimaki, K., Aoyagi, T., Tokuda, K., Kitagawa, M., Yano, H., Tamamura, H., Fujii, N. and Kaku, M. 2011. The increase in surface CXCR4 expression on lung extravascular neutrophils and its effects on neutrophils during endotoxin-induced lung injury. Cell Mol Immunol. ( In press).
- Yeh, C. C., Kao, S. J., Lin, C. C., Wang, S. D., Liu, C. J. and Kao, S. T. 2007. The immunomodulation of endotoxin-induced acute lung injury by hesperidin in vivo and in vitro. Life Sci. 80:1821–1831.
- Zhang, Q., Li, Q., Mao, B. L., Qian, G. S., Xu, J. C. and Chen, Z. T. 2004. Studies on the expression of mRNA of anti- and pro-inflammatory cytokines in acute lung injury induced by lipopolysaccharide in rat. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 16:585–588.